

1 Introduction
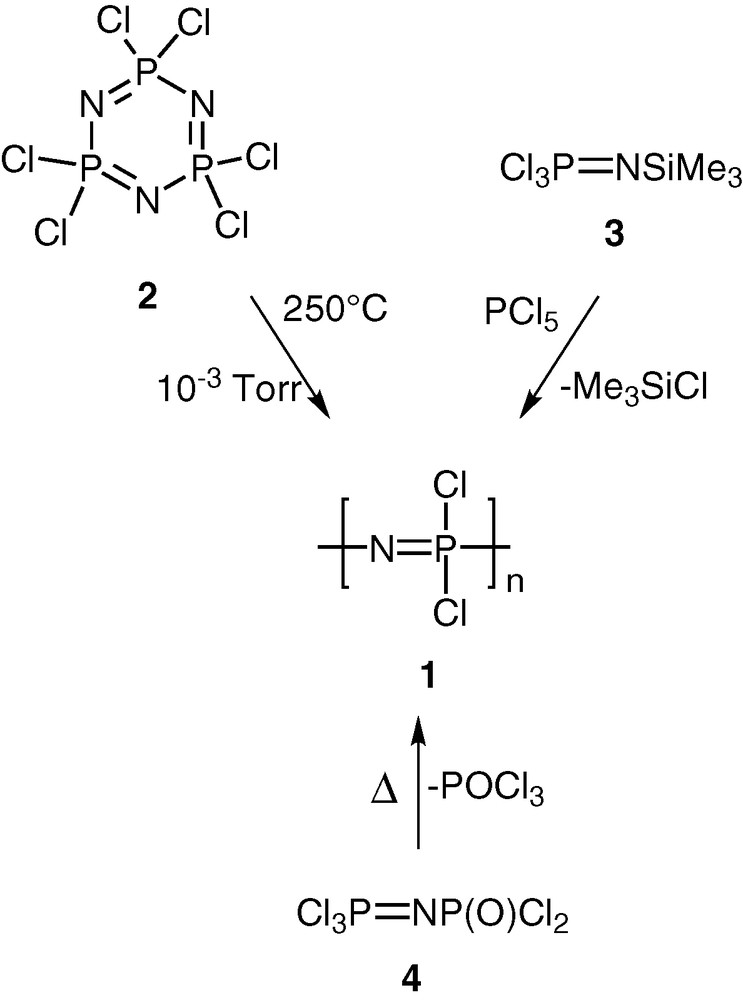
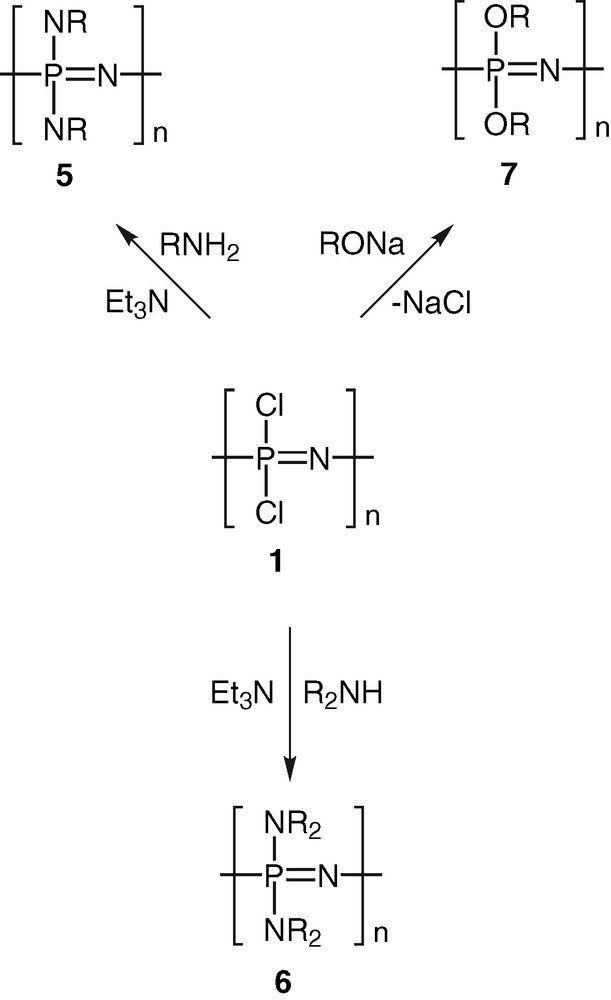
Polyphosphazenes, [N=PR2]n, are among the most important and well-studied hybrid polymeric systems [1–10]. Unlike organic polymers, these contain an inorganic backbone in the form of alternating phosphorus and nitrogen atoms. The side chains, which are usually organic groups, are exclusively present on the phosphorus centres. Most polyphosphazenes are derived from the parent polymer viz., polydichlorophosphazene [N=PCl2]n (1). The latter is obtained by any of the following two methods, (a) a ring opening polymerisation of the six-membered ring, N3P3Cl6 (2) [11,12], (b) condensation reactions involving the phosphoranimine Cl3P=NSiMe3 (3) [13,14] or Cl3P=NP(O)Cl2 (4) [15,16] (Scheme 1). Although polydichlorophosphazene [N=PCl2]n (1) is a high polymer with the repeat unit ranging up to n = 15 000, its hydrolytic sensitivity due to the labile P–Cl bonds renders this polymer useless for any practical applications. However, this major disadvantage can be turned around into an asset. Thus, the reaction of [N=PCl2]n with a variety of nucleophiles such as amines, alcohols or phenols results in the substitution of the P–Cl units and the formation of stable polyphosphazenes (Scheme 2). More than 800 polymers have been prepared by the utilization of this method. Polyphosphazenes containing P–C bonds cannot, however, be prepared in this way and have to be assembled by the use of an alternative route. This involves the thermolysis of phosphoranimine monomers such as R2(OR)P=N(SiMe3) [17–19] (Scheme 3).
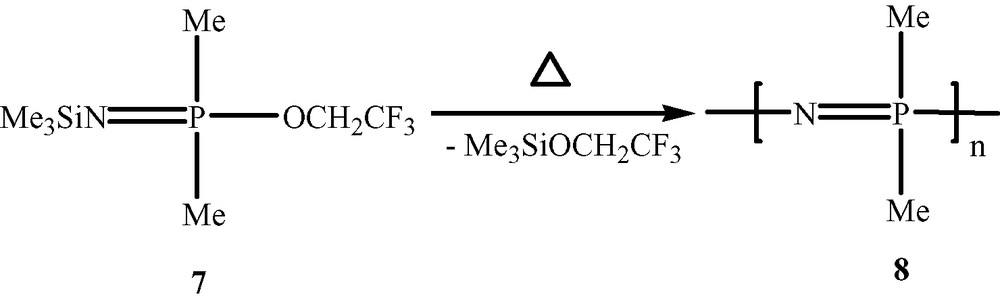
Another type of related hybrid polymers contains an intact cyclophosphazene ring as a pendant group attached at regular intervals to the backbone of an organic polymer [20–22]. These polymers can be prepared by polymerising the vinyl group in cyclophosphazene monomers such as N3P3R5(O-R′-CH=CH2) (9) (Scheme 4).
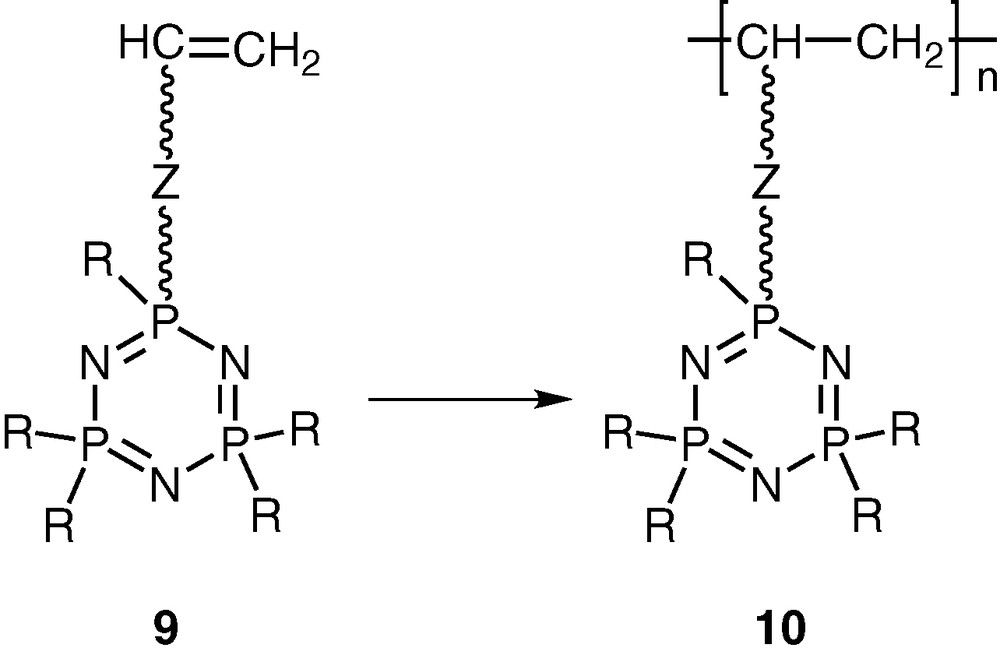
Although polymers such as 10 have been much less studied than polyphosphazenes, in principle they offer as much versatility as the former in terms of tunability of polymer function and property. In particular, these types of polymers are suitable for the design of a new family of polymeric ligands. This article summarizes our recent research efforts in this area.
2 Cyclophosphazene ligands
Cyclophosphazenes are potential ligands for coordination to transition metal ions [23–27]. The lone pair of electrons on the skeletal nitrogen atoms of the cyclophosphazene ring allows the possibility of using these rings for interaction with transition metal ions. The versatility of the cyclophosphazene ligands can be enhanced by incorporating suitable coordinating groups in the periphery of the inorganic ring system. A large variety of cyclophosphazene ligands with diverse coordination capabilities have been assembled using this approach (Schemes 5 and 6).
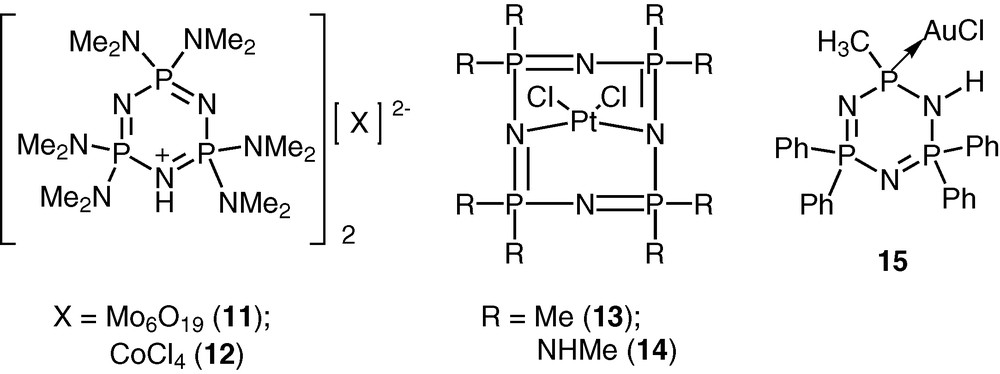
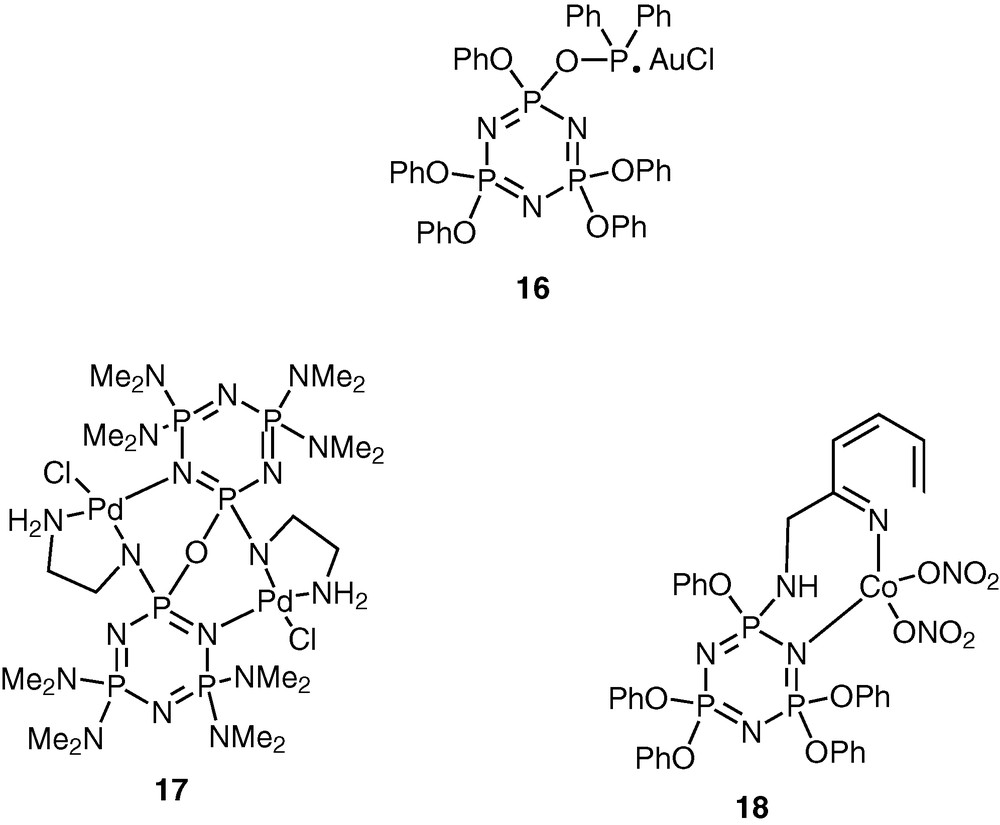
The advantage of the cyclophosphazene ligands containing exocyclic donor groups is that these can be readily adapted to the polymeric systems. This approach will be exemplified in the following sections.
3 Pyrazolyl cyclophosphazene–containing polymers
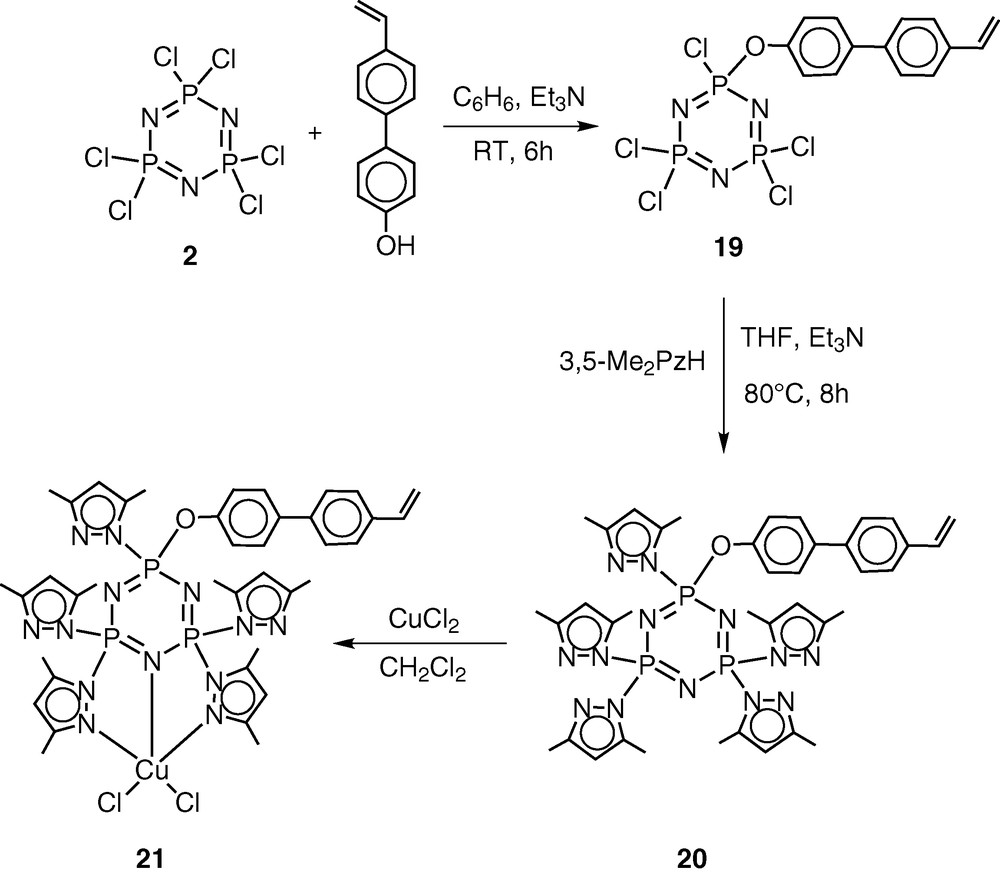
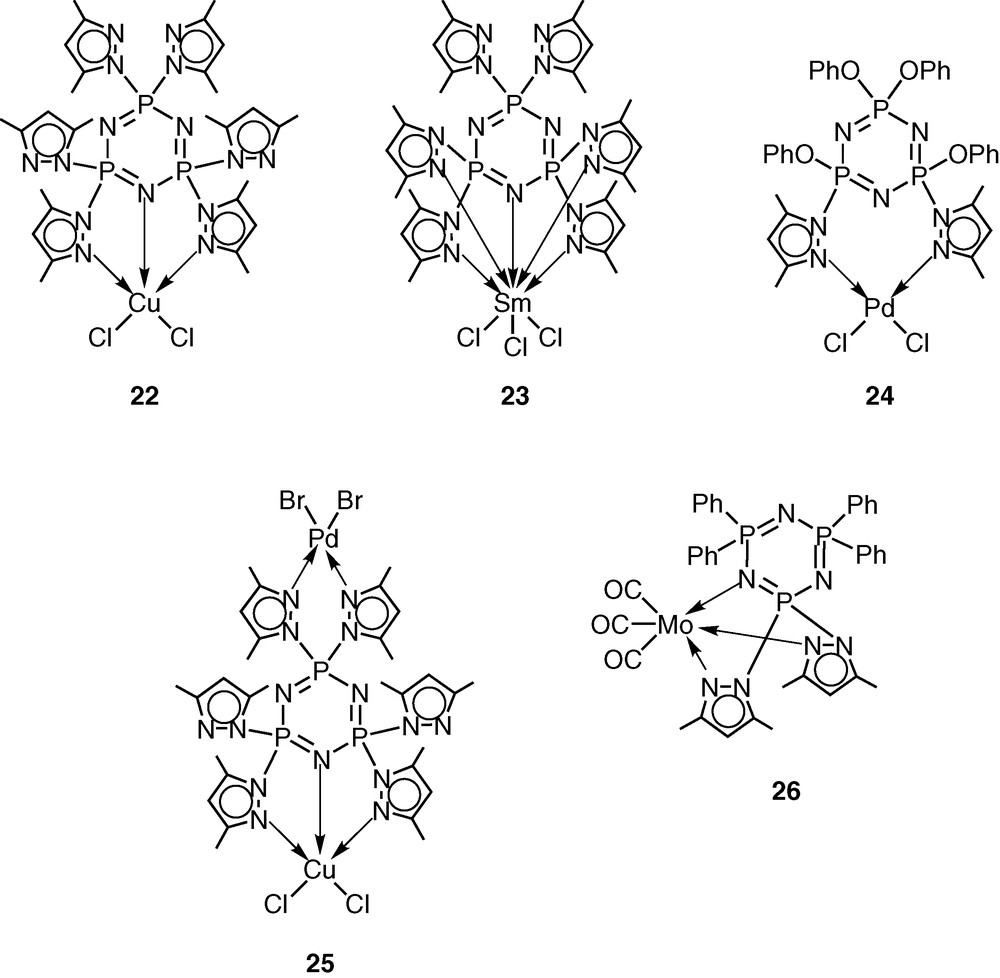
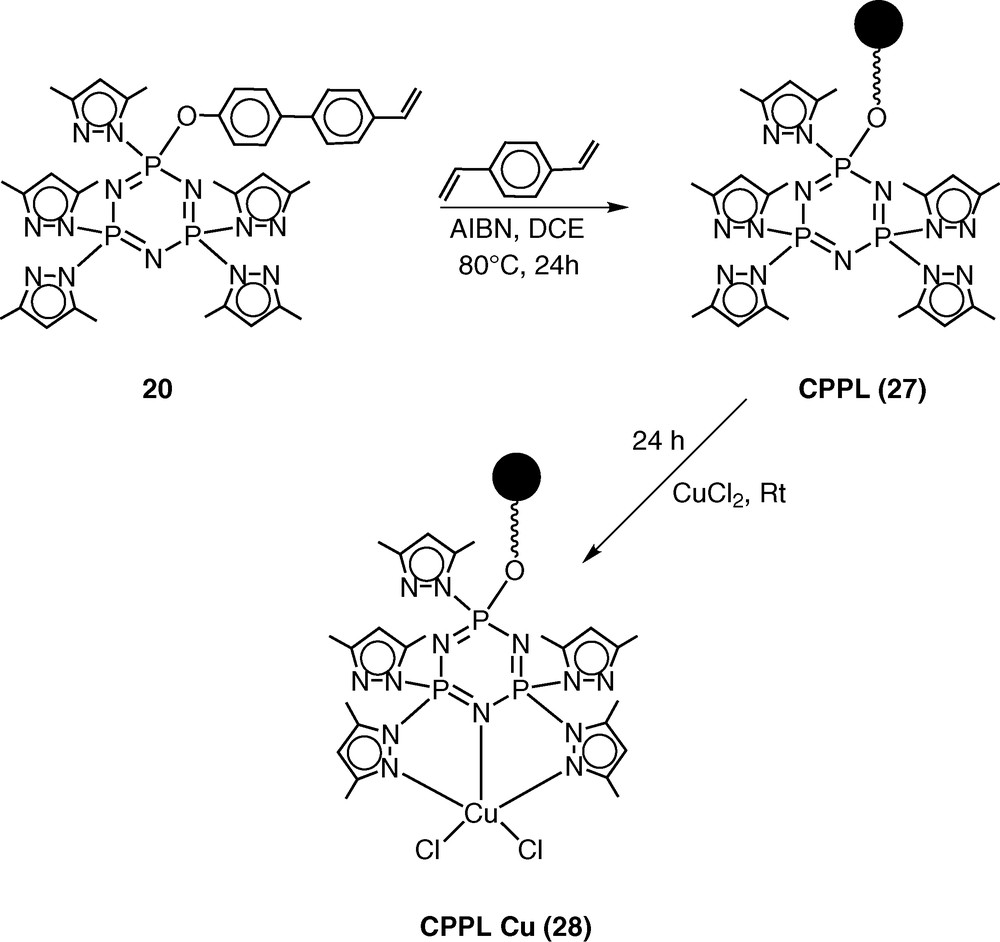
The pentachlorocyclotriphosphazene N3P3Cl5(O–C6H4–p-C6H4–p-CH=CH2) (19) has previously been shown by Inoue [28–30] and us [31] to be an excellent monomer in terms of its ease of preparation, hydrolytic stability and ready polymerisability. An additional advantage of this monomer is the presence of five reactive P–Cl bonds, which can be readily replaced by a variety of nucleophiles. Using this feature, we prepared the pyrazolyl derivative N3P3(3,5-Me2Pz)5(O–C6H4–p-C6H4–p-CH=CH2) (20) by the reaction of 19 with 3,5-dimethylpyrazole in the presence of triethylamine as the hydrogen chloride scavenger [32] (Scheme 7). It had been shown previously that pyrazolyl cyclophosphazenes such as N3P3(3,5-Me2Pz)6 or gem-N3P3Ph2(3,5-Me2Pz)4 are extremely versatile multi-site coordination ligands towards several types of transition metal ions as well as of metal carbonyls, such as Mo(CO)6 [33–37] (Scheme 8). In view of the proven coordination behaviour of these ligands, we have chosen to incorporate the pyrazolyl moiety in the monomer 19. The ligand 20 readily reacts with copper(II)chloride to afford the 1:1 complex N3P3(3,5-Me2Pz)5(O–C6H4–p-C6H4–p-CH=CH2).CuCl2 (21) (Scheme 7).
Monomer 20 has been co-polymerised in the presence of divinylbenzene to afford a cross-linked polymer CPPL (27) (Scheme 9). Metallation of CPPL (27) with copper(II)chloride occurs readily to afford the metalated polymer CPPL-Cu (28) [38] (Scheme 9).
3.1 Hydrolysis studies of pNPP, bNPP and hNPP with CPPL-Cu
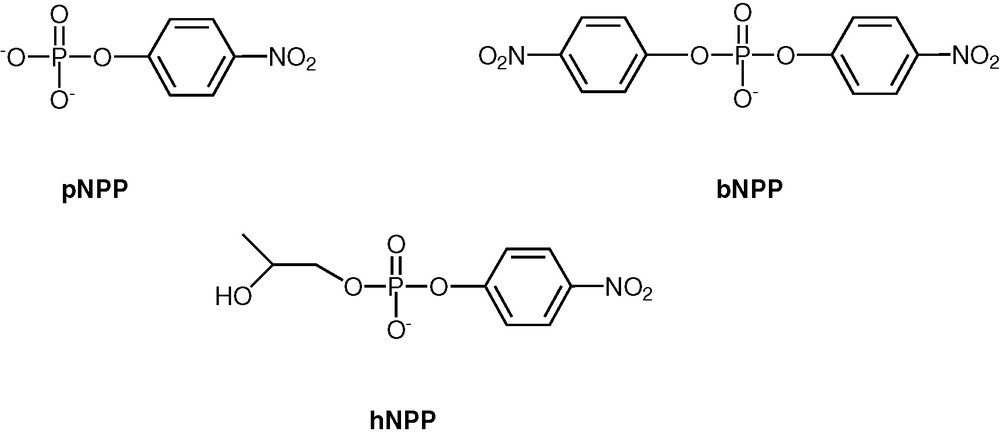
To screen the utility of CPPL–Cu on phosphoesterase activity, the hydrolysis reactions of three model substrates, p-nitrophenyl phosphate (pNPP), bis(p-nitrophenyl)phosphate (bNPP) and 2-hydroxypropyl–p-nitrophenyl phosphate (hNPP) (Scheme 10) were studied [38]. While pNPP is a model for phosphomonoester, bNPP acts as a phosphodiester substrate. Both of these are examples where the hydrolytic reaction is triggered in an intermolecular manner by an external nucleophile. The substrate hNPP is a phosphodiester and is also a model for RNA in that the 2′-OH position of the latter is structurally mimicked. Our results on this substrate show an intramolecular hydrolytic reaction, through the activation of an internal hydroxyl group (vide infra). Hydrolysis of all the three substrates pNPP, bNPP and hNPP in the presence of CPPL-Cu was followed in 50% aqueous methanol/0.01 M N-ethylmorpholine buffer at pH 8.0 by time-dependent release of the p-nitrophenolate anion at 400 nm.
Rate constants for the hydrolysis of all these substrates by CPPL–Cu were determined under pseudo-first-order conditions. The hydrolysis proceeds with considerable rate enhancement relative to that of the uncatalysed [39,40] reaction for all three substrates (Table 1).
Pseudo-first-order rate constants and krel for the hydrolysis of pNPP, bNPP and hNPP
| Substrate | kobs (min–1) | krel (kobs/kuncat) |
| pNPP | 4.42 × 10–4 | 8.98 × 102 |
| bNPP | 2.19 × 10–4 | 2.81 × 105 |
| hNPP | 5.31 × 10–3 | 2.68 × 103 |
A most promising feature of CPPL–Cu is that it is very robust and can be recycled several times [38]. Such behaviour augurs well for the design of other polymeric catalysts using this hybrid-polymer approach.
4 Phosphinylcyclotriphosphazene–containing pendant polymers
Heck arylation of olefins is an important C–C-bond-forming reaction and the use of polymer-supported catalysts for this reaction has not been very successful because of palladium leaching during the reaction. It was of interest to test the utility of hybrid polymers for this process. As shown above, 19 is a very good starting material for preparing coordinating ligands. It possesses five reactive chlorines and also a polymerisable vinyl group. In order to incorporate coordinating phosphine groups on the cyclophosphazene skeleton, the substrate HO–C6H4–p-PPh2 was selected for reaction. Accordingly, the reaction of 19 with HO–C6H4–p-PPh2 affords the fully substituted product 29 in very good yields (Scheme 11) [41].
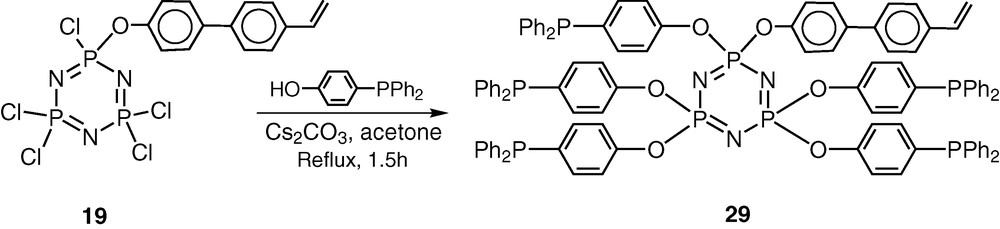
The choice of the reagent HO–C6H4–p-PPh2 to introduce the phosphino groups in the cyclophosphazene skeleton is based on the following considerations. The presence of the spacer group ensures that the coordinating phosphino groups are away from the cyclophosphazene ring. Secondly the structural motif –C6H4–p-PPh2 is similar to what is found in analogous Merrifield–Wang resins.
The monomer 29 has been co-polymerised in the presence of divinylbenzene to afford the cross-linked polymer CPPLP (30) (Scheme 12).
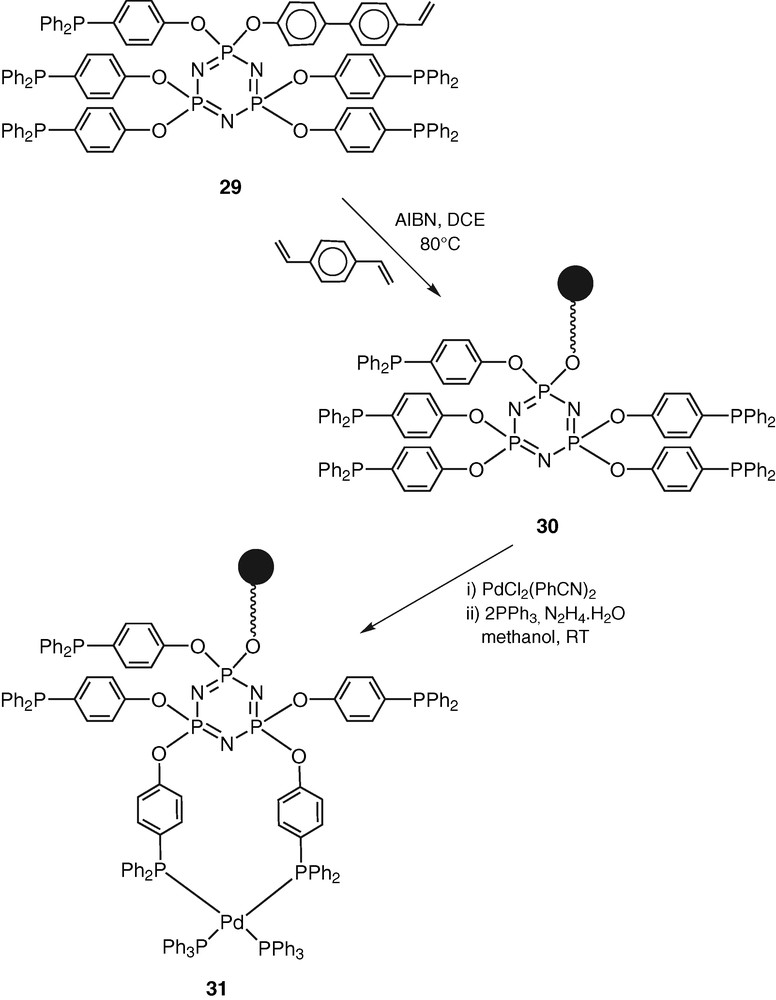
Compound 30 was used as a ligand for the preparation of a Pd(0) complex CPPLP-Pd (31) (Scheme 12). In order to ensure that the palladium incorporation in CPPLP-Pd is a result of coordination and not due to occlusion the latter was washed several times thoroughly with dichloromethane, methanol and acetone and subsequently dried in a vacuum oven [41].
4.1 Heck arylation with CPPLP–Pd
The catalytic activity of CPPLP-Pd towards the Heck arylation of the olefin reaction (Scheme 13) has been tested under the standard reaction conditions that are commonly employed for this reaction. We have carried out the reactions of iodobenzene with styrene (and with other vinyl substrates such as acrylonitrile or methacrylate) in the presence of tri-n-butylamine as the base and acetonitrile as the solvent while keeping the reaction temperature at 90 °C (Scheme 13). We have also tried using p-bromobenzene or p-chlorobenzene as aryl substrates. The results of our experiments are summarized in Table 2 [41].
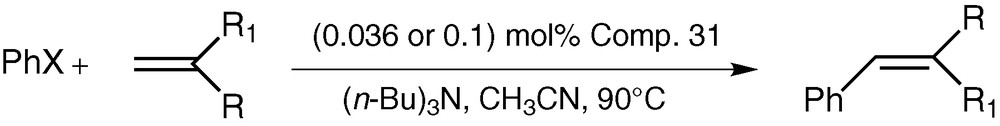
Heck arylation reaction catalysed by CPPLP–Pd
| S.No. | X | Catalyst (mol%) | R | R1 | Time (h) | Yield (%) (isolated) |
| 1 | I | 31 (0.036) | Ph | — | 4 | 93 |
| 2 | I | 31 (0.036) | COOMe | — | 5 | 99 |
| 3 | I | 31 (0.036) | COOEt | — | 5 | 98 |
| 4 | I | 31 (0.1) | COOMe | Me | 30 | 79 |
| 5 | I | 31 (0.1) | CN | — | 20 | no reaction |
| 6 | Br | 31 (0.1) | Ph | — | 70 | no reaction |
| 7 | Cl | 31 (0.1) | Ph | — | 48 | no reaction |
As seen from Table 2, although CPPLP–Pd is not effective with substrates such as acrylonitrile (entry 5) bromobenzene (entry 6) or chlorobenzene (entry 7); it was found to be quite efficient in catalysing the Heck reaction in other situations. The isolated yields of the products are quite high.
In order to optimise the reaction conditions, we have also carried out the reaction between iodobenzene and styrene in various solvents, in the presence of base (n-Bu)3N, at 90 °C. These results are given in Table 3.
Heck arylation reaction catalysed by CPPLP-Pd in various solvents
| S.No | Solvent | Time (h) | Isolated yield (%) |
| 1 | - | 4 | 78 |
| 2 | CH3CN | 4 | 93 |
| 3 | DMF | 4 | 96 |
| 4 | NMP | 6 | No reaction |
| 5 | Toluene | 6 | No reaction |
As can be seen from Table 3, the reaction proceeds very well in acetonitrile and in DMF (entries 2 and 3). In the absence of solvent, a yield of 78% was also recorded (entry 1). The reaction does not proceed when we use N-methyl pyrrolidine (NMP) or toluene as the solvent.
We have also tested the influence of various bases in the Heck arylation reaction in the presence of CPPLP-Pd. These results are summarized in Table 4. The yield of the reaction is quantitative when the base used is tri-n-butyl amine [(n-Bu)3N] or triethyl amine.
Heck arylation catalysed by CPPLP–Pd in the presence of various bases
| S.No | Base | Solvent | Time (h) | Isolated yield(%) |
| 1 | (n-Bu)3N | CH3CN | 4 | 93 |
| 2 | NaHCO3 | CH3CN | 8 | 11 |
| 3 | Na2CO3 | CH3CN | 12 | No reaction |
| 4 | Et3N | CH3CN | 4 | 86 |
| 5 | Na2CO3 | DMF | 8 | 43 |
| 6 | NaHCO3a | NMP | 12 | No reaction |
a The reaction was carried out at 120 °C.
We have also tested the possibility of recycling the CPPLP–Pd catalyst. It has been found that the heterogeneous catalyst CPPLP–Pd is robust with minimal palladium leaching. This polymeric catalyst can be recycled several times without affecting the catalytic activity.
5 Hydrazone-appended cyclotriphosphazene containing pendant polymers
The above methodology of the hybrid polymer system has been extended to synthesize hydrazone-based derivatives. We and others have shown that hydrazine-based phosphorus ligands are quite versatile in terms of their interaction with transition metals as well as of their behaviour as building blocks for dendrimers [42–49]. The reaction of 19 with 10 equiv of N-methylhydrazine results in the complete substitution of the chlorine atoms affording N3P3[N(Me)NH2]5[O–C6H4–p-C6H4–p-CH=CH2] 32 in an excellent yield (Scheme 14). This reaction is regiospecific; the N-methyl end of the difunctional reagent is found to exclusively attach to the phosphorus atom [42–45].
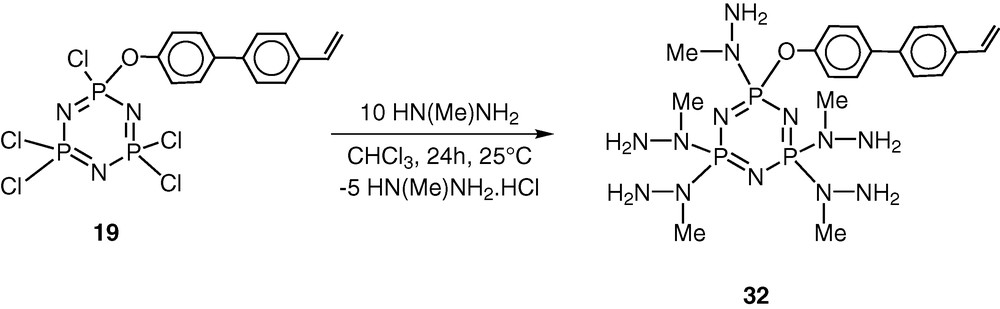
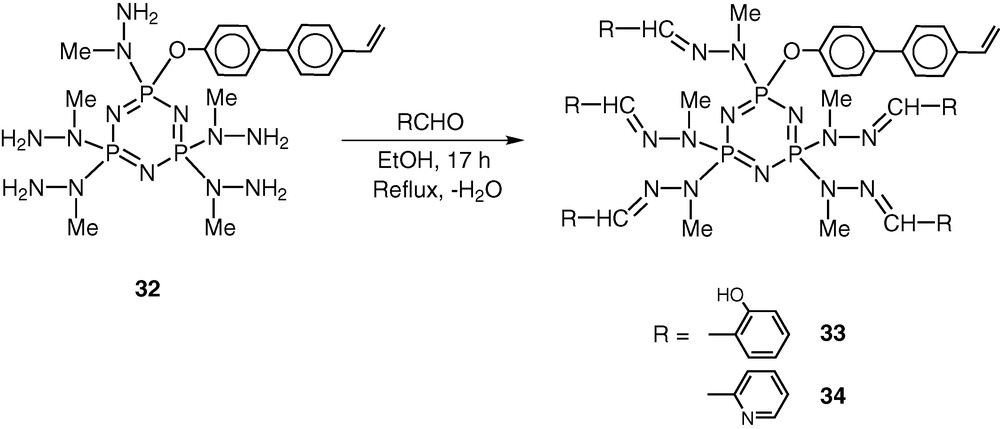
The most important feature of the reaction of 19 with N-methylhydrazine is that the product 32 still contains five reactive groups in the form of peripheral –NH2 units. Also, the polymerisable vinyl group remains intact in the conversion of 19 to 32. Condensation of 32 with 5 equiv of o-hydroxybenzaldehyde proceeds in boiling ethanol to afford the hydrazone 33. Similarly condensation of 32 with pyridine-2-carboxaldehyde affords the hydrazone 33 [45] (Scheme 15).
Compounds 33 and 34 can be readily homopolymerized in dichloroethane by using AIBN as the initiator to afford polymers 35 and 36 (Scheme 16).
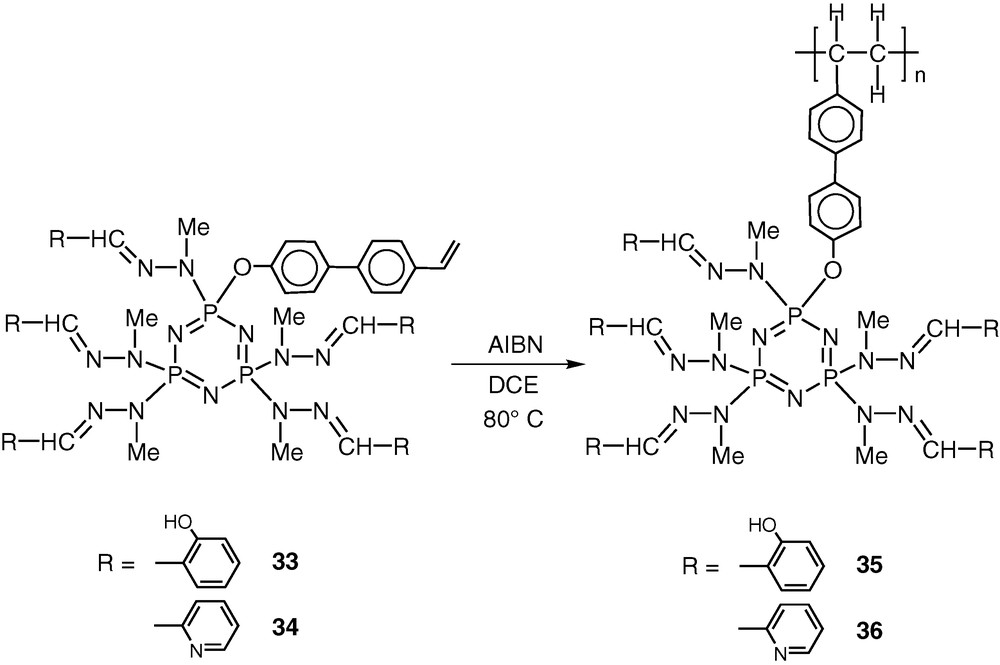
It is anticipated that these polymeric ligands would also be quite effective towards metalation reactions.
6 Conclusion
We have developed a general strategy for the preparation of polymeric ligands based on hybrid polymer systems. Our methodology utilizes the novel monomer N3P3Cl5(O–C6H4–p-C6H4–p-CH=CH2), which contains five replaceable chlorine atoms. Utilizing this compound, two different types of ligands, one containing pyrazolyl end groups and the other one containing phosphino end groups, have been prepared. These monomers can be converted into cross-linked polymeric resins by a co-polymerisation process involving 1,4-divinylbenzene as the co-monomer. These polymeric ligands have been metallated and the metal derivatives have been shown to be robust catalysts towards phosphate ester hydrolysis [Cu(II) complex] and the Heck arylation reaction [Pd(0) complex]. In view of the generality of this method, it is anticipated that a library of polymeric ligands based on this family should be readily assembled and utilized.
Acknowledgements
We thank MHRD (New Delhi) for financial support. VK and PT are thankful to CSIR (India) for SRF fellowships.