

1 Introduction
Although the direct co-condensation of unsaturated hydrocarbon ligands with metal vapours is a well-established route to generating organometallic compounds sometimes not normally accessible by more conventional techniques, we were the first to utilise the phospha-alkyne tBuCP to generate phospha-organometallic compounds.
Preliminary experiments [1]involving co-condensation of iron atoms with tBuCP yielded the already known pentaphosphaferrocene, [Fe(η5-P3C2tBu2)(η5-P2C3tBu3)] [2]. Interestingly, there was no evidence for the production of the known hexa-phospha-substituted compound [Fe(η5-P3C2tBu2)2], although both are easily prepared by treatment of FeCl2 with a mixture of LiP3C2tBu2/Li(P2C3tBu3) [2]. Similarly chromium atom co-condensation with tBuCP yields only the pentaphosphachromocene [Cr(η5-P3C2tBu2)(η5-P2C3tBu3)], even though the structurally related complex [Cr(η5-P3C2tBu2)2] can be prepared from the LiP3C2tBu2) salt [3]. We also have reported the fully structurally characterised paramagnetic vanadium compound [V(η5-P3C2tBu2)(η5-P2C3tBu3)] via metal-vapour synthesis (Fig. 1) [4].
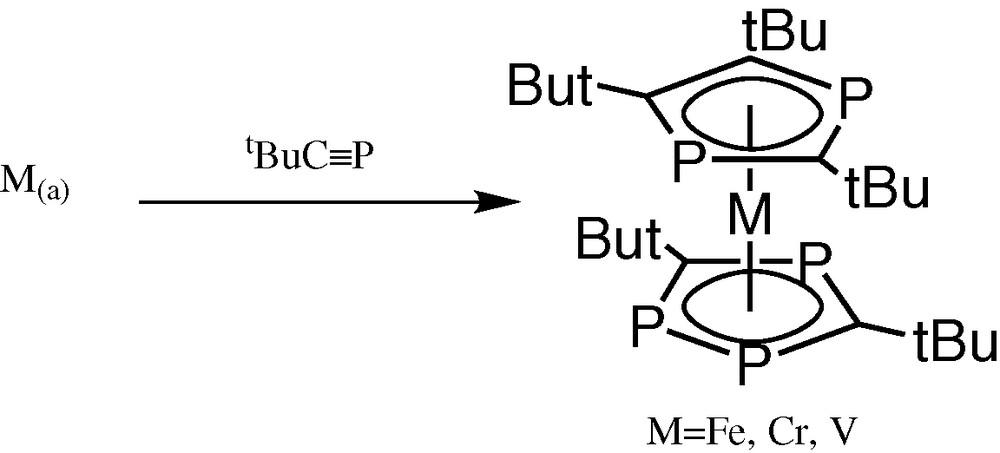
Metal-vapour synthesis of the paramagnetic vanadium compound [V(η5-P3C2tBu2)(η5-P2C3tBu3)].
Co-condensation of molybdenum or tungsten atoms with tBuCP on the other hand unexpectedly afforded red crystalline compounds of molecular formula [M(tBuCP)6] (M = W, Mo), indicative of the assembly of six rather than five phospha-alkyne units around the metal centre affording an 18-electron diamagnetic compound. Contrary to expectations, the compounds formed were not the bis(η−triphosphabenzene) sandwich complexes [M(η6-P3C3tBu3)3], but shown to be the homoleptic tris(1,3-diphosphacyclobutadiene) complexes [M(η4-P2C2tBu2)3] (M=W, Mo) (Fig. 2) [5].
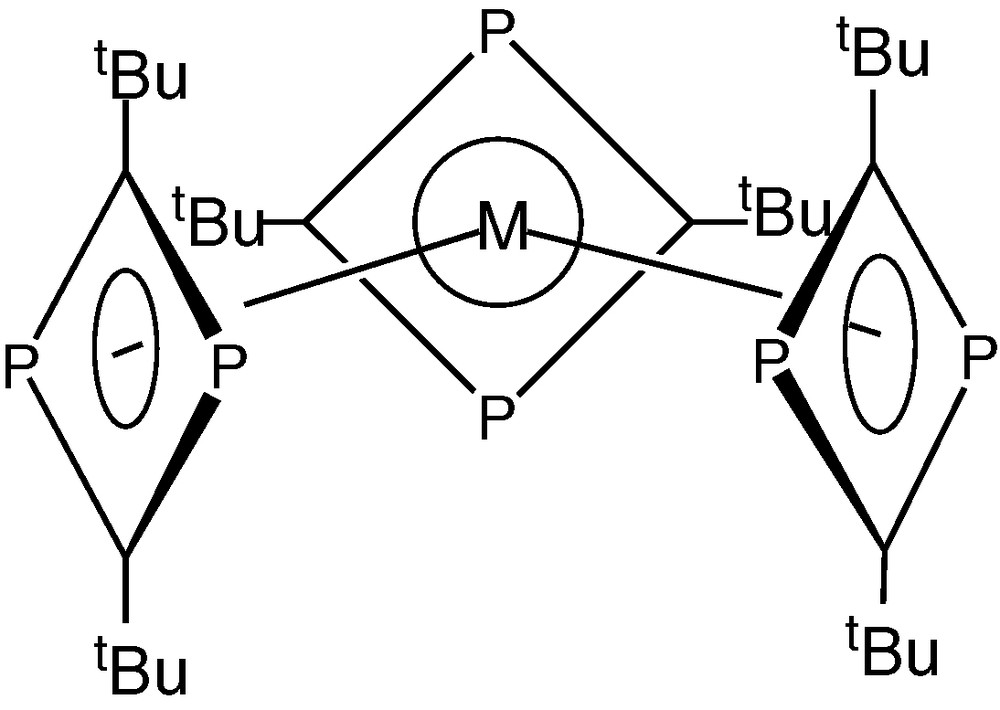
Molecular structure of [M(η4-P2C2tBu2)3] (M=W, Mo).
Subsequently, we prepared the first triple-decker sandwich complex containing the 1,3,5-triphosphabenzene ligand [Sc2(η5-P3C2tBu2)2(μ,η6:η6-P3C3tBu3)] by co-condensation of tBuCP with scandium vapour and the novel homoleptic complex [Sc(η5-P2C3tBu3)2] (Fig. 3) [7].
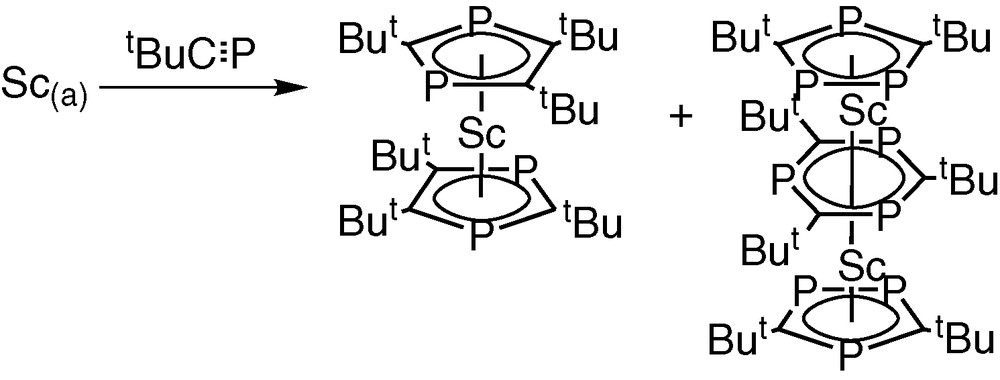
Preparation of the first triple-decker sandwich complex containing the 1,3,5-triphosphabenzene ligand [Sc2(η5-P3C2tBu2)2(µ,η6:η6-P3C3tBu3)] by co-condensation of tBuCP with scandium vapour and the novel homoleptic complex [Sc(η5-P2C3tBu3)2].
Since we were interested in a better understanding of the nature of the phosphorus ring products formed in these types of co-condensation of metal atoms with the phospha-alkyne we extended our studies to cobalt and nickel. We recently described the formation of three complexes [Co(η5-P3C2tBu2)(η5-P2C2tBu2)], [Co(η5-P2C3tBu3)(η4-P2C2tBu2)] containing both ligated 4- and 5-membered phosphorus ring systems, as well as the unusual [Co(η4-P4C4tBu4H)(η4-P2C2tBu2)], containing a protonated tetraphosphabarrelene ligand from the cobalt reaction [8]]. We now describe in a full paper our results using Ni, which leads to the first fully structurally characterised example of a complex containing the phosphirenyl cation (a preliminary report of part of this work has appeared [9]).
2 Results and discussion
Co-condensation of electron beam generated nickel atoms with tBuCP affords a black, petrol-soluble solid. Chromatographic work-up in hexane afforded roughly equal amounts of two reaction products 1 and 2, both having the molecular formula [NiP4C4tBu4], as evidenced by their mass spectra clearly indicating the involvement of four phosphaalkyne ligands in each case:
The simplicity of 1H and 31P{1H} NMR spectroscopic data for the orange crystalline solid 1, showed it to be the known sandwich compound bis(η4-2,4-di-tert-butyl-1,3-diphosphacyclobutadiene)nickel(0), made previously by Binger et al. by a completely different synthetic route involving the in situ reduction of a nickel(II) acac complex in the presence of tBuCP (Fig. 4) [10].
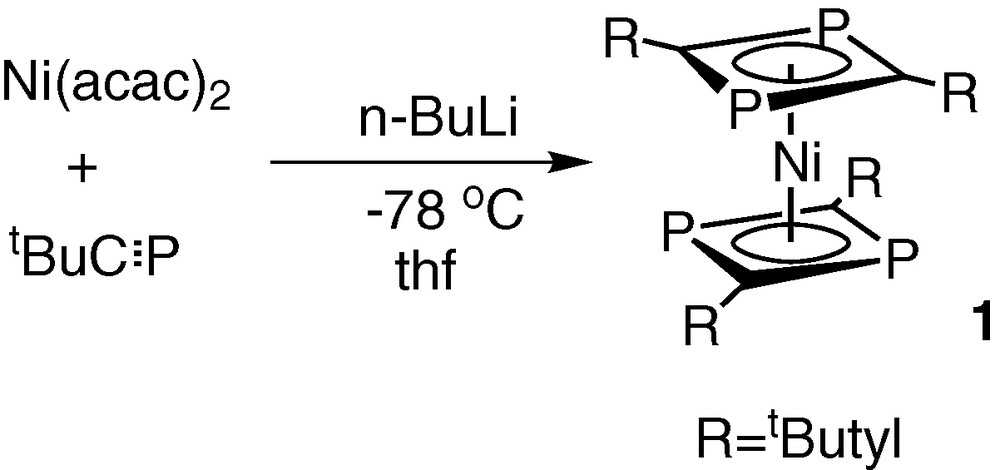
Synthetic route to the sandwich compound bis(η4-2,4-di-tert-butyl-1,3-diphosphacyclobutadiene)nickel(0) involving the in situ reduction of a nickel(II) acac complex in the presence of tBuCP.
The 1H NMR spectrum of 1 displays a singlet at δ = 1.2 ppm and 31P{1H} NMR spectroscopic data consists of a singlet at δ = 82.3 ppm, both in full agreement with the published spectrum [10].
The second nickel complex, 2, which was isolated as a red oil by sublimation (50 °C, 10–3 mbar), was found to be isomeric with 1. Mass spectral data show a characteristic mass ion at m/z 458 {[NiP4C4tBu4]}, but more interesting was the observation of the an ion at m/z 320, corresponding to the loss of a tBuCCtBu fragment that was not seen in the fragmentation pattern of 1.
The 13C{1H} NMR spectrum of 2 was particularly significant in the subsequent structural assignment, showing a complex multiplet centred at δ = 178.9 ppm, which was successfully simulated as an [AMXY] spin system (Fig. 5a and b), and confirmed the presence of the two equivalent ring carbons within a (P3C2tBu2)– ring anion.
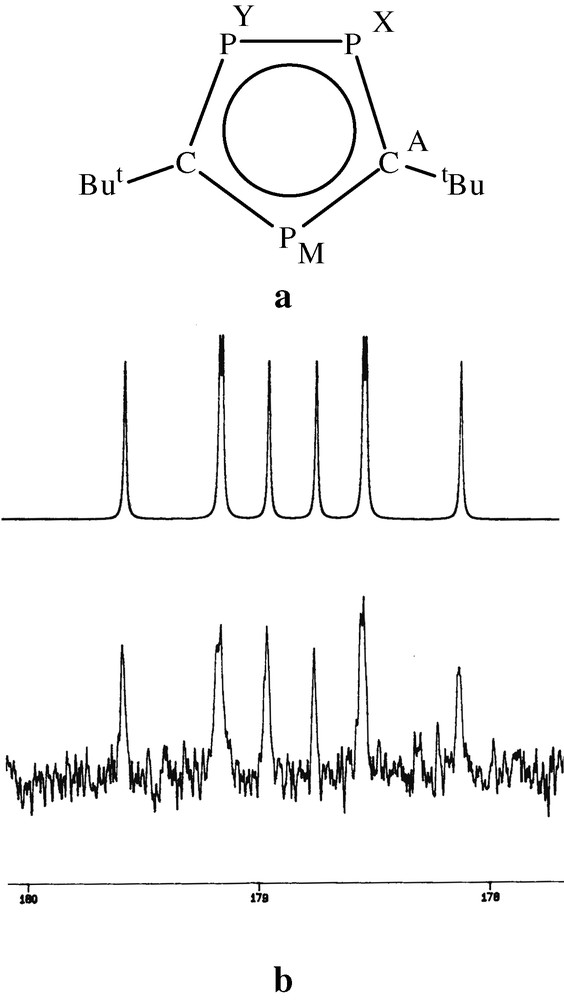
(a) NMR labelling scheme. (b) The A part of the AMXY spin system observed for complex 2. Simulated (top) and observed (bottom).
These observations strongly suggested that the remaining ligand attached to nickel in 2 was the novel PC2But2 phosphirenyl cation, leading to the structure [Ni(η3-PC2But2)(η5-P3C2tBu2)] shown in Fig. 6.
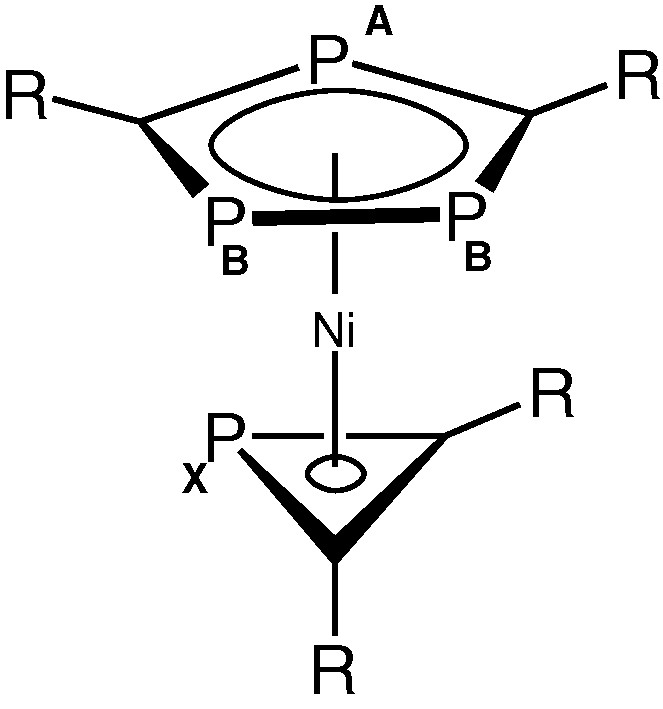
Proposed structure of 2. (R = tBu).
Further supporting evidence for the presence of an η3-ligated phosphirenyl cation in 2 comes from 13C{1H} NMR spectroscopic data. The spectrum contains a doublet at δ = 94.4 ppm (1JC-P = 70 Hz) that may be readily attributed to the ring carbon of this species. 31P{1H} NMR data for 2 can be interpreted as an [AB2X] spin system (Fig. 7). The [AB2] portion may be attributed to the phosphorus atoms of the 1,2,4-triphosphacyclopentadienyl ring and the [X] part to the phosphorus atom of the phosphirenyl cation. Of particular note is the chemical shift of the PX (–161.2 ppm), which lies very close to the reported value of δ = –161.3 ppm for the structurally related compound [Ni(η3-P3)(η-C5H3tBu2)] previously described by Scherer et al. [11].
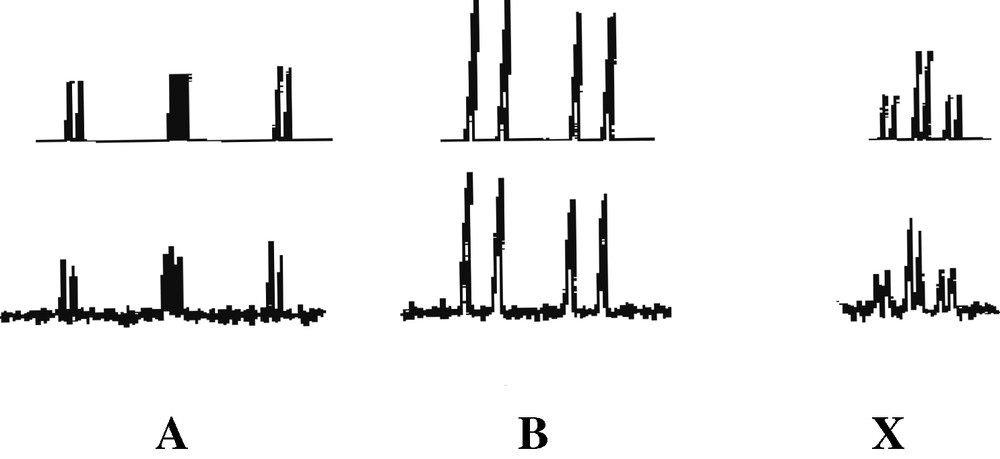
31P{1H} NMR spectrum of 2, observed (bottom) and simulated (top).
Compound 2 thus represents the first known complex to contain the phosphirenyl cation, involving the simplest possible 2π-aromatic species containing a phosphorus atom. Earlier studies concerning phosphirenium salts have shown that they do not exhibit aromatic behaviour [12] and this is also predicted for the isoelectronic silirene [13]. However, although it was proposed that a stronger 3pπ–2pπ interaction between P and C would be present in the phosphirenyl cation [14], all previous attempts to synthesise the latter species have been unsuccessful.
Mathey et al. [15] have commented on the suitability of 1-chlorophosphirenes as synthetic precursors to phosphirenyl systems but treatment of chlorophosphirenes with metal salts containing [BF4–] or [BPh4–] anions led instead to the production of the fluoro- or phenyl-substituted phosphirenes by abstraction reactions from the respective counteranions (Fig. 8). Previous studies by Mathey et al. of triphenylphosphirenes have mainly concerned their ligation and ring opening properties [16-18].
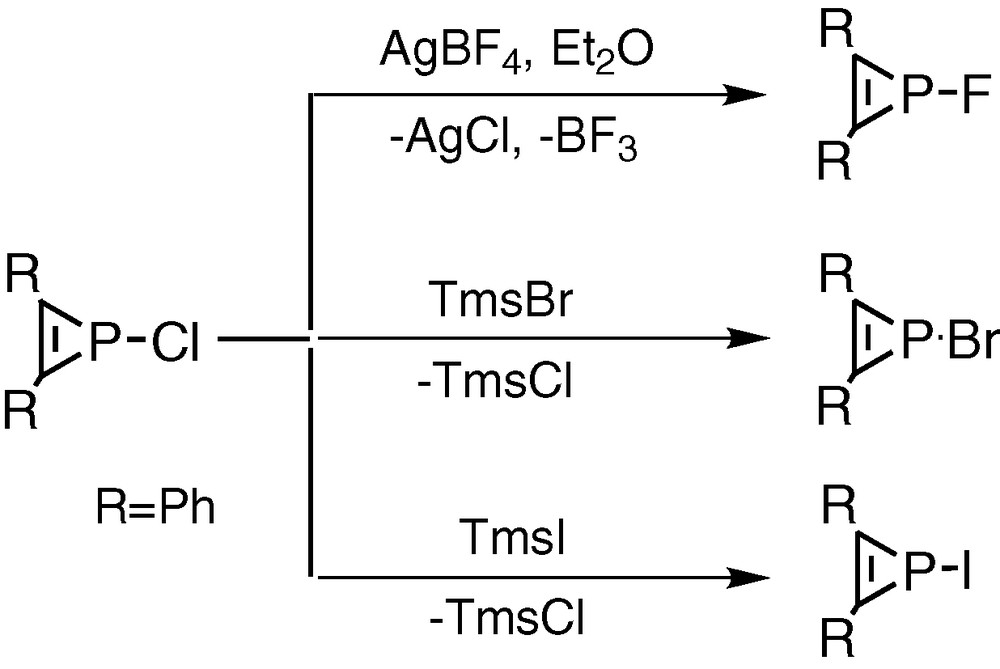
Treatment of chlorophosphirenes with metal salts containing or anions leading to the synthesis of the fluoro- or phenyl-substituted phosphirenes by abstraction reactions from the respective counteranions.
Regitz, Laali and their co-workers [19a,b] have studied several phosphirenyl cations and recent theoretical calculations by Regitz and Eisfield [19c] suggest a high degree of aromatic character. There is a single report [19a,b] of a persistent phosphirenyl cation whose formulation was solely based upon NMR evidence for the abstraction of the triflate group from the phosphirene R2C2P–OSO2CF3 by treatment with BX3 (X = –OSO2CF3), but no confirmatory structural data are available. The work described in this paper therefore represents the first example of a structurally authenticated phosphirenyl cation. Because it proved impossible to grow suitable single crystals of complex 2 suitable for X-ray diffraction studies, an attempt was made to derivatise 2 by further ligation to other metal ligand fragments utilising the P-lone pair electrons. This type of approach has been successfully employed by us in several other related complexes containing unsaturated phosphorus rings (vide infra).
2.1 Preparation of [Ni(η3-PC2tBu2)(η5-P3C2tBu2)W(CO)5] 3
The availability of several lone pair of electrons on the ring phosphorus atoms in 2 offers the possibility of further ligation from any one of these atoms to a coordinatively unsaturated metal centre [20–24]. Such reactivity is well established for phosphorus-containing ferrocenes such as [Fe(η5-P3C2tBu2)2W(CO)5], [Fe(η5-P2C3tBu3)(η5-P3C2tBu2)W(CO)5] and their ruthenium analogues [21,22]. Ligation to other moieties such as [Ru3(CO)10], [Fe(CO)4], [W(CO)5] or [PtCl2(PR3)] (R = Me, Et, Ph) provides a useful means of exploring the relative donor abilities of ring phosphorus atoms within complexes to act further as phosphine ligands [23,24]. More usefully, ligation invariably leads to changes in the solubility of the parent compound, which often facilitates formation of crystalline material from otherwise intractable reaction products.
Accordingly, the [W(CO)5] adduct 3 was readily prepared by treatment of 2 with [W(CO)5thf] in thf solution (Fig. 9), and obtained in crystalline form and its molecular structure determined both spectroscopically and by a single crystal X-ray diffraction study (vide infra).
Preparation of the [W(CO)5] adduct 3 by treatment of 2 with [W(CO)5thf] in thf solution (R = tBu).
Low-temperature 1H and 31P{1H} NMR studies on 3 (see Experimental section for details) showed clearly that the [W(CO)5] unit is preferentially bound to one of the two directly bonded phosphorus atoms of the η5-ligated P3C2tBu2 ring, while the η3-bound PC2tBu2 ring in 1 is unaffected. The assignment of the proposed structural formulation is based on the 31P{1H} NMR spectrum of 3 and especially the resonance assigned to the tungsten bound phosphorus at δ = 86.6 ppm, which is shown in Fig. 10.
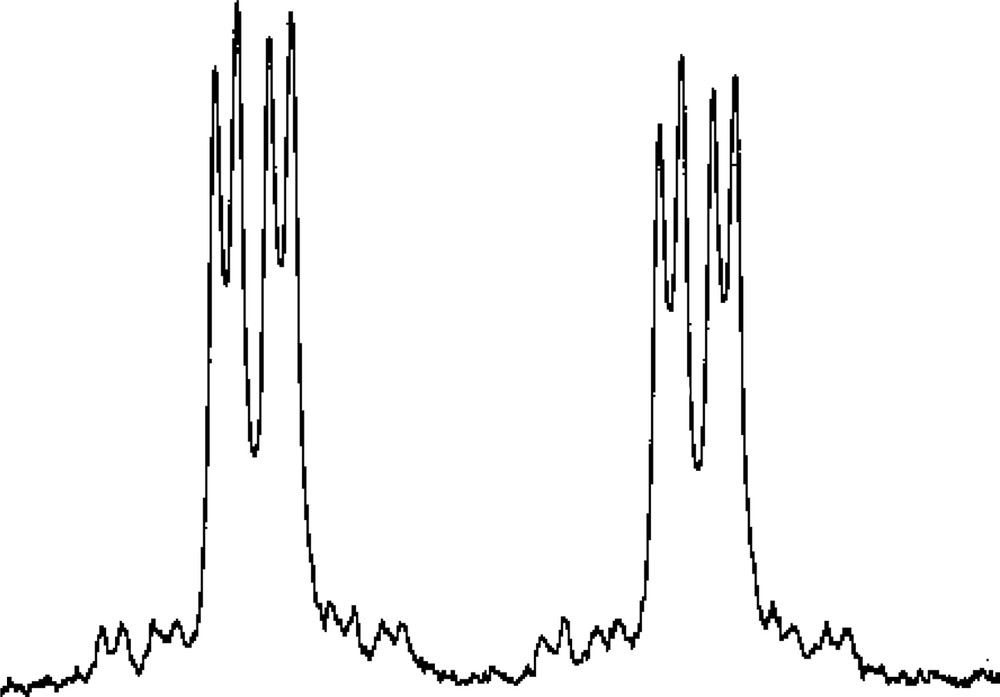
31P{1H} NMR resonance attributable to the tungsten-bound phosphorus atom of 3.
The observation of the 183W satellite structure {I = 1/2, 14% abundance} (1JP–W = 226.5 Hz) and the large magnitude of the 1JP–P coupling constant (439 Hz) is also fully consistent with ligation of the [W(CO)5] fragment to the (P3C2tBu2) ring. The magnitude of the additional inter-ring coupling to the single phosphorus of the phosphirenyl ring (22.3 Hz), strongly suggests [2,25] that the two rings are eclipsed and this was subsequently confirmed by a single crystal X-ray diffraction study on 3. The molecular structure of 3 is shown in Fig. 11 together with selected bond lengths and bond angles.
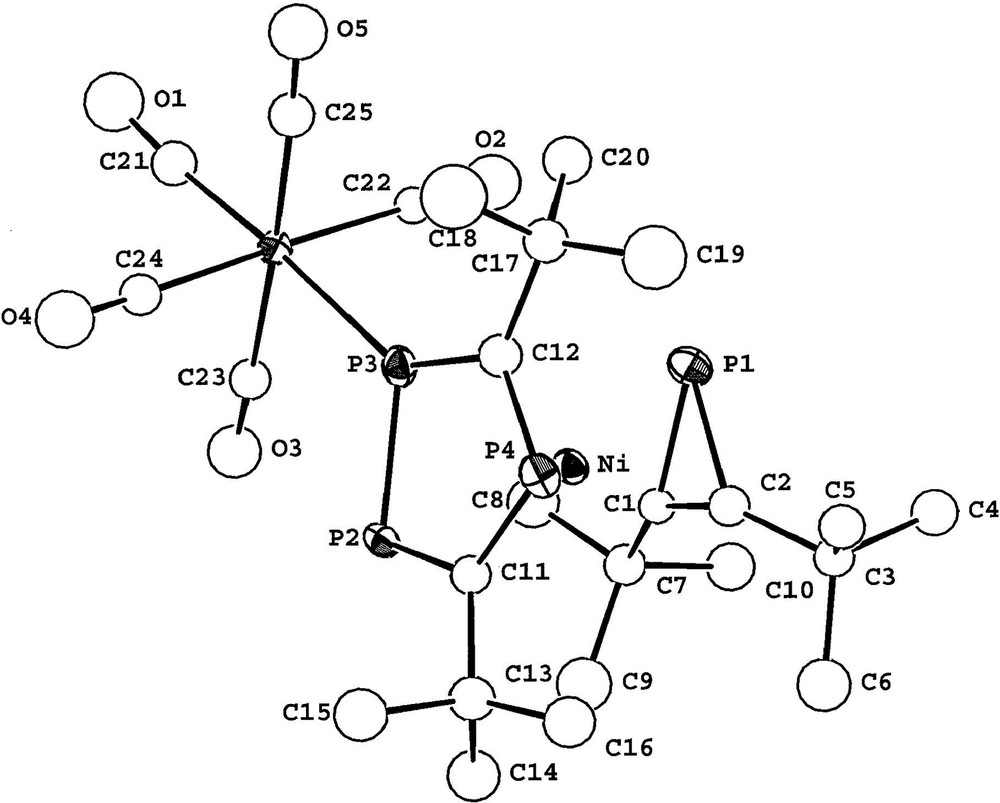
X-ray structure of 3 with selected bond lengths (Å) and bond angles (°). M1 and M2 are the centroids of the three– and five–membered rings, respectively. Ni–P1 2.207(5), Ni–P2 2.397(5), Ni–P3 2.354(5), Ni–P4 2.354(5), Ni–C1 2.02(2), Ni–C2 2.04(2), Ni–C11 2.24(2), Ni–C12 2.12(2), Ni–M1 1.860, Ni–M2 1.692, W–P3 2.517(5), P1–C1 1.78(2), P1–C2 1.82(2), P2–P3 2.111(7), P2–Cl1 1.74(2), P4–C12 1.76(2), C1–C2 1.33(3); M1–Ni–M2 176.0, P3–W–C21 176.7(7), Cl–P1–C2 43.3(8), P3–P2–C11 96.4(7), P2–P3–C12 101.6(7), C11–P4–C12 99.6(9), P1–C1–C2 70(1), P1–C2.C1 67(1).
The molecular structure of 3 confirms the sandwich-type structure and the presence of both an η3-bound phosphirenyl cation (PC2tBu2) and also the ligation of the [W(CO)5] moiety to the η5-bonded (P3C2tBu2) ring. The two rings lie essentially parallel to one another (dihedral angle 4°). Steric repulsion between tertiary butyl groups of both rings, and the presence of the [W(CO)5] fragment forces the molecule to adopt the staggered conformation that is seen clearly if viewed down the ring centroid-metal ring centroid axis (Fig. 12). This has the effect of bringing phosphorus atoms P1 and P3 close enough for a through-space coupling as discussed earlier. A similar ‘through-space’ interaction was proposed by us to explain the significant phosphorus–phosphorus inter-ring coupling observed in both the hexaphosphametallocenes [M(P3C2tBu2)2] (M = Fe, Ru) [2,25].
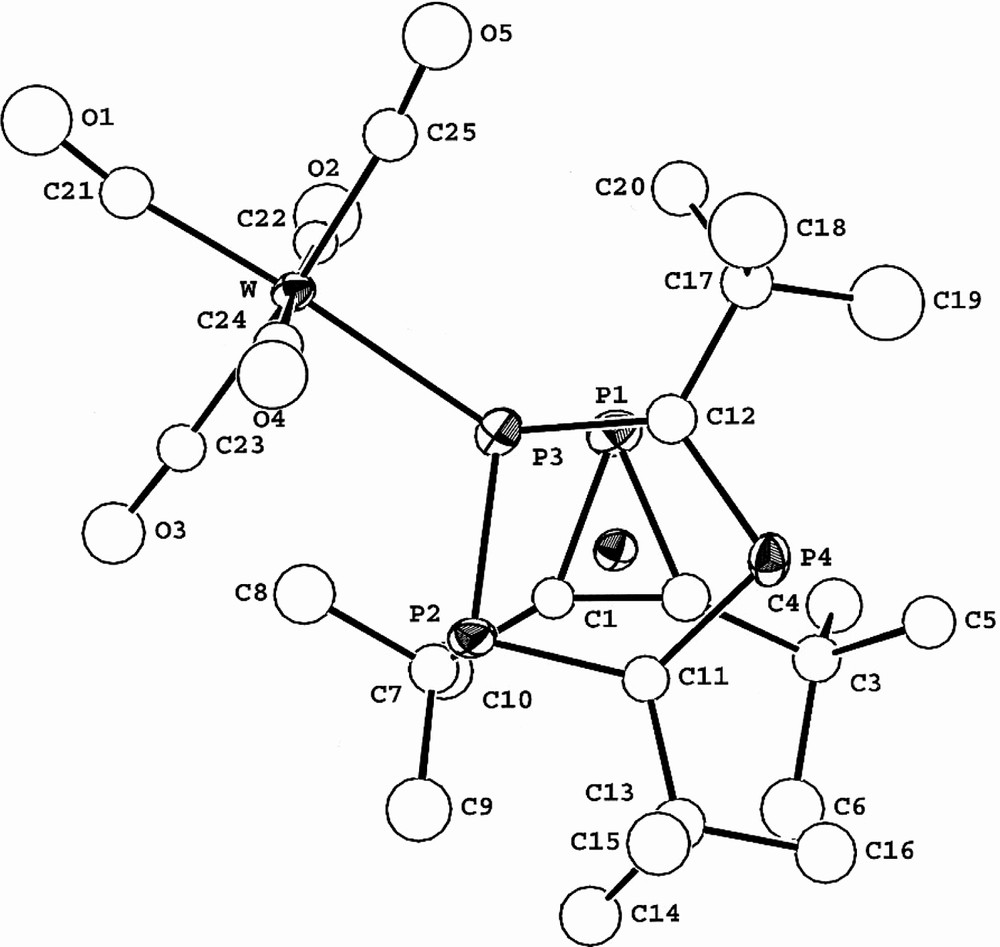
View of 3 showing the relative positions of P1 and P3.
Of special interest is the structure of the ligated phosphirenyl cation in 2 and 3. Two calculations [25,26] on the uncoordinated phosphirenyl cation PC2H2+ at the Hartree–Fock and MP3/6-31G* levels predict a geometry that suggests a strong aromaticity (P–C 1.70–1.73, C=C 1.34–1.35 Å). In 3, the C=C distance is 1.33(3) Å and the P–C distances are 1.78(2) and 1.82(2) Å, which suggests that there is no appreciable electron delocalisation within the ring but clearly that more structural information on this type of transition metal complex is desirable.
In a discussion of the structurally related cyclopropenium complex [Ni(η3-C3Ph3)(η5-C5H5)], the wide variation in Ni–C distances has also been noted for other heteroleptic sandwich complexes of nickel containing carbocyclic ring systems [27]. Only minor changes in the nickel-ring centroid distance are observed for a wide range of differing complexes reviewed, suggesting Ni-centroid distance to be the critical factor in controlling the molecular geometry. The lack of change in nickel-ring centroid distance is offset by fluctuations in Ni–C distances for the compounds studied, and is contrary to results obtained for [V(η5-C5H5)(η7-C7H7)] [28] for which V–C distances lie in the narrow range 2.23–2.25 Å.
Table 1 lists comparative data for 3 with those for [Ni(η3-C3Ph3)(η5-C5H5)]27 and the related triphosphenium complex [Ni(η3-P3)(η-C5H3tBu2)] [11]. The variation in metal-to-ring-centroid and metal-to-ring atom distances is not large, the most significant differences in bond lengths occurring between the P3 ring cation in [Ni(η3-P3)(η5-C5H3tBu2)] and the phosphirenyl cation-metal bond lengths in 3. Steric factors appear to be a likely cause of the elongation of bond lengths in 3, despite a reduction in the number of ring phosphorus atoms.
Comparative structural data
| Complex | |||
| A | 1.860 | 1.874 | 1.779 |
| B | 1.692 | 1.699 | 1.726 |
| C | 2.207 | 2.235 | n/a |
| D | 2.12-2.24 | 2.078–2.095 | 2.100 |
| E | 176.0 | 178.6 | 180 |
| Reference | This work | 11 | 27 |
2.2 Variable-temperature NMR spectroscopic study on complex 3
The solid-state structure of 3 is not wholly retained in solution. Dynamic behaviour of the [W(CO)5] fragment is apparent from the systematic changes observed in the variable temperature 1H and 31P{1H} NMR spectra as the tungsten pentacarbonyl unit undergoes an intra molecular 1,2 shift between the two directly bonded phosphorus atoms of the η5-ligated triphosphacyclopentadienyl ring (Fig. 13).
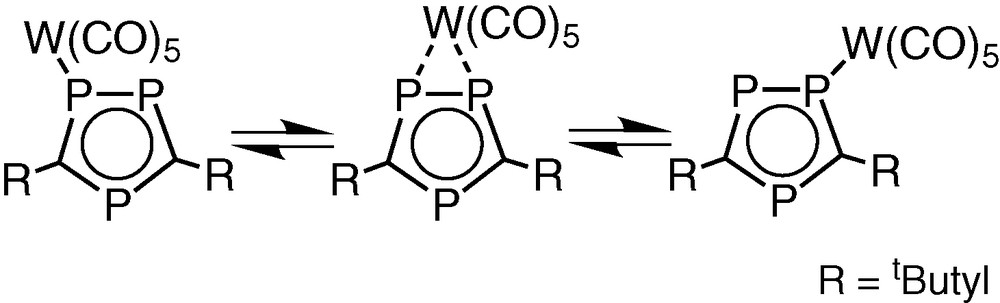
Proposed 1,2 shift of [W(CO)5] in 3.
This motion is established from changes in the 1H NMR spectrum (Fig. 14), which exhibits one singlet above room temperature attributable to the two non-equivalent tbutyl groups of the (P3C2tBu2) ring that collapses into two singlets as the sample is cooled to 253 K. Surprisingly no change is observed in the tBu resonance for the phosphirenyl cation, which remains as a singlet down to the freezing point of d8-toluene.
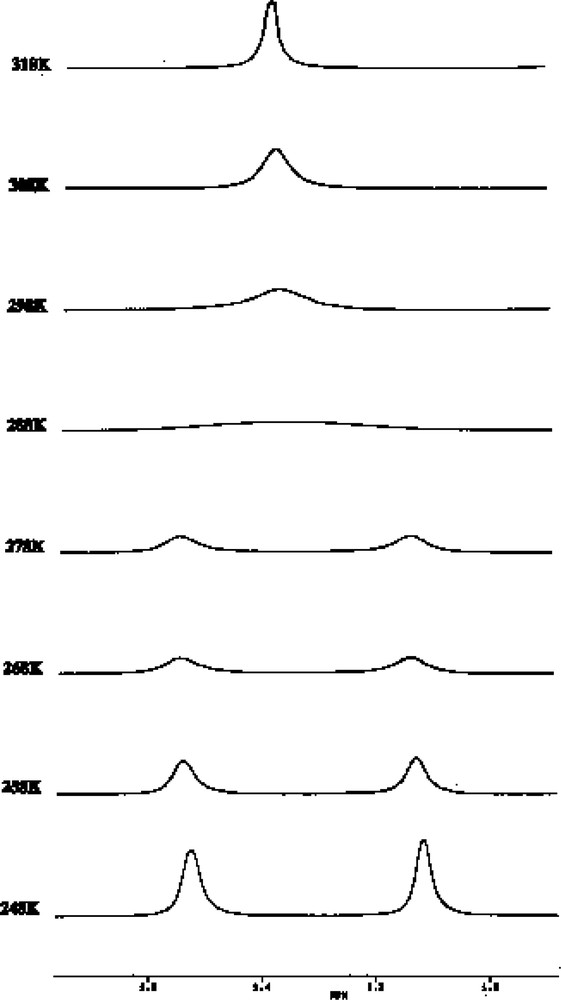
Variable-temperature 1H NMR spectra for 3 showing the resonance of the tBu protons of the η5- ligated P3C2tBu2 ring.
This type of dynamic behaviour has previously been observed for the known ruthenium and iron complexes [Fe(η5-P3C2tBu2)2W(CO)5], [Fe(η5-P2C3tBu3)(η5-P3C2tBu2)W(CO)5], [Fe(η5-P3C2tBu2)2Fe(CO)4], and [Ru(η5-C5H5)(η5-P3C2tBu2)W(CO)5] [29]. Previously we showed [30], by a detailed 195Pt and 31P{1H} NMR study of the complexes trans-[MCl(P3C2tBu2)(PR3)2] and trans-[M(P3C2tBu2)2(PR3)2], the first such 1,2-metal atom shifts for phosphorus-based systems.
2.3 Mechanistic aspects of the formation of [Ni(η3-PC2But2)(η5-P3C2tBu2)] 2 and [Ni(η4-P2C2But2)2] 1 from Ni atoms and tBuCP
The formation of the complex [Ni(η4-P2C2But2)2] 1 from Ni atoms and the phosphaalkyne, tBuCP, is readily understandable in terms of a [2+2] cycloaddition of the presumed initially formed bis-phosphaalkyne nickel(0) complex (most likely involving η2 rather than η1 ligation). At first sight, the simultaneous synthesis of [Ni(η3-PC2But2)(η5-P3C2tBu2)] 2 in the metal-vapour synthesis experiment suggests that cleavage of the P–C triple bond of the phospha-alkyne is occurring; however this high-energy process is considered unlikely.
Very recently, Zenneck and co-workers have made a series of important observations concerning the ‘Ambiphilicity reactivity principal’ of π-bound phosphorus heterocycles, which may throw new light on the mechanistic aspects of the metal-vapour syntheses of phospha-organometallic compounds from phospha-alkynes [31]. They showed in a series of variable temperature X-ray diffraction studies on [Mo(η4-P2C2tBu2)3] that, as the temperature is lowered, there is a steady interaction between two of the originally η4-bonded rings via a nucleophilic attack by one of the phosphorus lone pair electrons on the C atom of an adjacent ring, leading to a complex containing a saturated ring C atom and an η3-bonded 1,3,-diphospha allyl ring (Fig. 15). This transformation in the solid state was found to be reversible as the temperature is raised until at room temperature the structure is identical with that first reported by Nixon and Cloke et al. [5].
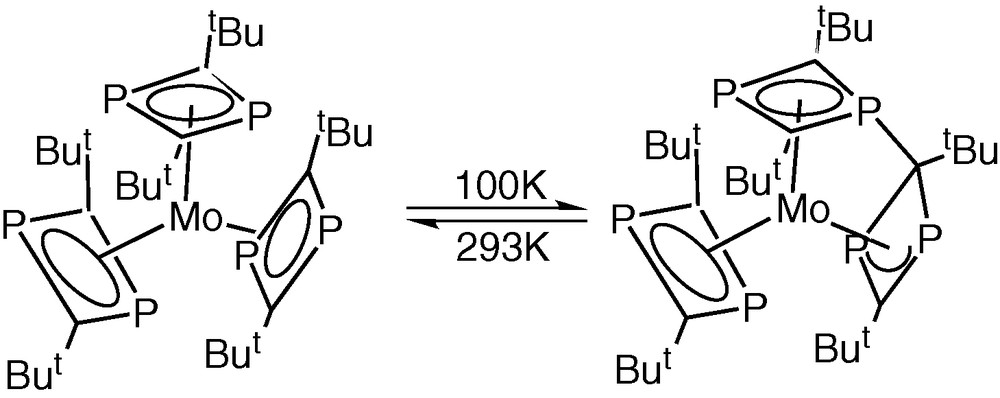
Influence of temperature on [Mo(η4-P2C2tBu2)3].
In subsequent theoretical calculations it has been established that the known exclusive formation of the pentaphosphaferrocene [Fe(η5-P3C2tBu2)(η5-P2C3tBu3)]2 from iron atoms and the phosphaalkyne, tBuCP, could result from a similar type of inter-ring nucleophilic attack involving the initially formed complex [Fe(η6-P3C3tBu3)(η4-P2C2tBu2)] in which the phospha-alkyne has been cyclotrimerised and cyclodimerised at the iron centre (Fig. 16) [32].
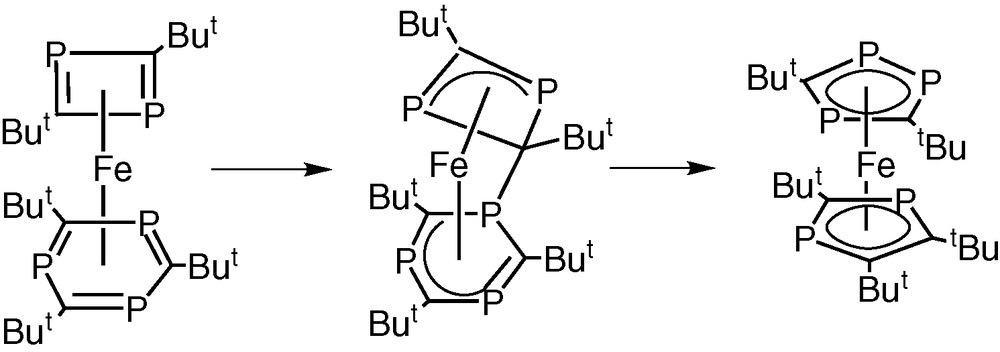
Inter-ring nucleophilic attack involving the complex [Fe(η6-P3C3tBu3)(η4-P2C2tBu2)] in which the phospha-alkyne has been cyclotrimerised and cyclodimerised at the iron centre.
This mechanistic proposal is much more energetically acceptable and immediately suggests that perhaps the [Ni(η3-PC2But2)(η5-P3C2tBu2)] complex, described in the present work, might be formed from the initial product [Ni(η4-P2C2But2)2] by transfer of a P atom from one 4-membered ring to the other. An obvious energetic advantage of this type of isomerisation would be the significant gain in aromaticity in the system since the resulting three-membered PC2But2 and five-membered P3C2tBu2 rings are both aromatic (2π and 6π electron systems respectively).
3 Experimental
All manipulations (unless otherwise stated) were conducted in an inert atmosphere of argon or dinitrogen. This was achieved using a dual vacuum/argon (or dinitrogen) line, employing conventional Schlenk line techniques, or by use of either a Miller–Howe or Braun glove box under an atmosphere of dinitrogen. All solvents were pre-dried by distillation under dinitrogen over the appropriate drying agent and subsequently degassed and stored in glass ampoules under argon in the over a potassium mirror. All glassware, cannulae and Celite were stored in an oven (> 373 K) and glassware and Celite were flame-dried in vacuo immediately prior to use. Nickel for the MVS experiments was degassed prior to use, by melting under vacuum. Deuteriated solvents were purchased from M.S.D. Isotopes, Ltd, and Goss Scientific. They were degassed by the freeze-thaw method, refluxed in vacuo over molten potassium or sodium (according to boiling point) and then vacuum transferred to a glass ampoule fitted with Young's tap prior to storage in the glove box.
Solution NMR spectra were recorded on Brüker ACP250, WM360, DMX300, DMX400 or AMX500 instruments at ambient temperature unless otherwise stated. Coupling constants are given in Hz, and chemical shift (δ) in ppm is relative to the residual proton chemical shift of the deuterated solvent (1H), the carbon chemical shift of the deuterated solvent (13C), and H3PO4 (31P).
Electron impact (EI) mass spectra were recorded on a Kratos MS80RF instrument in the School of Chemistry at the University of Sussex by Dr. A. Abdul-Sada.
Microanalyses were performed by Medac Ltd (UK) or Canadian Microanalytical Services Ltd (Canada).
3.1 Preparation of [Ni(η4-P2C2tBu2)2] 1 and [Ni(η5-P3C2tBu2)(η3-PC2tBu2)] 2
Cocondensation at 77 K of electron beam vaporised (400 W) nickel (0.99 g, 16 mmol) with an excess of tbutylphosphaalkyne (25 g, 250 mmol) over 4 h resulted in the formation of a deep red matrix that was allowed to warm to room temperature under vacuum. The apparatus was then opened to an external trapping system and unreacted tBuCP was collected in an external trap at 77 K. After 4 h, the reaction vessel was refilled with dinitrogen and the matrix washed into a Schlenk receiver with petroleum ether (b.p. 40–60 °C). The crude reaction mixture was then filtered through a flame-dried bed of Celite to remove colloidal metal and solvent removed in vacuo. The resultant black solid yielded two products after chromatographic work up (deactivated alumina {4% m/m H2O}, petroleum ether (b.p. 40–60 °C) as eluent). The primary fraction (yellow band) yielded orange crystals of 1 (200 mg, 2.7% yield) following recrystallisation from pentane at –50 °C.
1H NMR (d6-benzene): 1.2 (s, tButyl)
31P{1H} NMR (d6-benzene): 82.3 (s, ring P)
MS, 70 eV; 458 (40, M+), 258 (20, M+–C10H18P2)
The secondary fraction yielded 2 as a red oil (ca 200 mg, 2% yield) following purification by sublimation (10–3 mbar, 50 °C).
MS (70 eV) m/z (%) 458(14) [M+], 358(5) [M+–C10H18P2].
3.2 NMR Data (see Fig. 17 for labelling scheme)
1H NMR: δ 1.45 (d, 4JPXH = 0.9 Hz,18H, (C3H3)), 1.14 (s, 18H, (C6H3)). 31P{1H} NMR: AB2X, δ 155.4 (PA), 145.1 (PB), –161.2 (PX), JPAPB = 51.8 Hz, JPAPX = 5.1 Hz, JPBPX = 16.0 Hz. 13C{1H} NMR: δ 32.4 (s, C3), 32.9 (d, JCPX = 4.3 Hz, C2), 36.8 (A part of AMXX′, JAM = 9.3 Hz, JAX′ + JAX′ = 9.2 Hz, (A = C6, M = PA, XX′ = PB), 40.0 (A part of AMXX′, JAM = 18.5 Hz, JAX + JAX′ = 12.4 Hz, A = C5, M = PA, XX′ = PB), 94.6 (d, JCPX = 70.0 Hz, C2), 178.9 (A part of AMXY, JAM = 78.7 Hz, JAX + JAY = 104.4 Hz, ▵δXY = 31.6 ppb).
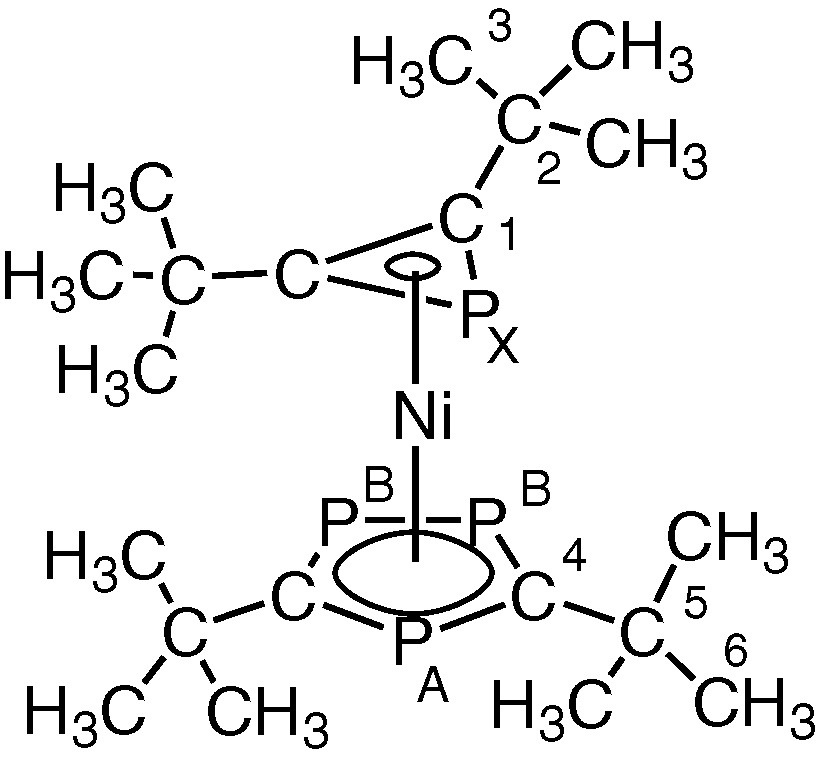
Structure of [Ni(η5-P3C2tBu2)(η3-PC2tu2)] 2.
3.3 Preparation of [Ni(η3-PC2tBu2)(η5-P3C2tBu2)(W(CO)5] 3
To [W(CO)5thf] generated in situ from photolysis of [W(CO)6] (0,10g) in thf (25 ml) was added [Ni(η3-PC2tBu2)(η5-P3C2tBu2)] (ca 0.05 g) and the mixture stirred for 4 h. The solvent was removed in vacuo to yield a dark oil. Extraction of the oil into petroleum ether (b.pt. 40–60 °C) followed by chromatographic work up on Kieselgel 60 GF254 with petroleum ether (b.p. 40–60 °C) as eluant afforded [Ni(η3-PC2tBu2)(η5-P3C2tBu2)(W(CO)5] 3 as a micro-crystalline powder on removal of solvent. Recrystallisation from pentane (–50 °C) afforded analytically pure red crystals (0.05 g, 58%), which were suitable for an X-ray crystal structure determination.
MS (70 eV) m/z (%) 782(17) [M+]
Microanalysis: found C 38.72, H 4.71%; calculated, C 38.35, H 4.63%.
3.4 NMR Data (see Fig. 18 for labelling scheme)
1H NMR (d8-toluene, 318 K): δ 1.12 (s, 18H, (C3H3)3), 1.38 (s, 18H, (C6H3)3); (d8-toluene, 248K): δ 1.08 (s, 18H, (C3H3)3), 1.26 (s, 9H, (C6H3)3), 1.47 (s, 9H, (C6H3)3). 31P{1H} NMR (d8-toluene, 253K): δ –144.7 (d, 2JPAPD = 22.3 Hz, PA), 86.6 (ddd, 1JPDW = 226.5 Hz, 1JPDPC = 439.3 Hz, 2JPDPB = 52.7 Hz, 2JPDPA = 22.3 Hz, PD), 129.9 (dd, 1JPCPD = 439.3 Hz, 2JPCPB = 46.6 Hz, PC), 149.1 (dd, 2JPBPD = 52.7 Hz, PB). 13C{1H} NMR (d8-toluene, 253 K): δ 31.4 (s, (C3H3)), 31.9 (s, (C3H3)) , 32.1(s, C(C6H3)), 32.3 (s, C(C2H3)), 35.5 (dd, 3JCP = 5.4 Hz, 3JCP = 11.0 Hz, C6), 35.9 (dd, 3JCP = 8.9 Hz, 3JCP = 8.9 Hz, C6), 39.57 (dd, 1JCP 15.9 Hz, C5), 39.61 (dd, 1JCP = 22.2 Hz, C5), 97.7 (d, 1JCP = 69.6 Hz, C1), 100.9 (d, 1JCP = 71.1 Hz, C4), 159.9 (ddd, 1JCP = 83.6, 1JCP = 64.3 Hz, 2JCP = 9.5 Hz, C4), 176.4 (dd, 1JCP = 78.9 Hz, 1JCP = 91.8 Hz), 196.4 (d, 2JCPD = 2.5 Hz, 1JCW = 126.6 Hz, cis C8O), 198.8 (d, 2JCPD = 31.0 Hz, trans C7O).
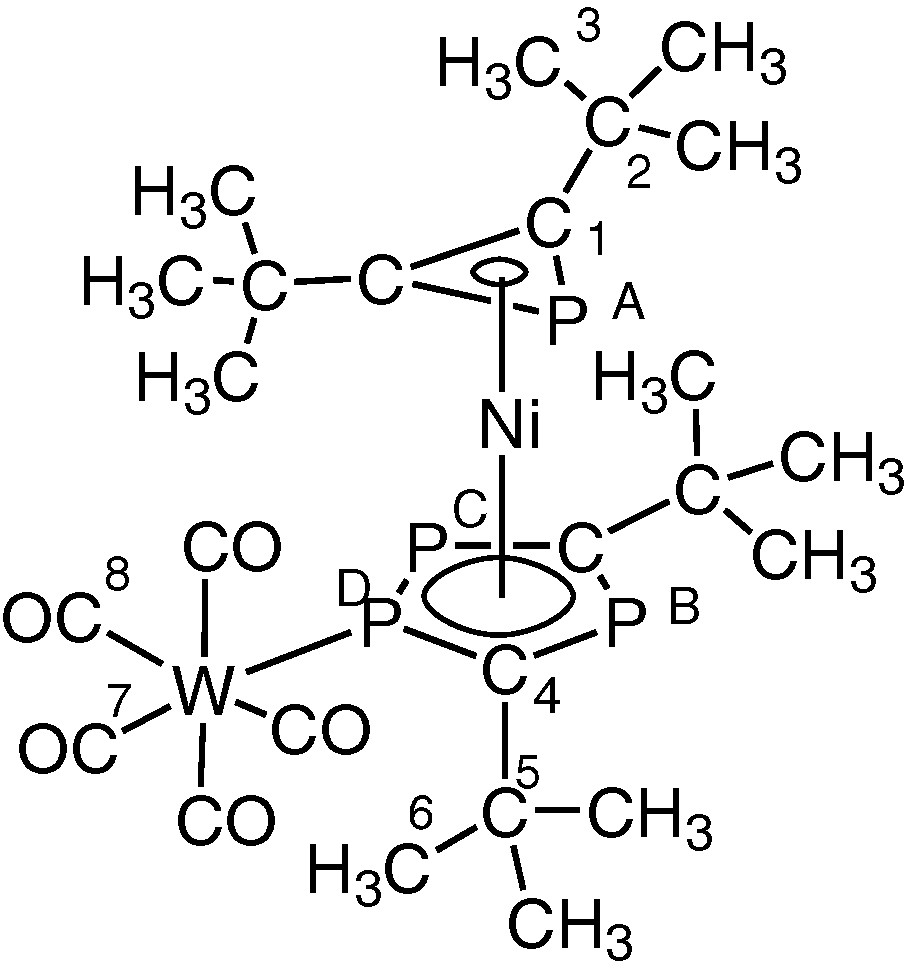
Structure of [Ni(η3-PC2tBu2)(η5-P3C2tBu2)(W(CO)5] 3.