

1 Introduction
The heartwood of Quercus petraea and Q. robur contains ca 10% by weight of C-glucosidic ellagitannins [1–3]. These hexahydroxydiphenoyl (HHDP) esters, the main source of wood's durability [4], help protect it against fungal or bacterial decay [5] and impart an astringent taste to the wood extract [6]. The study of ellagitannins is important because this group of hydrolysable tannins is used in various industrial and agri-food applications [7] and is partially responsible for the colour of oak wood, which in turn contributes to its market quality [8,9]. Ellagitannins also greatly affect the quality and composition of wines [10,11] or spirits [12] kept in oak barrels.
Monomeric and oligomeric forms of ellagitannins are well known [1,13]. The major constituents of oligomeric ellagitannins from heartwood extracts are vescalagin (1) and castalagin (2) (Fig. 1). Many publications have reported the occurrence of polymeric condensed tannins [14]. For ellagitannins, the few partial studies conducted excluded any participation of microbial degradation of polyphenols in plant tissues [15]. These studies evidenced insolubilization [16] and hydrolysis [3,17] during heartwood ageing. A tentative partial characterization of the polymeric fraction of ellagitannins was presented by Klumpers et al. [18].
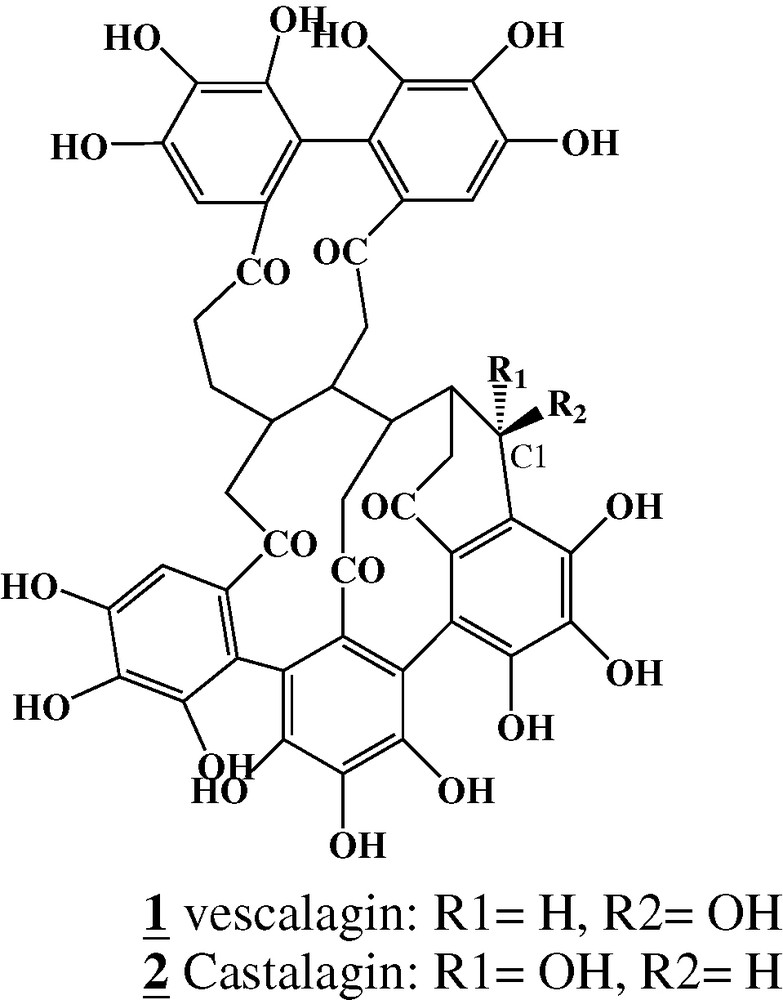
Molecular structures of vescalagin (1) and castalagin (2).
The duraminization process and oxidation are the main reactions occurring during polymeric changes in heartwood. This report concerns ellagitannin polymerisation and its impact on heartwood qualities, on colour in particular.
2 Materials and methods
2.1 General
Castalagin was isolated and purified from the duramen of Q. robur, under conditions described by Vivas et al. [13] Gallic and ellagic acids were supplied by Acros™.
All solvents employed were of analytical grade.
2.2 Plant material
The oak samples were made up of heartwood from approximately 175-year-old trees, from homogeneous and appropriately maintained forest compartments. Only the first quarter of the trunk, the cooperage grade timber, was used for the study. The wood was chopped or sawed and then naturally dried for 24 months. The different samples were planed and then crushed to sawdust in liquid nitrogen, before being strained so as to keep only particles smaller than a 60 mesh size. The samples were freeze-dried, stored and analysed within a period of 2 months.
2.3 Colour measurements
The colour of the wood pieces was measured using a Cielab Minolta 508™ chromatic system. Measurements of the yellow hue of the solution were made in a liquid medium (extracts of fractions) with an Anthelie Secomam™ spectrophotometer.
2.4 Polyphenol extraction
1 g of sawdust (60 mesh) was extracted with 100 ml of acetone/water (70:30, v/v) for 12 h at room temperature (about 20 °C) under stirring (150 rpm). The resulting extract was then filtered through 0.45-μm membranes and freeze-dried.
2.5 Analytical high performance liquid chromatography (HPLC)
The HPLC analyses were performed according to Scalbert et al. [2]. Samples were analysed in the reversed-phase on a HPLC Waters™ with an Interchrom™: C18 column (250 × 4.6 mm, dp: 10 μm). The elution program was performed at a constant flow of 1 ml min–1, passing from 0 to 10% B in 40 min, and then rising to 100% B in 25 min (solvent A: H2O/H3PO4 = 999:1, solvent B: MeOH/H3PO4 = 999:1). Injection volume: 20 μl. Detection was performed by UV 280 nm.
2.6 Size-Exclusion Chromatography (SEC) analysis
Study of the Mp (peak-average molecular weight) distribution of samples was performed using the acetyl derivatives. Samples (10 mg) of freeze-dried material were acetylated with pyridine-acetic anhydride (1:1) for three days at room temperature. The precipitate obtained by pouring the mixture into cooled water was recovered by centrifugation, and consecutively washed with distilled water, methanol and finally chloroform. It was dried, dissolved in 0.5 ml tetrahydrofuran (THF) and filtered before analysis by SEC.
SEC analysis was performed using a Thermo Quest™ instrument equipped with three columns (300 × 7.8 mm): TSK™ Gel G 1000 HXL, TSK™ Gel G 2000 HXL, TSK™ Gel G 2500 HXL, in series, protected with a guard column of the same material. Analytical conditions were: THF as the eluent, flow-rate: 1 ml min–1, injection volume: 20 μl and analysis time: 45 min. The calibration curve was obtained with polystyrene standards.
Detection was made at 280 nm, by an UV detector (Spectra Series™ UV-150) and a refractometric detector (Spectra Series™ RI-150). PL Caliber™ software was used for data acquisition.
2.7 Purification of soluble polymeric ellagitannins
Polymeric soluble ellagitannins were extracted from the acetone/water extract of wood samples. We chose three distinct purification steps:
- • fractionation of the soluble and insoluble fractions in ethanol (EtOH) 90% vol. after 12 h at 4 °C; only the soluble fraction contained ellagitannins (the precipitate represented the polysaccharide–ellagitannin complex);
- • with low-pressure chromatography using Sephadex LH 20 gel, we collected a fraction eluted with methanol (MeOH) and rejected fractions eluted respectively with water (hydrolytic products of vescalin and castalin, some vescalagin, castalagin and gallic acid) and with 30% MeOH (eluate monomers and ellagitannin dimers);
- • thirdly, residual ellagic acid, was removed by liquid extraction of the fraction with EtOEt and represented pure Eps.
2.8 Ellagitannin estimation
2.8.1 Soluble ellagitannin quantification
The proposed method was based on the one perfected by Peng et al. [16]. It is based on acid hydrolysis of the ellagitannins in a water-bath, followed by HPLC quantification of free ellagic acid. 2.5 mg of dried sample were introduced in a hydrolysis tube fitted with a Teflon seal, the contents solubilized by 5 ml of 2 N methanol/ hydrochloric acid. We measured the ellagic acid present, and, after two hours of acid hydrolysis (oil bath at 100 °C), the resulting ellagic acid. The difference between the two values corresponds to the ellagic acid released by the ellagitannins. The HPLC quantitative determination was analysed in the reverse phase on a HPLC Waters™ with an Interchrom™ column: C18 (250 × 4.6 mm, dp: 10 μm; the elution program was performed at a constant flow of 1 ml min–1, passing from 0 to 30% B in 3 min, and then rising to 50% B in 2 min, to 70% B in 5 min, to 80% B in 5 min, and finally to 100% B in 2 min. (Solvent A: H2O/H3PO4 pH adjusted to 2.4; solvent B: MeOH/H3PO4 pH adjusted to 2.4). Injection volume: 20 μl. Detection UV: 370 nm.
2.8.2 Insoluble ellagitannins quantification
The method was identical to the one above, hydrolysis was not performed on the acetone/water extract but on the total sawdust after extraction.
2.9 Phenol determination
The total phenolic compounds of the wood extract were determined either by the Folin–Ciocalteu reagent method (FC) or by measuring UV absorption at 280 nm of the extracts diluted at 1:100 [19]. The FC reaction was performed by introducing 0.2 ml of fractions, 1 ml of FC reagent, completed to 20 ml with sodium carbonate (Na2CO3) at 4%. After 20 min at 70 °C, the intensity of the colour was measured at 750 nm. The FC index corresponds to the O.D. value.
These two methods produced comparable results but are not specific to ellagitannins.
2.10 IR spectra
IR spectra were performed on pastilles of fraction A with potassium bromide (KBr). IR spectrometer was a Perkin Elmer™. The IR bands were assigned according to work by Puech [20], Faix [21], and Collier et al. [22].
2.11 Nuclear magnetic resonance (NMR) experiments
Solid NMR was measured by the cross polarisation of the magic-angle spinning method (CPMAS 13C NMR). The experiments were performed on a Bruker™ DPX 400. Experimental conditions have been described by Davis et al. [23]. The samples consisted of well-dehydrated sawdust sieved at 40 mesh. The sawdust was packed in a NMR tube.
2.12 In vitro chemical oxidation assay
Three sterile aqueous solutions of castalagin were prepared: castalagin at 1 g l–1 with 8 mg l–1 of oxygen; castalagin at 1 g l–1 with 8 mg l–1 of oxygen in equivalent hydrogen peroxide (H2O2), and the same solution containing 1 mg l–1 of copper sulphate (CuSO4). The O.D. was followed at 420 nm over time.
2.13 Thioacidolysis reaction
The method applied is described in detail by Rolando et al. [24]. It consists of a thioacidolysis of the lignin (0.2 M boron trifluoride diethyl (BF3) etherate in dioxane/ethanethiol; 8.75:1). The degradation products, presented in the form of thioethyled derivatives, were analysed by GC/MS after sylilation (BSTFA/TMCS). Only the reagents measures stipulated by the authors were modified: 30 mg of EPs were added to 14 ml of the thioacidolysis reagent. The final residue was redissolved in 2 ml of dichloromethane; 1 ml of the solution was trimethylsilylated with 500 μl of N,O-bis-(trimethylsilyl)-trifluoroacetamide (BSTFA) + 1% trimethylchlorosilane (TMCS) and 50 μl of GC-grade pyridine in a 2-ml reaction vial fitted with a Teflon-lined screwcap placed at ambient temperature; the silylation was completed after 4 h.
2.14 Gas chromatography–Mass spectrometry analysis
GC analysis was performed on a DBXLB column (30 m × 0.25 mm × 0.25 μm). The temperature was programmed from 80 °C (1 min) to 120 °C at a rate of 50 °C min–1, to 260 °C at a rate of 4 °C min–1 and to 320 °C at a rate of 8 °C min–1. Injector temperature: 240 °C; 1 μl was injected in splitless mode. Detection was accomplished with a VG-Autospec EQ™ magnetic spectrometer operating in electron impact mode with a ionisation energy of 70 eV; the temperature of the transfer line was 240 °C.
3 Results
3.1 Increase in ellagitannins during wood ageing
We collected different samples of sapwood and heartwood (Fig. 2) from two series of approximately 175 years old Q. petraea trees. Dry extract, total phenols, soluble and insoluble ellagitannins were recorded (Table 1). Both series led to the same observations.
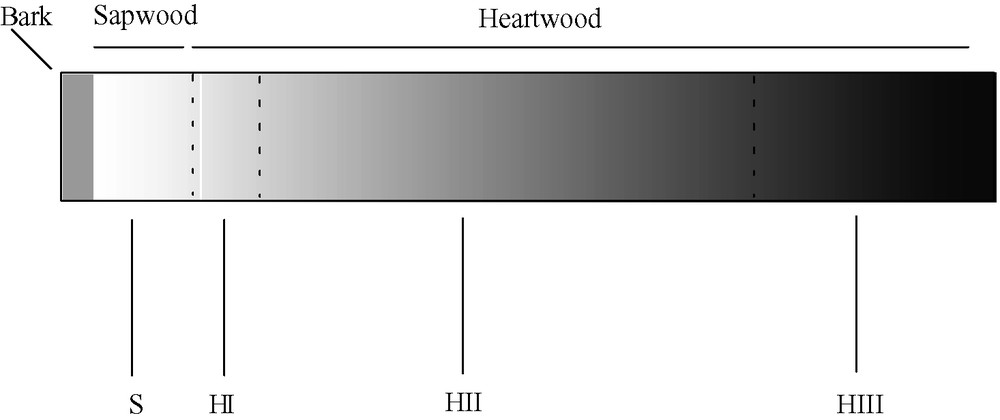
Sapwood and heartwood samples' localisation on a cross section. S: Sapwood (rejected samples); HI: early heartwood (first 10 years); HII: outer heartwood; HIII: inner heartwood (darkly coloured inner heartwood).
Evolution of total phenols. Dry extract and ellagitannins contents of different samples of sapwood and heartwood (acetone/water extract of Q. petraea wood). Unless otherwise indicated, results are expressed in mg g–1 wood
| Dry extract | Total phenols' indexb | Soluble ellagitannins (a) | Insoluble ellagitannins (b) | (a)/(b) | |
| First seriesa | |||||
| Sapwood | 32 ± 8.4 | 5 ± 0.8 | 11 ± 2.5 | trace | — |
| Heartwood Ic | 138 ± 11.5 | 24.5 ± 5.2 | 112 ± 14.3 | 22 ± 7.3 | 5.1 |
| Heartwood IIc | 146 ± 13.2 | 27 ± 2.5 | 108 ± 12.7 | 28 ± 7.5 | 3.8 |
| Heartwood IIIc | 95 ± 9.6 | 19.5 ± 1.4 | 89 ± 9.5 | 37 ± 7.4 | 2.4 |
| Second seriesa | |||||
| Sapwood | 51 ± 5.3 | 3 ± 1.2 | 5 ± 1.3 | 5 ± 3.2 | 1 |
| Heartwood Ic | 118 ± 12.6 | 22 ± 3.5 | 126 ± 13.8 | 19 ± 9.4 | 6.6 |
| Heartwood IIc | 125 ± 11.7 | 20 ± 2.7 | 109 ± 17.2 | 24 ± 7.8 | 4.5 |
| Heartwood IIIc | 105 ± 10.8 | 16,5 ± 1.7 | 64 ± 10.3 | 29 ± 7.3 | 2.2 |
a Trees approximately 175 years old.
b Folin–Ciocalteu index (O.D. g–1wood).
c See Fig. 1 for legend.
On the one hand, the duraminization process transformed sapwood into heartwood, thus increasing the amount of the acetone/water extract in relation to phenols, principally ellagitannins, accumulation. On the other hand, the ageing process led to a decrease in ellagitannin monomers and dimers quantified by high-performance liquid chromatography (vescalagin, castalagin, grandinin, roburins A–E). Insoluble ellagitannins increased regularly from HI to HIII. Correlated with insoluble ellagitannins accumulation, the colour of the wood deepened, becoming darker and redder, as evaluated respectively by decrease of L and increase of a* to CIELAB chromatic system (Fig. 3).
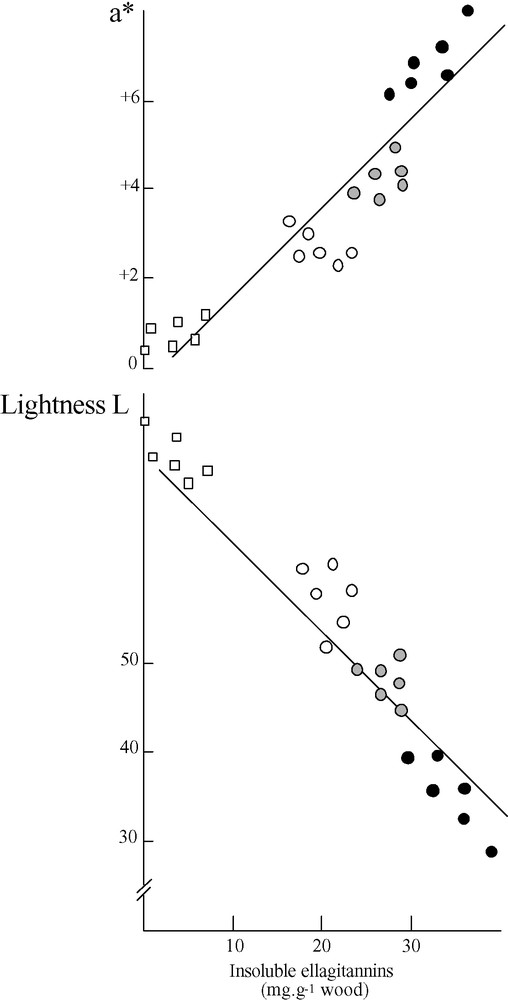
Sapwood (white square) and heartwood (HI: white circle, HII: grey circle, HIII: black circle) coloration in relation to insoluble ellagitannin contents (IE). The colour was recorded with the CIELAB system. No significant relationship was observed between IE and b* parameter. L/IE, r = –0.89 (significant at p < 0.01) and a*/IE, r = 0.94 (significant at p < 0.01).
But the proportion of soluble ellagitannins cannot account for the whole dry extract fraction; in the same way, the increase in insoluble ellagitannins (+ 15, + 10 mg g–1 for the first and second series) does not correspond to the decrease in soluble ellagitannins (–23, –62 mg g–1, respectively, for the first and second series). We suggest that there may be an intermediate form of soluble polymeric ellagitannins (EPs) before total insolubilisation occurs.
3.2 Partial characterization of soluble polymeric ellagitannins
Polymeric soluble ellagitannins were extracted from an acetone/water extract of wood samples. The SEC of samples HI-HIII was also recorded (Fig. 4). This technique provided a good separation of the number-average of molecular weight (Mn) polymers, oligomers and low-Mn phenols. Fresher heartwood (HI) presented mainly monomers and dimers; in accordance with previous work [3], during the duraminization process we noted a partial hydrolysis reaction of the ellagitannins identified by the increase of peaks attributed to ellagic acid and vescalin and castalin (HII-HIII). In old heartwood (HIII), one major reaction was the formation and accumulation of polymeric soluble ellagitannins. We chose three purification steps for EPs (for details, see experimental section). First, fractionation of the insoluble and soluble fraction in EtOH 90% vol. after 12 h at 4 °C; only the soluble fraction contained ellagitannins, while the precipitate represented a polysaccharide–ellagitannin complex [11]. Secondly, with low-pressure chromatography using Sephadex LH 20 gel, we collected a fraction eluted with MeOH (HIIc, Fig. 5) and rejected fractions eluted respectively by water (hydrolytic products of ellagitannins vescalin and castalin, monomers vescalagin, castalagin and gallic acid) and 30% vol. MeOH (monomeric and dimeric of ellagitannins). The content of these fractions was analysed by reverse-phase HPLC. Purification quality was estimated by SEC, as shown in Fig. 5 for HII samples. Thirdly, residual ellagic acid was removed by liquid extraction of the fraction with EtOEt and represented pure EPs. Then, by repeated injections, we collected enough HIIc and HIIIc fractions for a partial characterization. SEC on TSK gel gave an average Mn of 2700 ± 375 for HIIc and HIIIc.
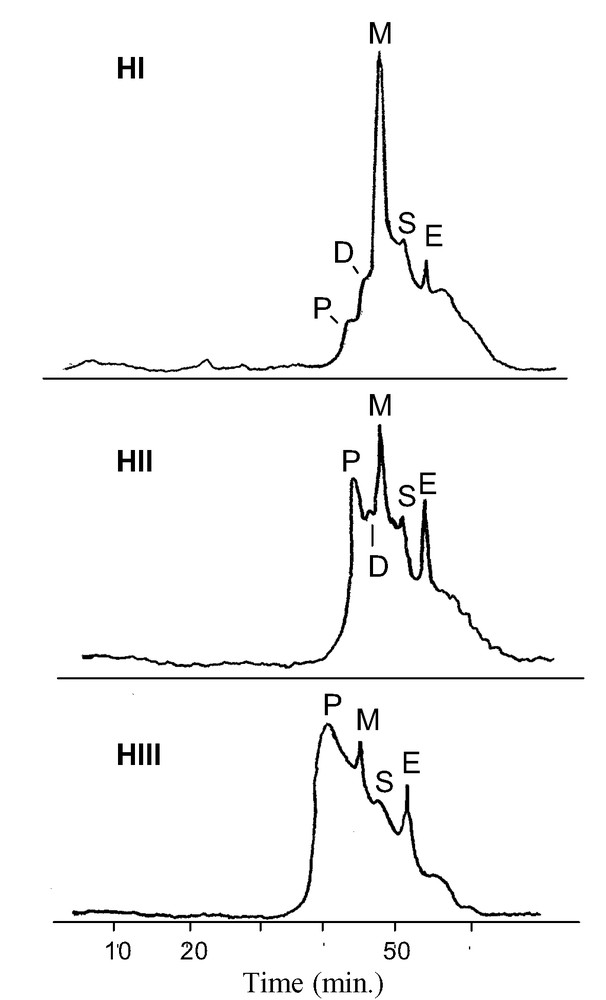
Size exclusion chromatograms of heartwood extracts. E: Ellagic acid; S: vescalin, castalin; M: monomeric ellagitannins; D: dimeric ellagitannins; P: polymeric ellagitannins (for HI-HIII see Fig. 1).
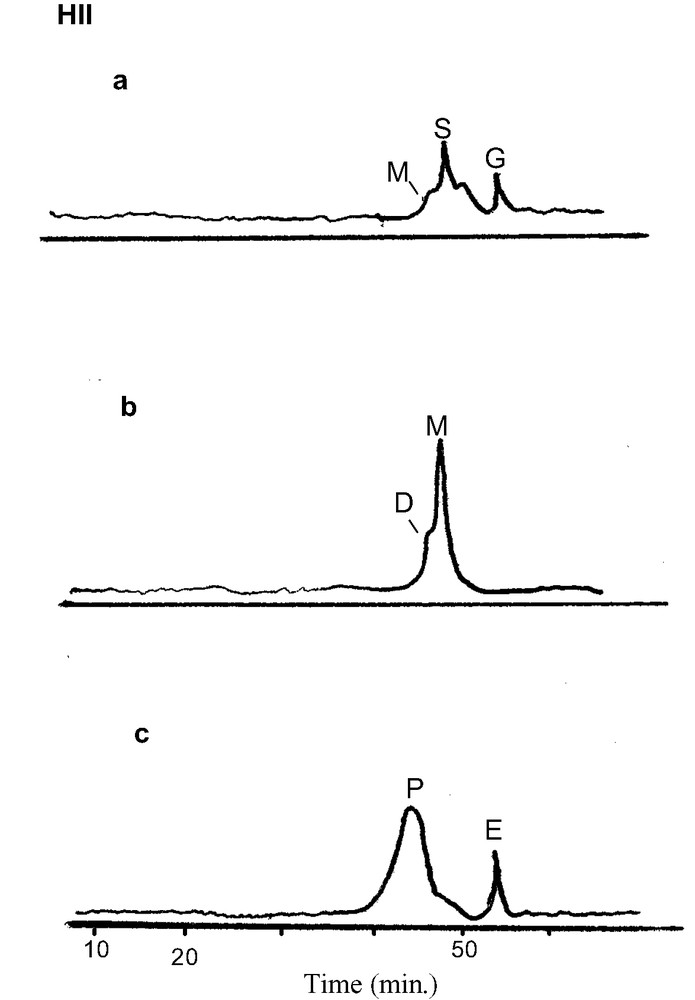
SEC chromatograms of fractions HII(a), (b), (c) obtained by low-pressure chromatography using sephadex LH 20 gel (a: elution with water; b: 30% vol. MeOH; c: MeOH). G: Gallic acid; E: ellagic acid; S: vescalin, castalin; M: monomeric ellagitannins; D: dimeric ellagitannins; P: polymeric ellagitannins e.g. EPs fraction (HII: outer heartwood).
EPs and total extract of HI-HIII were assayed by acid degradation in MeOH–HCl, and released ellagic acid was estimated by reverse-phase HPLC; also total phenols were estimated by the Folin–Ciocalteu method. The ellagitannin/phenol ratio varied in HI–HIII extractives, probably with the relative content of EPs and seemed constant for EPs main compounds of HIc–HIIIc (Table 2). This can be adequately explained by the formation during heartwood ageing of covalent linkages involving hexahydroxydiphenyl units of C-glucosidic ellagitannins. This structural modification affects ellagic acid formation during acid degradation more than the reducing capacities of EPs with the Folin–Ciocalteu reagent.
Evolution of the ellagitannins/total phenols ratio for acetone/water extract of HI–HIII and for purified fractions of soluble polymer ellagitannins (EPs) (results are expressed in mg g–1 wood in gallic acid equivalents for phenols and vescalagin equivalents for ellagitannins)
| Total extract | EPs | |||||
| HI | HII | HIII | HIc | HIIc | HIIIc | |
| Ellagitanninsd/phenolse | 0.68 | 0.43 | 0.17 | 0.02 | 0.03 | 0.02 |
d Acid degradation and ellagic acid determination.
e Folin–Ciocalteu assay.
Purity and polymeric aspects of EPs were investigated by NMR experiments. With solid-state CPMAS 13C NMR, it was possible to estimate the relative content of some of the main groups of components [23]: i.e. cellulose (δ 65–62, 75–73, 89, 84, 105 ppm), hemicellulose (δ 21, 172 ppm), lignins (δ 56, 120, 137, 148, 153 ppm) and ellagitannins (δ 145 ppm). EPs showed a wide signal only at δ 145 ppm. EPs thioacidolysis released no typical lignins products and oses. This polymer was not attributed to heterocomplex forms with ellagitannins, lignins and polysaccharides.
3.3 Chemical reaction involved in polymer formation
To obtain a satisfactory model of the polymeric reaction in the heartwood of live trees, we chose castalagin as monomeric substrate and sterile water with various oxidative factors added (oxygen, hydrogen peroxide, copper). During the experiment, we compared the development of the typical yellow/brown colour of EPs, the chromatographic formation of polymers and spectral characteristics compared to those of natural EPs. To obtain coloured castalagin polymers, a Fenton reaction with H2O2 destroyed by Cu2+ (Fig. 6) was used. The formation of EPs was only found – by SEC – in these conditions. This suggested that the reaction required oxidation by a scavenging agent:
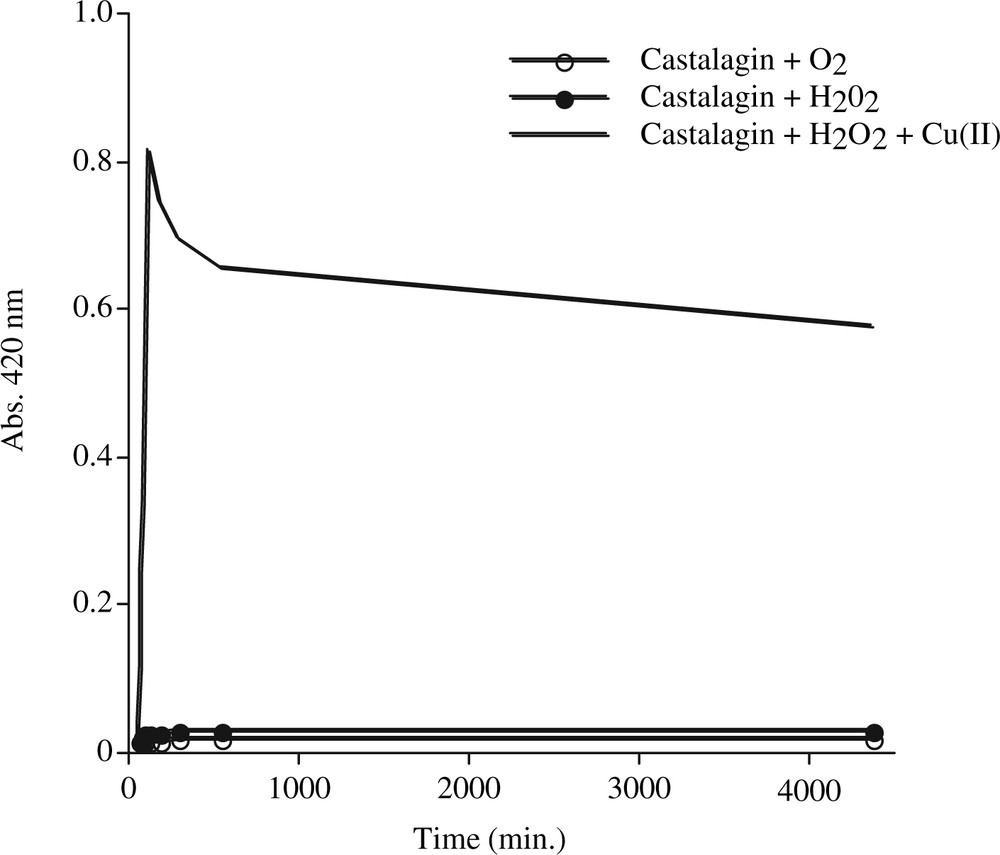
Colour development (420 nm) of castalagin solutions containing: O2 and H2O2 (± Cu2+). —○—: Castalagin + O2; —•—: castalagin + H2O2; ——: castalagin + H2O2 + Cu(II).
The synthetic EPs were collected and isolated in the same way as natural EPs, and the spectral data compared. We named synthetic EPs the product formed by in vitro chemical oxidation of vescalagin, and purified by MeOH LH20 fractionation (HIIIc fraction). This polymer has the same UV spectra as castalagin without a maximum between 250 and 350 nm, only a shoulder at 280 nm. In comparison, lignins present a distinct maximum around 280 nm and a shoulder at 310–320 nm [25]. The bathochromic shifts in the MeOH solution were similar to those observed for castalagin (Fig. 7): with AlCl3 the maximum at 230 nm changed to +3 nm for castalagin and did not change for synthetic and natural EPs, the shoulder at 371 nm shifted to + 22 nm for castalagin and to + 9 nm for EPs. By adding HCl the initial value was recovered in all cases. Compared with ellagic acid and gallic acid spectra, UV absorption at 230 nm can be attributed to the trihydroxymonophenic part of gallic acid and absorption at 371 nm was attributed to the hexahydroxydiphenic part of ellagic acid. The different wavelengths were identified in the spectra of pure acids. The infrared data are collected in Fig. 8. We noted a broad similarity between synthetic and natural EPs (HIIIc) and castalagin and oligomeric ellagitannins (HIIIb). The following results confirm that oxidative reactions occurred during the formation of EP structures. Firstly, compared to castalagin and HIIIb, we noted that the best resolution for synthetic and natural EPs was in the 3500–3200 cm–1 range. All the vibration bands correspond to the OH function. It was presumed that the oxidative polymerisation process enabled an association of a part of OH. Secondly, after oxidation, the vibration bands at 1740 cm–1, due to saturated esters, disappeared, while the band at 1715 cm–1 appeared. This may correspond to the C=C bonds. Thirdly, the characteristic bands of native ellagitannins (1445–1455, 1500–1510, 1615–1620, and 1740 cm–1) decreased or disappeared. The oxidative transformations not only enabled polymerisation between molecules, but also induced major modifications in the structure of the native products. Fourthly, the presence in the two oxidised products of bands at 1250 cm–1 and in the 830–890 cm–1 zone was characteristic of the occurrence of epoxy structures.
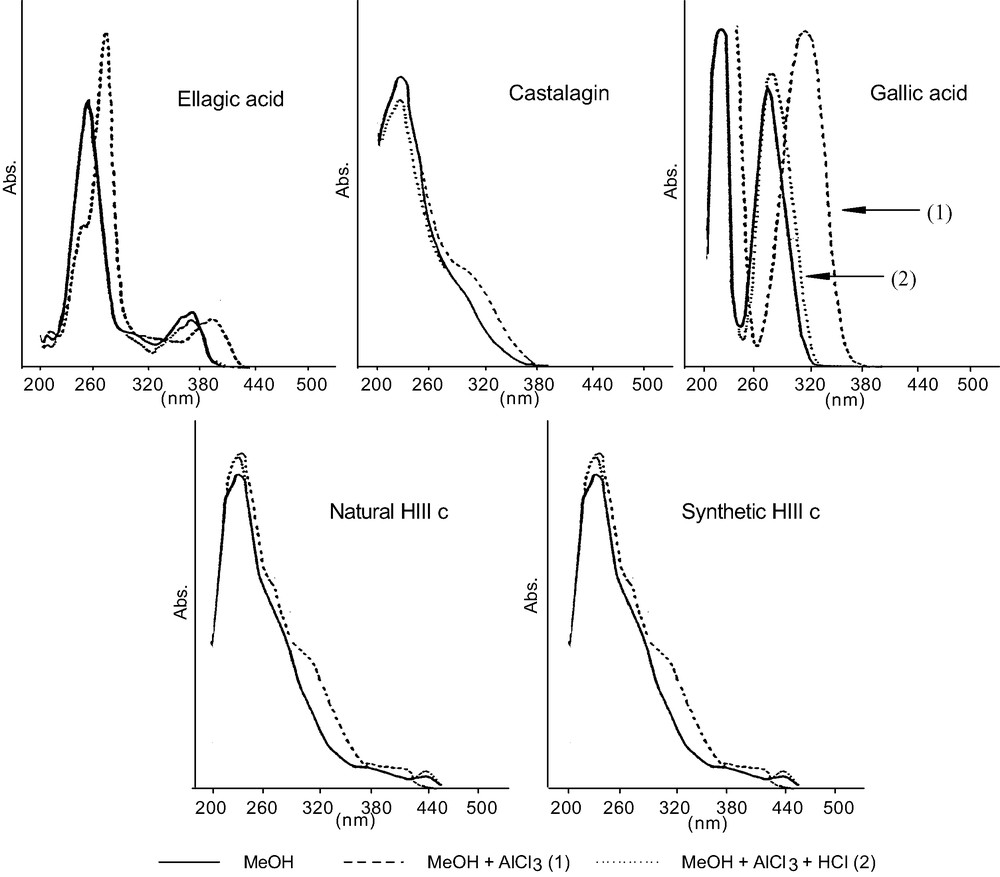
UV–Visible spectrum and bathochromic shifts characteristic of ellagic acid, castalagin, gallic acid, natural HIIIc and synthetic HIIIc.
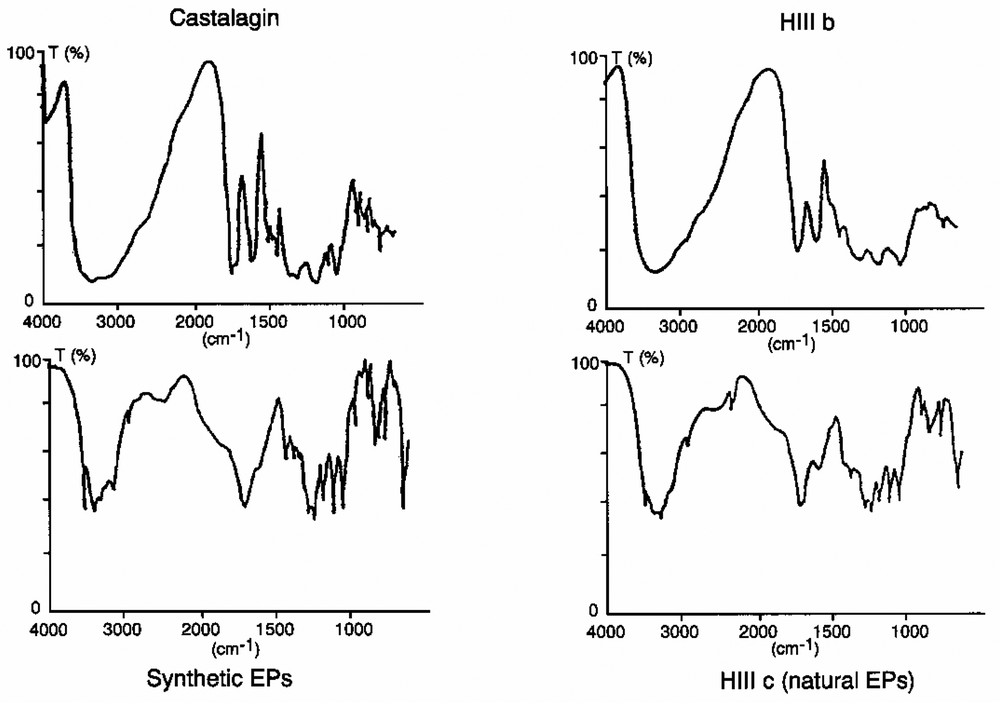
IR spectra of castalagin, HIIIb fraction, synthetic soluble polymeric ellagitannins, HIIIc fraction (natural soluble polymeric ellagitannins).
4 Discussion
In this work, we monitored the formation and conditions for the formation of polymeric forms of ellagitannins from the heartwood of Q. robur and Q. petraea. We observed an accumulation of polymeric forms of ellagitannins during wood ageing. The analyses focused only on the extractible part of EPs, so the study concerns the intermediate stage of EPs normal development towards total insolubilization [18]. In these final conditions, the polymeric forms cannot be extracted and studied because the polymeric level is too high and associations with the wood components of the cells are the cause of this insolubilization.
Our results in in vitro conditions showed that the formation of EPs requires a scavenging reaction (Fenton reaction). Presumably there are similar reactions in wood in an aqueous medium with some catalytic effects of metallic ions. The presence of metallic ions was quantified in a previous work [26]. The presence of EPs in inner heartwood was demonstrated by Viriot et al. [3]; in addition, the authors noted that dimerisation and hydrolysis during wood ageing were associated with polymerisation reactions. Considering those results and the new ones, polymerisation appeared to be the main reaction.
Analytically, the study of purified polymers was difficult albeit possible, for example after a specific depolymerisation reaction like thioacidolysis for lignins and proanthocyanidols [24,27]. For EPs, most of the chemical reactions often used to characterize the polymer composition yielded no results because of the origin of Ep formation. In addition to the normal linkage of ellagitannins polymers, we found a specific linkage due to oxidative reactions affecting all the chemical properties of the molecules. Also because of the complexity of the polymers, identification in NMR signals was not satisfactory.
A complementary analytical study clearly showed that the EPs fraction presented no polysaccharides or lignins associated to ellagitannin polymers.
Vous devez vous connecter pour continuer.
S'authentifier