

1 Introduction
One of the most intriguing properties of zeolites is their ability to generate spontaneously organic radical cations upon adsorption of organic electron donors. The basis of the spontaneous ionization phenomena upon sorption in zeolites was recently reviewed [1]. This review goes back to the early incorporation studies and covers extensively the literature until mid-2000. The rigid microporous solids serve as excellent matrices, stabilizing otherwise reactive or unstable radical cations. The restricted mobility within zeolite pores limits the tendency of free radicals to dimerize and prevents access of reagents that typically would cause their decay in solution. Accordingly, radical cations have increased lifetimes and can be studied by conventional spectroscopic techniques. The radical cations sequestered in zeolites have attracted interest in their own right. The reactions of primary radical cations and their conversions into well-defined secondary intermediates or products have been attracting ever-growing interest. Radical cations have been obtained from a wide range of substrates, including aromatics, polyaromatics, alkenes, cycloalkenes, alkynes, and even selected alkanes and cycloalkanes. A plethora of organic radical cations can be generated spontaneously in zeolites [1]. In contrast, there is limited experimental information to identify the fate of ejected electron, which is the second part of the ionization phenomenon [2]. The composition and structure of the centers responsible for the electron acceptor ability of zeolites are still subject to controversy and debate. Initially, the activity of zeolites was ascribed to impurities, such as iron(III) or extraframework species. A more widely accepted explanation links the ionization ability of zeolites to their acidity, in analogy to the formation of radical cations from aromatics or heterocycles by liquid or solid Brønsted or Lewis acids. A consensus appears to have been reached about the inability of Al-free zeolites to generate ionization. However, recently, alkali metal addition to pure silica zeolites generates substantial ionization and releases relatively free electrons [3]. Understanding the nature of the electron trapping sites may provide control over their population and strength and makes it possible to ‘tune’ zeolites to produce optimal yields of ionization. These features have obvious implications in the area of catalysis in the petrochemical industry.
In this short review, we present selected recent contributions of our research group devoted to the spontaneous ionization of polyaromatic molecules by sorption in ZSM-5 zeolites. In these works, the electron paramagnetic resonance (EPR) spectroscopy, diffuse reflectance UV–visible absorption (DRUVv) spectroscopy and Raman spectroscopy were used to investigate extensively the sorption of anthracene (ANTH), naphthalene (NPH) and biphenyl (BP) in acidic and non acidic ZSM-5 zeolites. We lay the emphasis on the fate of ejected electron and subsequent electron transfer. Applying pulsed EPR techniques, we were able to characterize the surroundings of unpaired electron species of unusual long-lived electron-radical cation or electron–hole pairs in high yield. From all the results, we were also able to propose a comprehensive mechanism of the spontaneous ionization of polyaromatics upon sorption in ZSM-5 zeolites. The proposed mechanism includes size and ionization potential of sorbates, aluminum content of framework and extraframework cations of MFI hosts as well as Lewis acid sites generated during the calcination of the zeolites.
2 Experimental section
Crystals (several μm in size) of as-synthesized silicalite-1 (Si/Al > 1000) and aluminum MFI (Si/Al ~95, 47, 31, 23) were synthesized in high purity according to the fluoride medium procedure in the ‘Laboratoire des matériaux minéraux’, UMR-CNRS 7016, ENSC Mulhouse, France [4]. An industrial source of ZSM-5 zeolites (less than 2 μm in size) was also used. The NH4+-exchanged ZSM-5 samples (Si/Al = 13, 27) were obtained from VAW aluminum (Schwandorf, Germany). The elementary analyses correspond to the following formula: [NH4]3.4(SiO2)92.6(AlO2)3.4·n H2O and [NH4]6.6(SiO2)89.4(AlO2)6.6·n H2O. The elementary analyses as well as spectroscopic investigations indicate iron impurities at trace levels in the as-obtained zeolites. The BP (C12H10), NPH (C10H8), ANTH (C14H10), and 9,10-dimethyl anthracene (DMANTH, C16H16) were used after dehydration procedure. Pure and dry Ar and O2 gas were used.
All the zeolite samples were prepared by a calcination procedure. The as-synthesized and as-obtained zeolites were dried in flowing air from 300 to 400 K over 6 h, then the samples were calcined under pure dry O2 at 800 K for 12 h. The calcined samples were cooled slowly to room temperature under moisture, washed with water, filtered and stocked under wet atmosphere. The H+ cations of HnZSM-5 zeolite samples have been completely exchanged by Li+, Na+, K+, Rb+, Cs+, Mg2+, Ca2+. The exchange was carried out according to the process reported previously [5].
Before each sorption experiment, the Mn(AlO2)n(SiO2)96–n zeolite samples were dried at 573 K for 2 h under vacuum and then dehydrated under Ar at 400 K for 6 h. The Mn(AlO2)n(SiO2)96–n zeolite samples were also calcined under Ar or O2 for 12 h at 473, 573, 673, 773, 873, 973, and 1023 K. Then, the sample was cooled to room temperature under dry argon and held under vacuum for 1 h and held under dry Ar. The vacuum-argon treatment was repeated three times. The powder XRD patterns, 29Si, 27Al MAS-NMR, IR absorption and Raman spectra of bare MFI zeolites used in this present work were recorded to check the crystallinity and purity of the samples. The Brønsted and Lewis acidity of the calcined Hn(AlO2)n(SiO2)96–n samples were characterized by FTIR measurements using pyridine as probe molecule [6].
Weighted amounts of polyaromatics were introduced under dry Ar into the cell containing Mn(AlO2)n(SiO2)96–n (powder of microcrystals sizing around 1 μm) and the powder mixture was shaken. The loading value was expected to correspond to 1 sorbate per host unit cell (UC) unless otherwise indicated. The mixtures were transferred under dry argon in quartz glass Suprasil cuvette for Raman and diffuse reflectance UV–visible experiments, respectively. For EPR experiments, the sample was loaded into cylindrical quartz tube under argon. The tube was flame sealed.
All the other experimental conditions: instrumentation, data processing and calculations were detailed in previous works [5–10].
3 Results
3.1 Sorption of anthracene and 9,10-dimethyl anthracene in MnZSM-5 (n = 0, 3, 3.4, 6.6; M = H+, Li+, Na+, K+, Rb+, Cs+)
3.1.1 Molecular modeling. Effect of sorbate size
The framework structure of purely siliceous ZSM-5 (n = 0) zeolite or silicalite-1 (SiO2)96 contains two types of intersecting channels, both formed by rings of 10 oxygen atoms, characterizing them as a medium-pore zeolite. One channel type is straight and has a nearly circular opening (0.53 × 0.56 nm2), while the other one is sinusoidal and has an elliptical opening (0.51 × 0.55 nm2). Monte Carlo simulations and energy minimization procedures of the nonbonding interactions between rigid molecules and fixed zeolite framework provide a reasonable structural picture of polyaromatic molecules occluded in silicalite-1 [8]. ANTH was found to be able to penetrate into the internal void space of the zeolite and the preferred locations lay in straight channels in the vicinity of the intersection with zigzag channel; the three cycles run along the b direction of the cell, (Fig. 1). The main structural role of the zeolite framework appears to constrain the molecular orientation of ANTH to be along the b direction of the cell because of the tight fit between the sizes of rod shape guest and straight channel of host. Molecular dynamics calculations indicate slow down the ANTH mobility in the void space over 1 ns simulation time [8]. However, slow ANTH diffusion can occur in the straight channel of MFI zeolites.
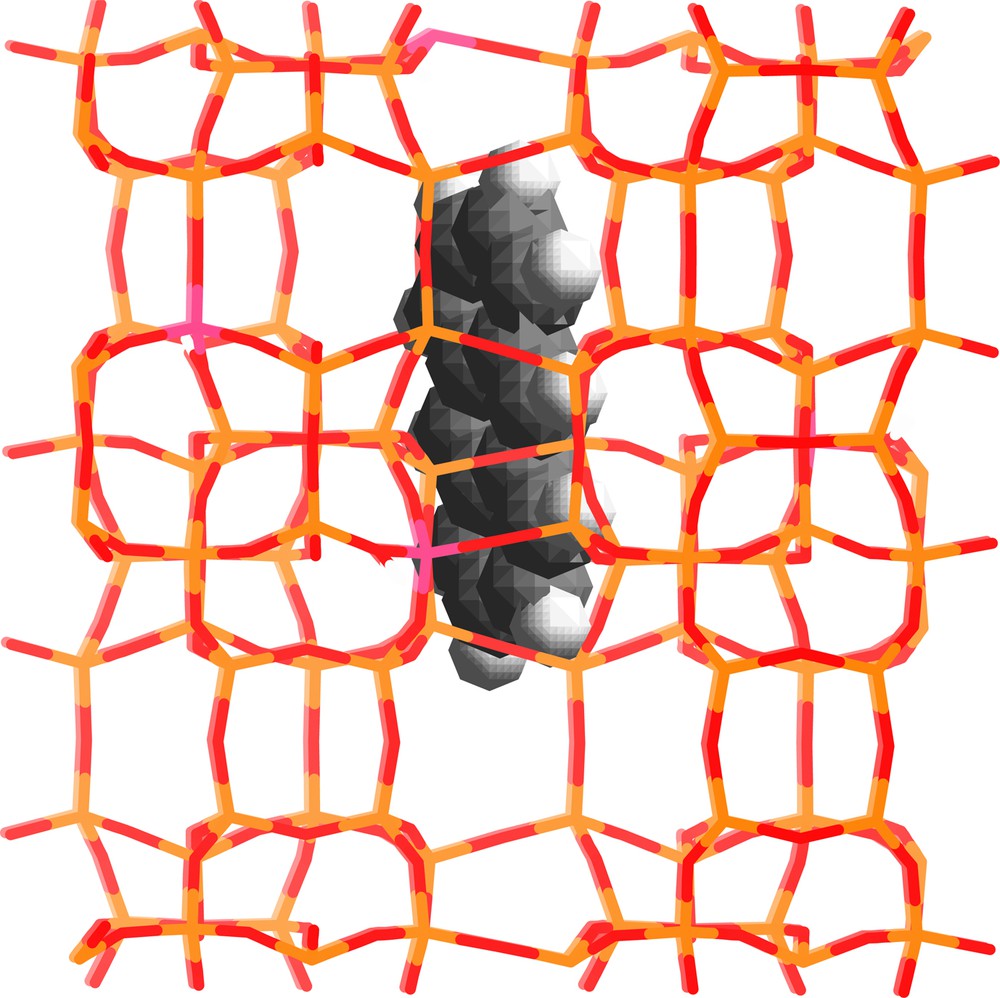
Modeling of the structure of ANTH occluded in the straight channel of silicalite-1. Light and dark grey sticks represent the Si and O atoms of the framework, respectively. The white and dark grey balls represent the H and C atoms, respectively.
Molecular modeling shows the energetically more favorable adsorption sites of DMANTH. Because of the large molecular size and sterical constraints of DMANTH (Fig. 2) DMANTH molecules are capable of plugging the pore opening. It is reasonable to assume that the remaining molecules lie flat on the external surface in non-specific adsorption sites. DMANTH was not found to be able to diffuse into the channel system of silicalite-1 [8].
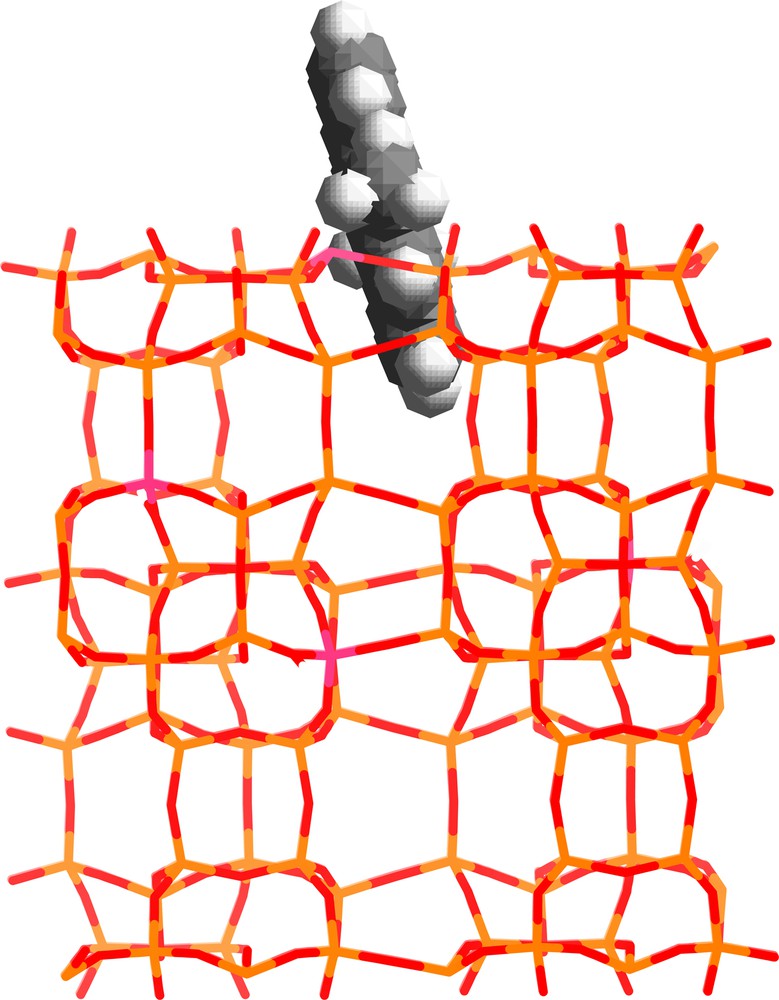
Modeling of the structure of DMANTH adsorbed at the openings of the pores of silicalite-1. Light and dark grey sticks represent the Si and O atoms of the framework, respectively. The white and dark grey balls represent the H and C atoms, respectively.
Modeling of the ANTH sorption in MnZSM-5 (n = 4; M = H+, Li+, Na+, Cs+) indicates a facial interaction between occluded ANTH and the counterbalancing cation as shown for NPH in the Fig. 7 of the Ref. [6].
CW-EPR spectra recorded at room temperature during the course (2 months) of ANTH sorption into dehydrated non acidic [Li6.6(AlO2)6.6(SiO2)89.4] zeolite. Solid ANTH (1 ANTH per UC loading) and Li6.6ZSM-5 dehydrated at 573 K were mixed under Ar.
3.1.2 Sorption of anthracene in acidic HnZSM-5 (n = 0, 3, 3.4, 6.6). Ionization
3.1.2.1 Diffuse reflectance UV–visible absorption (DRUVv) spectrometry
The exposure under argon atmosphere and at room temperature of solid ANTH to calcined silicalite-1 microcrystals did not generate any color change. However, DRUVv spectral changes indicated the sorption occurred, but did not provide any evidence of ionization. In contrast, the exposure in the dark under argon atmosphere at room temperature of dry ANTH to acidic Hn(AlO2)n(SiO2)96–n (n = 3, 3.4, 6.6) freshly calcined under Ar or after mere dehydration turned immediately the powder from white to green. In accordance with these observations, DRUVv spectra showed rapidly characteristic bands of ANTH+• radical cation between 500 and 750 nm.
The Fig. 3 shows the DRUVv absorption spectra recorded at room temperature for different times after the mixing of solids. Data processing using the SIMPLISMA program of numerous DRUVv recorded during the course of ANTH sorption provided evidence of ANTH•+ in high yield [8]. The absorption band at 709 nm was assigned to the 2Au ← 2B2g 0–0 electronic transition with vibronic bands at 652, 620 and 606 nm. The ionization went to completion at room temperature over several weeks. No clear spectroscopic evidence of trapped electron was detected in the UV–visible absorption spectra except a weak band at 450 nm.
| (1) |
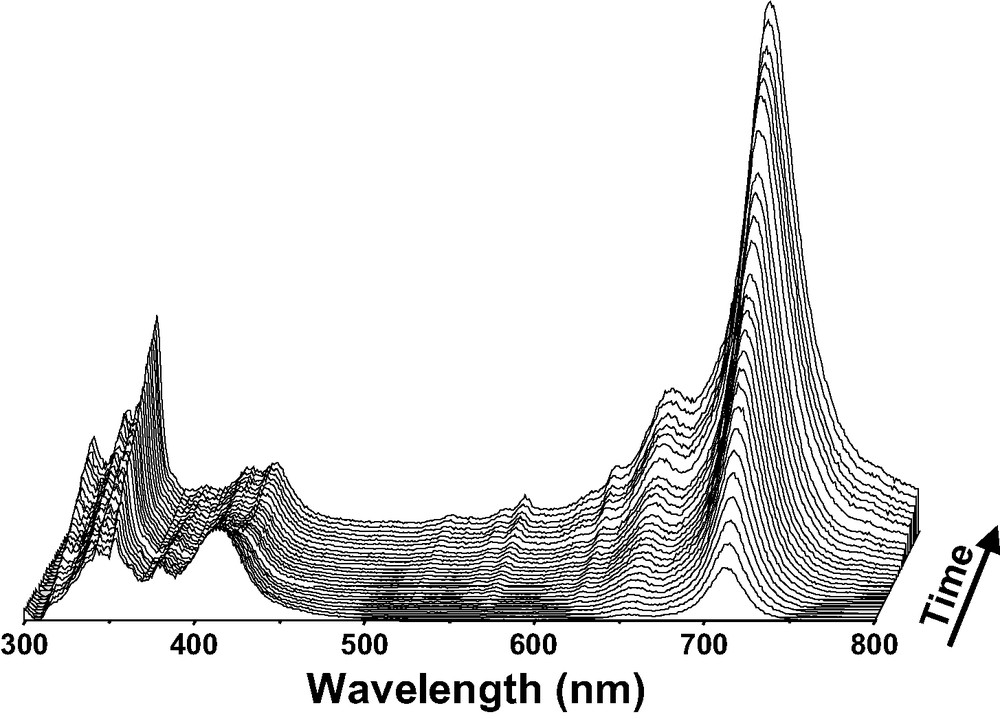
DRUVv spectra recorded at room temperature during the course (6 days) of ANTH sorption into dehydrated acidic [H3.4(AlO2)3.4(SiO2)92.6] zeolite. Solid ANTH (1 ANTH per UC loading) and H3.4ZSM-5 dehydrated at 573 K were mixed under Ar.
The exposure of solid DMANTH to calcined silicalite-1 microcrystals did not generate any color change. No evidence of DMANTH radical cation was observed in the DRUVv spectra after the mixing of DMANTH and Hn(AlO2)n(SiO2)96–n (n = 3, 3.4, 6.6) freshly calcined under Ar or O2. Due to its bulky size, the radical cation cannot take advantage of the stabilization within the void space of ZSM-5.
3.1.2.2 Continuous wave electron paramagnetic resonance (CW-EPR) spectrometry
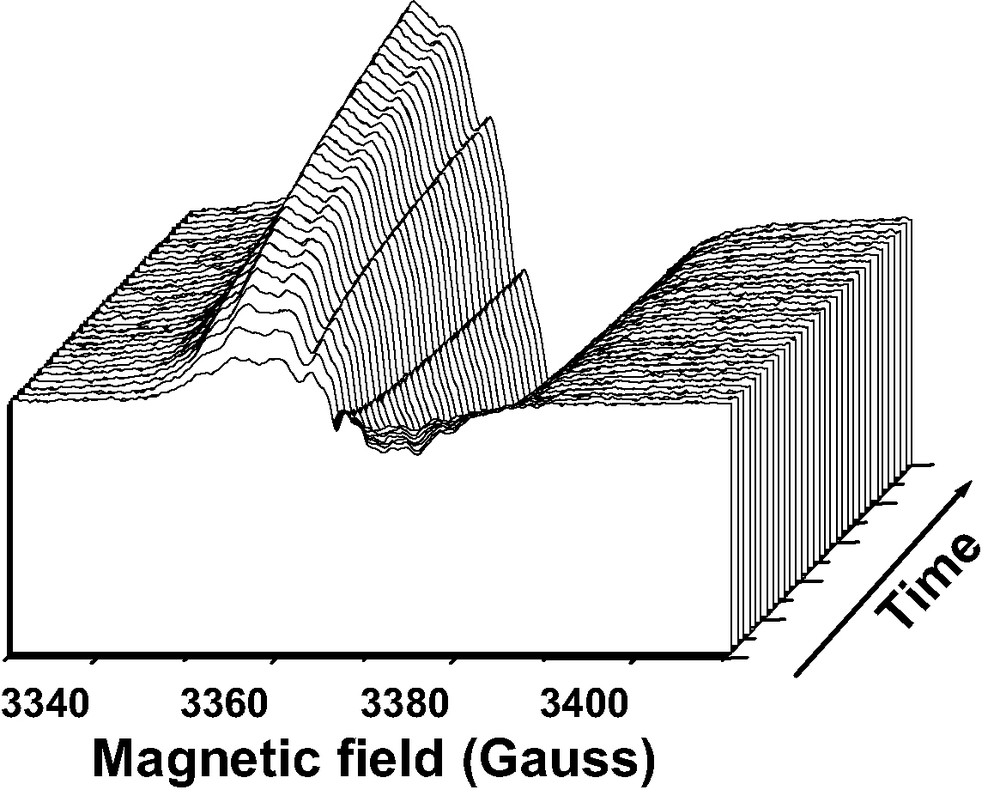
The examination of the CW-EPR spectra recorded after the mere mixing of solid ANTH to calcined HnZSM-5 in the same experimental conditions as described above, indicated sharp signals superimposed over a broad signal (10 G) (Fig. 4). The sharp and broad signals were assigned straightforwardly to ANTH•+ and to trapped electron, respectively [10].

CW-EPR spectra recorded at room temperature during the course (6 days) of ANTH sorption into dehydrated acidic [H3.4(AlO2)3.4(SiO2)92.6] zeolite. Solid ANTH (1 ANTH per UC loading) and H3.4ZSM-5 dehydrated at 573 K were mixed under Ar.
The double integration of the EPR signal in the g = 2 region indicated that the ionization is nearly complete after 3 months according to reaction [1] and was found to generate a spin quantity corresponding to two unpaired electrons per UC. No such behavior was observed with DMANTH as sorbate with analogous experimental conditions. The analysis of the spin process was performed by pulsed EPR techniques.
3.1.2.3 Pulse electron paramagnetic resonance (EPR) spectrometry
The advantages of pulse EPR compared to conventional continuous-wave CW-EPR include the large variety of experimental schemes and the multi-dimensionality, which allow for a much more detailed investigation of paramagnetic compounds. However, in contrast to NMR, CW and pulsed EPR are still to a large extent complementary. Field-swept EPR techniques reveal information about the electron Zeeman interaction (the electronic state of the material under study), and about the fine structure (coupling between unpaired electrons), and about strong hyperfine structure (interaction between the unpaired electron and the surrounding nuclei). A strong hyperfine interaction is usually observed when the nucleous is in close vicinity of the unpaired electron. The interactions with more distant nuclei can be investigated by means of electron spin echo envelope modulation (ESEEM) spectroscopy. Furthermore, a number of techniques have been invented to study distances between different paramagnetic centers. With ESEEM techniques, the spectral resolution can be improved by orders of magnitudes. Among all the available pulse techniques, the two-pulse ESEEM spectroscopy used the most simple pulse sequence. The Spin Echo Correlation SpectroscopY (SECSY) and Hyperfine Sublevel Correlation spectroscopy (HYSCORE) used multiple-pulse sequences and Fourier transformation of the time-domain data results in a two-dimensional (2D) frequency domain spectrum. They are largely used in the present work.
The excitation of the complex CW-EPR signal obtained after complete ionization as ANTH+•@Hn(AlO2)n(SiO2)96–n–• with a two-pulse echo sequence yields two spin process phenomena. Effectively, jointly to the echo generation a strong Free Induction Decay (FID) process is observed due to the presence of a homogeneous spin packet. These two jointly observed phenomena result from the presence in the sample of two non equivalent chemical magnetic species in ANTH+•@Hn(AlO2)n(SiO2)96–n–•. Performing spin lattice T1 measurements using, respectively, FID inversion recovery with 16 step phase cycling to suppress echo process and echo inversion recovery yields to, respectively, very long T1 for both processes with threefold higher values for the echo process [10].
The T1 values recorded at room temperature were found to be 100 and 310 μs for FID and echo processes, respectively. Such kinds of behaviors give us the possibility to identify the paramagnetic species that are responsible for two distinct spin magnetic processes. Recording 2D T1 versus field sweep for the FID signal allows us to observe along the field direction the typical CW ANTH+• spectrum with numerous lines due to the proton hyperfine coupling. The spectrum of Fig. 5 left is indicative of the formation of ANTH+• in accurate agreement with DRUVv and Raman spectroscopy [10]. This spectrum consists of at least twenty split components. Comparison of these 1H hyperfine splittings with those of a pure ANTH+• isotropic spectrum recorded in solution reveals the disappearance of some 1H hyperfine splittings due to an anisotropic broadening effect. This feature has also previously been observed for ANTH+• generated on a silica-alumina catalyst surface and during oxidation by poly-oxometalate [11,12].

Left. Projection slice spectrum (T1 = 150 μs) characteristic of the pure ANTH+• as observed along the field direction of 2D spin lattice relaxation time T1 versus field sweep measurements recorded at room temperature of ANTH+• radical cation/electron moiety occluded in H3ZSM-5. The FID inversion recovery with 16 step phase cycling to suppress echo processes was used. Right. Projection slice spectrum (T1 = 310 μs) characteristic of the pure trapped electron as observed along the field direction of 2D spin lattice relaxation time T1 versus field sweep measurements recorded at room temperature of ANTH+• radical cation/electron moiety occluded in H3ZSM-5. The Echo inversion recovery with 8 step phase cycling to suppress FID process was used. Masquer
Left. Projection slice spectrum (T1 = 150 μs) characteristic of the pure ANTH+• as observed along the field direction of 2D spin lattice relaxation time T1 versus field sweep measurements recorded at room temperature of ANTH+• radical cation/electron moiety ... Lire la suite
Similar experiments with echo detection only yield the observation of the echo shape along the field direction. The Fig. 5 right exhibits the projection slice spectrum (T1 = 310 μs) as observed along the field direction of 2D spin lattice relaxation time T1 versus field sweep measurements recorded at room temperature. The Echo inversion recovery with 8 step phase cycling to suppress FID process was used. The echo signal is relevant to the electron trapped in the zeolite framework but needs supplementary pulsed EPR experiments [10].
The differences in T1 values allow us to characterize separately ANTH+• and the trapped electron by SECSY spectroscopy. So, we have performed at room temperature and 4.2 K the analysis of the structure of these two species by using SECSY for the FID (Fig. 6 left) and echo SECSY for the echo signal (Fig. 6 right). The SECSY spectra give the usual EPR spectrum along the f2-axis and the nuclear frequencies along the f1-axis, respectively. This method provides the distribution of nuclear modulation frequency included in inhomogeneous EPR spectrum. In addition, the characterization of the echo signal was completed by HYSCORE experiments (not shown). It has been shown that these HYSCORE experiments offer an excellent resolution of the hyperfine splitting [10].

SECSY spectra of 1 ANTH+• @H3ZSM-5–•. Left. Two-pulse FID SECSY spectrum of ANTH+•. Right. Two-pulse echo SECSY spectrum of H3ZSM-5–• trapped electron. The spectra were recorded at 4.2 K at the maximum of CW-EPR signal. Two-pulse SECSY spectrum was recorded with pulse lengths of 12 ns for π/2 pulses and a 16 step phase cycling (π/2–t1–π/2–t1–t2–echo) was applied. (Reprinted with permission from Ref. [10]. Copyright (2003) Wiley-VCH Verlag).
Firstly 2D SECSY of the FID process was achieved (Fig. 6 left) and we observe along the diagonal the signature of proton/electron coupling of the ANTH+• as observed on the projection along CW domain of the Fig. 5 left. Moreover, we can observe that the diagonal signal measured at the resonance is not completely symmetric and that on both negative and positive quadrants some cross-peaks appear and occur from Heisenberg spin exchange mechanisms. This result confirms that pairing effect should exist between ANTH+• and trapped electron. Secondly, due to the strong FID process, a 16 step phase cycling SECSY sequence was used to suppress completely the anti echo FID process. The 2D echo SECSY contour plot shows in both positive and negative f1 quadrants the double quantum peak (2ν 13C = 7.4 MHz) of 13C nuclear modulation (ν 13C = 3.7 MHz) centered at 0 MHz in the f2 domain, (Fig. 6 right). This is consistent with an unpaired isolated trapped electron coupled with carbon of ANTH+•. Additional proton pattern centered at the carbon nuclear frequency is observed with ν 1H = 14.5 MHz and combination lines at 2ν 1H = 29 MHz [10]. Such changes were also observed in the HYSCORE spectrum (not shown) where the 1H anisotropic constant measured is 9.8 MHz and a weak carbon coupling is observed in the (+, +) f2 domain with an anisotropic constant of 2.1 MHz and a positive sign for the hyperfine splitting constant. Moreover no signal feature from zeolite interaction through 29Si or 27Al was observed in the HYSCORE spectrum. The same patterns of HYSCORE and SECSY are observed for room temperature and 4.2 K measurements. Due to its quadrupolar moment, 27Al can only be observed at 4.2 K. Consequently, the HYSCORE pattern observed at 3.7 MHz at room temperature and 4.2 K can only be attributed to 13C modulation and not to 27Al nucleus for which nuclear modulation frequency is 3.8 MHz [10]. Such results seem to indicate that the electron is trapped in close proximity of occluded ANTH+• with some pairing effect. It should be noted that the trapped electron is stabilized in the zeolite framework with no visible coupling with 27Al or 29Si nuclei and probably in close proximity of oxygen. The life time of the ANTH+•@Hn(AlO2)n(SiO2)96–n–• moiety was found to be more than several months. It should be noted that in solution the life time of radical cation-electron pair was reported to be less than the nano-second.
3.1.3 ANTH+•@Hn(AlO2)n(SiO2)96–n–• (n = 3, 3.4, 6.6). Temperature-dependent reversible electron transfer
Upon heating of ANTH+•@Hn(AlO2)n(SiO2)96–n–• sample equilibrated at room temperature, the hyperfine structure of the CW spectra disappears and only a featureless signal was obtained after 1 h at 473 K. The hyperfine splitting reappeared upon cooling to room temperature after an equilibration period of several hours. Identical phenomenon was observed by DRUVv spectrometry. The intense absorption bands found around 300 and 700 nm characteristic of ANTH+• disappeared gradually upon heating. The development went to completion at 473 K. The resulting spectral maxima (383, 364, 347 nm) were found to be analogous to those exhibited by occluded ANTH in the ground state (384, 364, 347 nm) except a weak and broad band absorption around 500 nm, spectrum (c) of Fig. 1 left of Ref. [10]. The broad EPR signal and the absorption at 500 nm were straightforwardly attributed to occluded electron–hole pairs with respect to ANTH occluded in ground state.
| (2) |
The warming of the electron–hole pair ANTH@H3ZSM-5+•–• did not provide any decrease of the double integrated EPR signal and did not accelerate the charge recombination. This electron transfer was found to be reversible when the sample was cooled to room temperature.
The broadness of the CW-EPR spectrum implies that the signal is attributed to the electron–hole pair because the magnetic interactions of the exchange and dipolar interactions electron and hole can make the EPR spectrum broader [13]. The two-pulse echo experiment recorded for the electron–hole pair shows the complete disappearance of the FID process in accordance with the disappearance of ANTH+•. Moreover, T1 echo measurements yield the same value as that measured for the trapped electron of the ANTH+•/electron pair (see above), demonstrating identical relaxation times for trapped electron of the ANTH+•/electron pair and for unpaired electrons involved in the electron–hole pair.
Both the 2D echo SECSY contour plot and the HYSCORE pattern of electron–hole pair were found to be analogous to those recorded for the ANTH+•/electron moiety (Fig. 6 right). In particular, the 3.7-MHz pattern observed in the HYSCORE spectrum is assigned to 13C nuclei. Such results indicate that the electron–hole pair interacts with occluded neutral ANTH. Unfortunately, no evidence was found for coupling between unpaired electrons and the 27Al or 29Si nuclei of zeolite framework. The electron–hole pairs are probably stabilized in close proximity of O nuclei. Unfortunately, the low 17O natural abundance did not permit any coupling in the HYSCORE pattern. The EPR signal broadness implies probably weak spin interaction through exchange and dipolar interactions [13].
3.1.4 Sorption of anthracene in non-acidic MnZSM-5 (M = Li+, Na+, K+, Rb+, Cs+, Mg2+, Ca2+). Effect of extraframework cation on ionization
The exposure in the dark under argon atmosphere at room temperature of dry ANTH to Li6.6ZSM-5 freshly dehydrated or calcined under Ar turned immediately the powder from white to green. In accordance with these observations, UV–visible absorption spectra show rapidly characteristic bands of ANTH+•. The examination of the CW-EPR spectra recorded after the exposure of solid ANTH to calcined Li6.6ZSM-5 indicates sharp signals superimposed over a broad signal of 10 G (Fig. 7).
The double integration of the EPR signal in the g = 2 region with respect to a standard indicates that the ionization is not complete after 3 months according to reaction [3] and was found to generate spin quantity corresponding to 0.6 unpaired electron per UC. It should be noted that the ANTH sorption was found to be quantitative using DRUVv experiments. Approximately 30% was sorbed as ANTH+•@Li6.6(AlO2)6.6(SiO2)89.4–• and 70% was sorbed as ANTH@Li6.6(AlO2)6.6(SiO2)89.4. The ionization yield was found to be very weak (2%) with Na+ as extraframework cation. No spontaneous ionization was observed upon sorption of ANTH in MnZSM-5 (M = K+, Rb+, Cs+) with analogous experimental conditions. In contrast, when the exchanged cations are Mg2+, Ca2+ spontaneous ionization occurs in good yield (~0.5 electron per UC) according to following reaction [3].
| (3) |
3.2 Sorption of naphthalene in activated acidic HnZSM-5 (n = 3, 3.4, 6.6). Ionization and electron transfer
3.2.1 Diffuse reflectance UV–visible absorption (DRUVv) spectrometry
No color change was observed after mixing solid NPH and non acidic MnZSM-5 (n = 0, 3.4, 6.6; M = Li+, Na+, K+, Rb+, Cs+) dehydrated or calcined under Ar or O2. No intense color was observed after the mixture of NPH with acidic HnZSM-5 dehydrated under Ar at 573 K. In contrast, several minutes after the mixing of solid NPH with HnZSM-5 (n = 3.0, 3.4, 6.6) zeolites calcined at 773 K under O2, the powder turned blue. Several months at room temperature after mixing, the blue powder turned to dark pink. Analogous dark pink powders were obtained after several days under gentle warming (400 K). DRUVv spectra (not shown) recorded after the mixing of NPH with non acidic MnZSM-5 (n = 0, 3.4, 6.6; M = Li+, Na+, K+, Rb+, Cs+) calcined under O2 at 773, 873 or 973 K exhibit marked increasing of the prominent band at 270 nm. This feature was attributed to sorption of NPH within the void space of porous material. The DRUVv spectra (not shown) recorded after the mixing of solid NPH with HnZSM-5 dehydrated at 573 K under Ar exhibited NPH sorption as intact molecule but did not generate NPH+• radical cation in high yield.
In contrast, the DRUVv spectra recorded after the mixing of solid NPH with HnZSM-5 zeolites (n = 3.0, 3.4, 6.6) calcined at 673, 773 and 873 K under O2 exhibit supplementary NPH+• absorption bands (Fig. 8) generated according to the following reaction [14–16].
| (4) |

DRUVv spectra recorded at room temperature during the course (2 months) of NPH sorption into calcined acidic [H3.4(AlO2)3.4(SiO2)92.6] zeolite. Solid NPH (1 NPH per UC loading) and H3.4ZSM-5 calcined under O2 at 773 K were mixed under Ar.
The characteristic absorption bands of NPH•+ disappeared approximately after 1 month at room temperature with concomitant appearance of ill-defined large and broad bands in the visible region. After several months at room temperature, the shape of the bands broadened to large background with maximum in the red region.
Under gentle warming at 400 K during several hours, the appearance of broad bands was observed at 470 and 620 nm concomitantly with the disappearance of NPH+•. The Fig. 9 shows some of the DRUVv spectra recorded under warming at 400 K of a sample obtained 24 h after the mixing of NPH (1 NPH per UC) with H3.4ZSM-5 calcined at 773 K. The broad absorption bands obtained after the disappearance of the NPH•+ bands were relevant to electron–hole pairs generated by electron transfer between NPH•+ and the zeolite framework (reaction [5]).
| (5) |
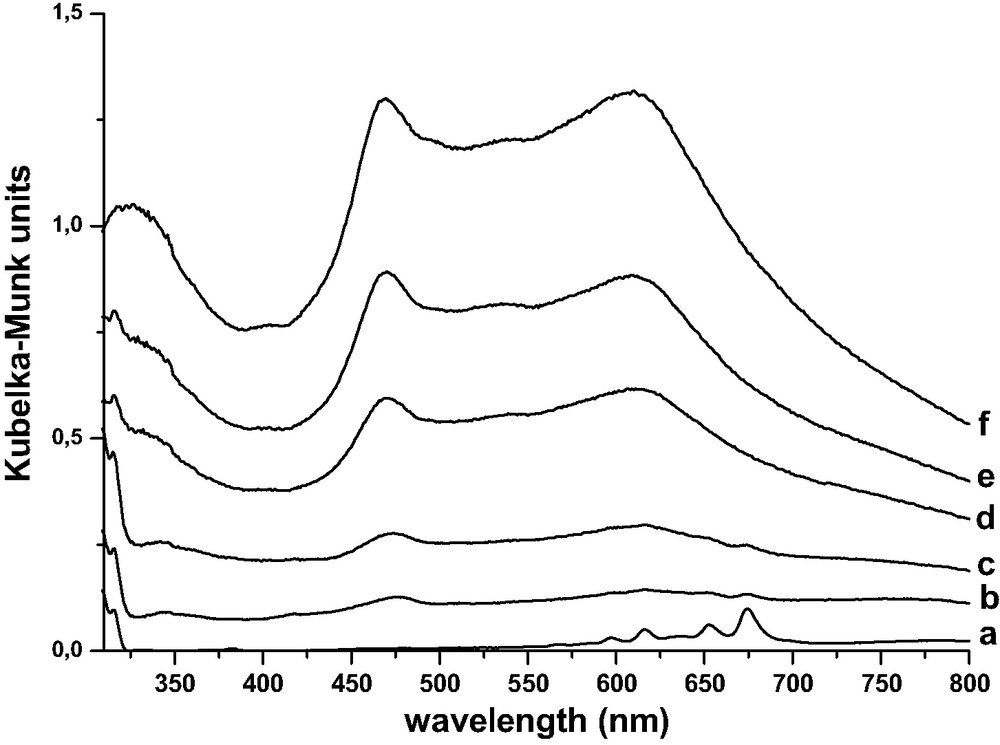
DRUVv spectra recorded for different times (b–f) under heating at 400 K of (a) 1 NPH@H3.4(AlO2)3.4(SiO2)92.6 sample obtained 24 h after the mixing under Ar of solid NPH and acidic H3.4(AlO2)3.4(SiO2)92.6 calcined under O2 at 773 K. (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society).
After several days at 400 K, the bands at 470 and 620 nm disappeared. This fact corresponds to the charge recombination.
| (6) |
3.2.2 Continuous wave electron paramagnetic resonance (CW-EPR) spectrometry
MnZSM-5 (n = 3.0, 3.4, 6.6; M = H+, Li+, Na+, K+, Rb+, Cs+) zeolites activated at 773 K under O2 after evacuation under vacuum and subsequent admission of Ar, did not exhibit any EPR signal at 100 or 300 K [17]. No supplementary signal was detected for non acidic MnZSM-5 (n = 3.4, 6.6; M = Li+, Na+, K+, Rb+, Cs+) activated under O2 and mixed with solid NPH (1 NPH per UC) in our experimental conditions. Very weak features were observed in the 2 g-values for aluminated HnZSM-5 (n = 3.0, 3.4, 6.6) zeolites calcined at 773 K under Ar and loaded with NPH (1 NPH per UC). In contrast, intense signal was detected immediately after exposure of solid NPH to HnZSM-5 (n = 3.0, 3.4, 6.6) samples calcined under O2 at 773 K (Fig. 10). Numerous EPR spectra were recorded at different times during the course of NPH sorption in H3.4ZSM-5 (Fig. 10).

CW-EPR spectra recorded at room temperature during the course (2 months) of NPH sorption into calcined acidic [H3(AlO2)3(SiO2)93] zeolite. Solid NPH (1 NPH per UC loading) and H3.4ZSM-5 calcined under O2 at 773 K were mixed under Ar. (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society).
In addition to narrow lines, the EPR spectra contain broad overlapping feature, which increases in intensity during the course of the sorption (Fig. 10). The data processing of the spectra set provides evidence of two spectra [6,10,18]. One spectrum (A) with resolved hyperfine structure was readily assigned to NPH•+. The characteristic spectral line shape is caused mainly by the hyperfine anisotropy of the ring protons [19]. The other one (B) with broad structureless signal was tentatively attributed to other unpaired electrons of HnZSM-5–• and HnZSM-5+•–• moieties produced by reactions [4] and [5]. The EPR signal broadness implies probably weak spin interaction through exchange and dipolar interactions. The possibility of electron trapping by H+ of Si–OH–Al group can be ruled out [20]. The EPR pattern was in accurate agreement with the ionization reactions [4] and [5]. The double integration of A and B signals provides spin density values with respect to a standard as a function of time, respectively [6].
The spin quantity was expressed in spins per UC. Fig. 11 exhibits the spin quantity corresponding to A and B components, respectively, detected over 1 day after the exposure at room temperature of solid NPH to H3.4ZSM-5 activated at 773 K under O2.
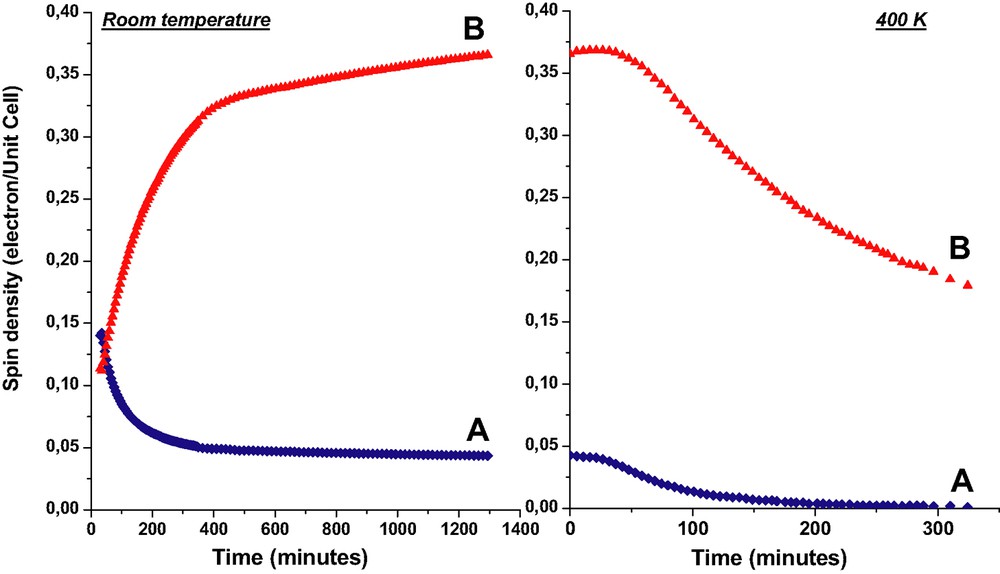
Spin quantities corresponding to NPH+• (A) and to unpaired electrons and holes (B) detected as a function of time. Left: over 24 h after the exposure at room temperature of solid NPH (1 NPH per UC loading) to H3ZSM-5 calcined at 773 K under O2. Right: upon heating at 400 K of the equilibrated sample at room temperature (24 h). (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society).
From Fig. 11, one can observe that A and B intensities are practically equivalent 30 min after the mixture of solids and correspond to 0.14 spin per UC. The NPH•+ signal decreases to 0.06 spin per UC during the first 3 h and reaches a plateau at 0.05 in about 6 h. In contrast, the broad signal increases markedly from 0.12 to 0.33 spin per UC during the first 5 h and increases hardly to 0.38 spin after 24 h. At room temperature, the disappearance of NPH•+ goes hardly to zero over 1 month after the mixing of the solids, whereas the broad signal is still prominent after several months. Upon heating at 400 K of an equilibrated sample at room temperature (24 h) the disappearance of NPH•+ was found to be complete over 6 h whereas the broad signal decreases from 0.48 to 0.24 over the same period and goes to zero over several days at 400 K (Fig. 11). Cooling to room temperature stopped the decrease of the signal and trapped the paramagnetic moieties within the zeolite framework.
In the same way, numerous EPR spectra were recorded at different times during the course of NPH sorption in H6.6ZSM-5 activated at 773 K under O2, with a loading corresponding to 1 NPH per UC. The EPR results (not shown) were found to be qualitatively analogous at room temperature and 400 K to that described above. The maximum values of NPH•+ (A signal) and electron and hole (B signal) spin densities measured by CW-EPR measurements during the NPH sorption were listed in Table 1 of Ref. [6] according to the calcination and loading conditions of the HnZSM-5 zeolites. The maximum quantity of spin of NPH•+ (A signal) generated by sorption depends highly on the temperature of calcination under O2 and on the calcination time. The ionization yield can reach 30% of occluded NPH after thermal treatment of H6.6ZSM-5 over several days under O2 at 873 K. It is difficult to estimate the maximum of spin quantity of the electrons and holes (B signal) generated at room temperature because of the slowness of the migration electrons and holes and competitive charge recombination. The maximum of spin density is better determined after gentle warming at 400 K and reaches 0.5 per UC starting from 0.15 per UC of NPH•+. The spontaneous ionization rate and yield increase as the calcination temperature increases. In the same way, the electron–hole pair yield increases as the ionization yield increases. Finally, complete charge recombination occurs over several days at 400 K.
3.2.3 SECSY and HYSCORE EPR measurements
Pulse EPR experiments were performed in order to obtain a detailed picture of the chemical environment of spins generated during the primary spontaneous ionization (Fig. 12) and after the disappearance of NPH•+ and subsequent formation of electron–hole moieties (Fig. 13). Both joined two-pulse SECSY and HYSCORE experiments were carried out at room temperature and at 4.2 K. In a first step, these experiments were carried out during the primary spontaneous ionization of NPH (Fig. 12). This chemical situation was obtained on sample obtained 1 day after the mixing of solid NPH with H3.4ZSM-5 calcined at 773 K under O2 (see experimental section). The CW-EPR spectrum of this sample is characterized by the proton hyperfine splitting of NPH•+ overlapped on a broad signal attributed mainly to a trapped electron with probably some amount of electron–hole moiety (see the CW-EPR paragraph). SECSY is a reliable technique that can be used for studying nuclear modulation pattern. The collection of the echo shape at the top of echo peak for each t1 as function of t2 leads to record CW spectrum along f2 domain after Fourier transform procedure.
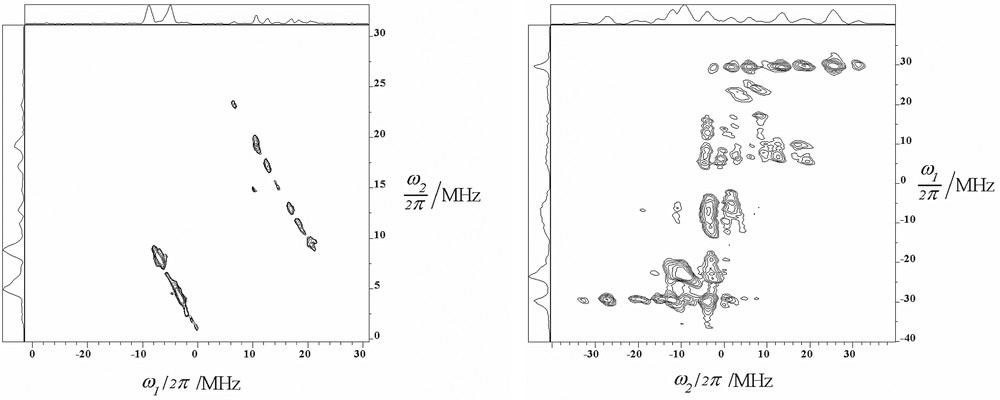
HYSCORE spectrum (left) and two-pulse SECSY spectrum (right) recorded 1 h after the mixing at room temperature of solid NPH and H3ZSM-5 calcined at 773 K under O2. The HYSCORE spectrum was achieved at 4.2 K with a τ = 256 ns and pulse lengths of 12 ns for π/2 pulses and 24 ns for π pulse were used and a four-step phase cycling (π/2–τ–π/2–t1–π–t2–π/2–τ-echo) was applied to suppress unwanted echo. Two-pulse SECSY spectrum was achieved at 4.2 K with pulse lengths of 12 ns for π/2 pulses and a 16-step phase cycling (π/2–t1–π/2–t1t2-echo) was applied. (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society). Masquer
HYSCORE spectrum (left) and two-pulse SECSY spectrum (right) recorded 1 h after the mixing at room temperature of solid NPH and H3ZSM-5 calcined at 773 K under O2. The HYSCORE spectrum was achieved at 4.2 K with a τ = 256 ns and ... Lire la suite
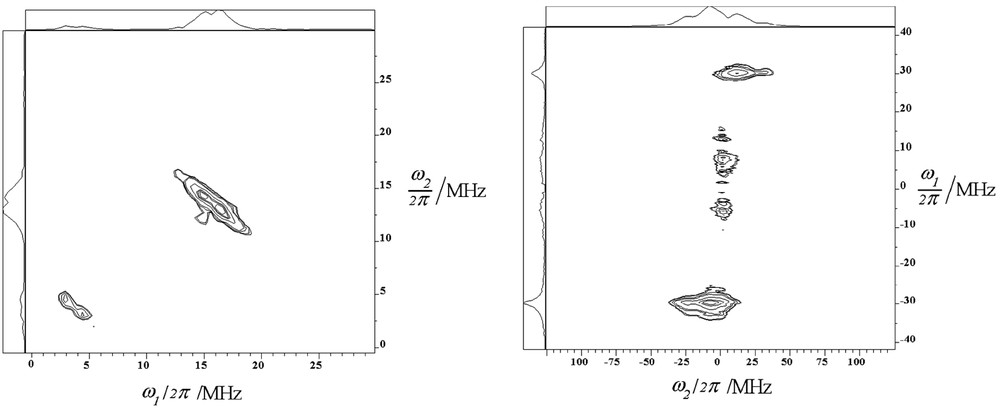
HYSCORE spectrum (left) and two-pulse SECSY spectrum (right) recorded 12 months after the mixing at room temperature of solid NPH and H3ZSM-5 calcined at 773 K under O2. The HYSCORE spectrum was achieved at 4.2 K with a τ = 256 ns and pulse lengths of 12 ns for π/2 pulses and 24 ns for π pulse were used and a four-step phase cycling (π/2–τ–π/2–t1–π–t2–π/2–τ-echo) was applied to suppress unwanted echo. Two-pulse SECSY spectrum was achieved at 4.2 K with pulse lengths of 12 ns for π/2 pulses and a 16-step phase cycling (π/2–t1–π/2–t1t2-echo) was applied. (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society). Masquer
HYSCORE spectrum (left) and two-pulse SECSY spectrum (right) recorded 12 months after the mixing at room temperature of solid NPH and H3ZSM-5 calcined at 773 K under O2. The HYSCORE spectrum was achieved at 4.2 K with a τ = 256 ns and ... Lire la suite
We show in the Fig. 12 right of SECSY 2D contour plot the double frequency of nuclear modulation from matrix protons which appeared along f1 domain in both positive and negative quadrants at the nuclear Zeeman frequency ωH/2π = 29 MHz covering 25 MHz along the t2 CW domain spectra. This result is close to the maximum Ax component measured of –24.1 MHz of the 1,4,5,8 protons in the CW NPH cation radical previously published [19]. The ωH/2π = 29 MHz observed results from the combination lines. The 2D SECSY spectrum also depicts on the f1 negative quadrant a peak centered at 13C nuclear Zeeman frequency of 3.7 MHz. On the positive quadrant another peak is observed at 7.4 MHz that can be attributed to the number of carbon I = 1/2 coupled with the spin S = 1/2. Moreover on the negative quadrant additional 1H pattern around 25 MHz is also observed resulting from Heisenberg spin exchange process from the weak coupled hydrogen 2,3,6,7 with the hydrogen 1,4,5,8. The corresponding HYSCORE spectrum (Fig. 12 left) with a τ value of 256 ns displays a strong 1H modulation pattern with an anisotropic constant of 14 MHz. This observed anisotropic coupling constant is close to those found for the proton 1,4,5,8 for the Az component of –17.4 MHz [19]. On the negative quadrant ω1 frequency domain nuclear frequency 13C is detected at 3.7 MHz and the double quantum 2ν 13C is present in the spectrum. These findings were found to provide further information to previous electron nuclear double resonance (ENDOR) and ESEEM results concerning NPH•+ photogenerated in H-ZSM-5 and CFCl3 matrix at low temperature [19]. Interactions of unpaired electron of NPH•+ with 1H of diaromatic moiety were demonstrated in the previous and present works. The present SECSY and HYSCORE results (Fig. 12) demonstrate supplementary interactions between trapped electrons with 13C of NPH•+. The two paramagnetic species formed primary after sorption of NPH in HnZSM-5 (n = 3.4, 6.6) calcined above 573 K under O2 interact differently with the surrounding magnetic nuclei. However, no evidence was found for interaction of trapped electron with 29Si or 27Al of zeolite framework [9,18]. It is possible that these latter interactions could be detected at very low temperature by pulsed EPR techniques.
Comparatively, the HYSCORE (Fig. 13 left) and SECSY (Fig. 13 right) spectra recorded on the broad one line CW electron EPR spectrum assigned to electron–hole pairs show drastic changes of the nuclear pattern. This chemical situation was represented by a sample obtained 1 month after the mixing of solid NPH with H3.4ZSM-5 calcined at 773 K under O2 and the complete disappearance of the proton hyperfine splitting of NPH•+ in the CW-EPR spectrum. The 2D SECSY contour plot shows in both positive and negative f1 quadrants the double quantum peak of 13C nuclear modulation centered at 0 MHz in the f2 domain (Fig. 13 right). This is consistent with a free electron in the matrix coupled with carbon and the combination lines 2ν 1H pattern are centered at the carbon nuclear frequency. Such changes were also observed in the HYSCORE spectrum (Fig. 13 left) where the 1H anisotropic constant is decreased to 7 MHz and a weak carbon coupling is observed in the (+, +) f2 domain with an anisotropic constant of 3.5 MHz and a positive sign for hyperfine splitting constant. Moreover no signal feature from zeolite interaction through 29Si or 27Al was observed in the HYSCORE spectrum. The same patterns of HYSCORE and SECSY are observed for room temperature and 4.2 K measurements so the 3.7 MHz pattern observed in the HYSCORE spectrum cannot be attributed to 27Al nucleus for which nuclear modulation frequency are close to carbon one. Such results seem to indicate that the electron and hole are surrounding and remain close to the occluded NPH [6]. In contrast, from previous pulse EPR results concerning sorption of BP in identical zeolites the electron and hole appear efficiently trapped in the neighboring side pockets of the channel containing aluminum atom and occluded BP through the observation of interactions between unpaired electron and 29Si or 27Al at low temperature [9].
3.2.4 Raman scattering study of naphthalene sorption in HnZSM-5
The sorption process of NPH in HnZSM-5 (n = 0, 3.4, 6.6) zeolites at loading corresponding to 1, 2, and 4 NPH per UC was monitored as a function of time using both FT-Raman spectrometry with exciting wavelength at 1064 nm and dispersive Raman technique with excitation wavelength at 632.8 nm.
The data processing using the SIMPLISMA approach of all the FT-Raman spectra recorded over several weeks in the mid-frequency region during the course of the sorption in HnZSM-5 (n = 0) or silicalite-1 provides evidence of one Raman spectrum. This spectrum was found to be identical to Raman spectrum of NPH in solution; this spectrum was attributed to NPH occluded in chemical environment with weak electrostatic effects. In the same way, the analysis of spectra sets obtained during the sorption of NPH in acidic HnZSM-5 (n = 3.4, 6.6) dehydrated under Ar provides evidence of two Raman spectra. The first one was straightforwardly assigned to residual NPH and the second one with a prominent band centered at 1612 cm–1 was assigned to NPH occluded in close proximity of H+ cation. The reconstructed spectra obtained from the extracted spectra of individual species were found to be in agreement with the experimental spectra.
The SIMPLISMA procedure was carried out with FT-Raman spectra set recorded over 2 weeks after the mixture of the solid NPH with HnZSM-5 (n = 3.4) activated under O2 at 773 K. This analysis yields three typical spectra exhibited in Fig. 7a–c of Ref. [6]. These spectra were identified to be residual solid NPH, occluded NPH associated with electron–hole pair NPH@H3.4ZSM-5+••– and occluded NPH@H3.4ZSM-5. NPH•+ was identified by resonance Raman scattering using 632.8 nm excitation wavelengths (Fig. 7d of Ref. [6]). The NPH•+ amount was found to be minor.
All the results obtained after Raman scattering investigations of the NPH sorption in HnZSM-5 (n = 6.6) exhibited analogous trends with those shown above for HnZSM-5 (n = 3.4).
Fig. 14 exhibits the relative amounts of species corresponding to the extracted spectra exhibited in Fig. 7a–c of Ref. [6] as a function of time during 15 days after the mixture of NPH with HnZSM-5 (n = 3.4) activated under O2 at 773 K. The curve 14a exhibits the continuous decrease of solid NPH amount, while the curve 14c indicates the continuous increase of occluded NPH amount. The amount of NPH@H3.4ZSM-5+••– (Fig. 14, curve b) was found to be maximum 24 h after the mixing of solids and decreased slowly over the 15 following days. The NPH+• amount was found to be weak.

Relative contribution (Raman spectral concentration) as a function of time of pure species deduced by data processing of FT-Raman spectra recorded after the mixing at room temperature of solid NPH and H3.4ZSM-5 calcined at 773 K under O2: (a) NPH associated with electron–hole pair, (b) NPH occluded close to –OH group of zeolite. (c) solid NPH, (Reprinted with permission from Ref. [6]. Copyright (2003) American Chemical Society).
3.3 Sorption of biphenyl in activated acidic HnZSM-5 (n = 3, 3.4, 6.6). Effects of aluminum content and thermal treatment upon ionization
3.3.1 Diffuse reflectance UV–visible absorption (DRUVv) spectrometry
No color change was observed after mixing solid BP and non acidic MnZSM-5 (n = 0.0, 1.0, 2.0, 3.0, 3.4, 4.0, 6.6; M = Li+, Na+, K+, Rb+, Cs+) calcined under Ar or O2. No intense color was observed after the mixture of BP with acidic HnZSM-5 dehydrated under Ar at 573 K. In contrast, several minutes after the mixing of solid BP with HnZSM-5 (n = 2.0, 3.0, 3.4, 4.0, 6.6) zeolites calcined at 773 K under O2, the powder turned blue. One day at room temperature after mixing, the blue powder turned to pink. DRUVv spectra recorded after the mixing of NPH with non acidic MnZSM-5 (n = 0, 3.4, 6.6; M = Li+, Na+, K+, Rb+, Cs+) calcined under O2 at 773, 873 or 973 K exhibit marked increasing of the prominent band at 255 nm [5]. This feature was attributed to sorption of BP as intact molecule within the void space of porous materials. The DRUVv spectra recorded after the mixing of solid BP with HnZSM-5 dehydrated at 573 K under Ar exhibited BP sorption as intact molecule but did not generate BP•+ radical cation in high yield [7].
In contrast, the DRUVv spectra recorded rapidly after the mixing of solid BP with HnZSM-5 zeolites (n = 3.0, 3.4, 6.6) calcined at 673, 773 and 873 K under O2 exhibit supplementary BP•+ absorption bands (Fig. 15) [21]. BP•+ was generated according to reaction [7].
| (7) |
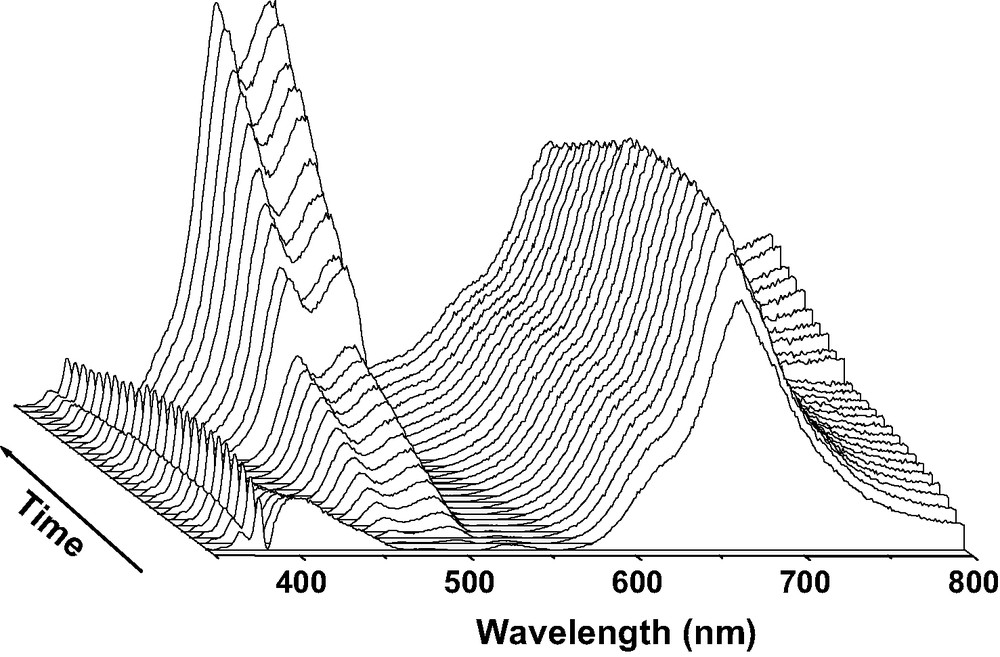
DRUVv spectra recorded at room temperature during the course (1 day) of BP sorption into calcined acidic [H3.4(AlO2)3.4(SiO2)92.6] zeolite. Solid BP (1 BP per UC loading) and H3.4ZSM-5 calcined under O2 at 773 K were mixed under Ar.
The characteristic absorption bands of BP•+ disappeared approximately after 1 day at room temperature with concomitant appearance of broad bands in the visible region [7,10,18].
| (8) |
The broad absorption bands obtained after the disappearance of the BP•+ bands were relevant to electron–hole pairs generated by electron transfer between BP•+ and the zeolite framework according to Eq. [8].
The yields of the initial ionization [7] with respect to the BP loading represented by BP•+/BP were determined according to previously reported procedure. The initial BP ionization yields were measured according to the calcination conditions of the zeolites with 1 BP per UC loading. Fig. 16 (▲) exhibits a very weak yield when H3(AlO2)3 (SiO2)93 zeolite samples were activated under Ar at 473, 573, 673, and 773 K. In contrast, Fig. 16 (♦) shows marked increasing of yield as temperature of calcination under O2 increases up to 1023 K. Fig. 17 (▲) exhibits no BP ionization phenomenon for all the Hn(SiO2)96–n(AlO2)n zeolite samples when the calcinations were done under Ar at 773 K. The calcination under O2 generates weak but significant ionization for 1 and 2 Al atoms per UC, whereas important yields were determined for 3, 3.4 and 6.6 Al per UC as shown in Fig. 17 (♦).
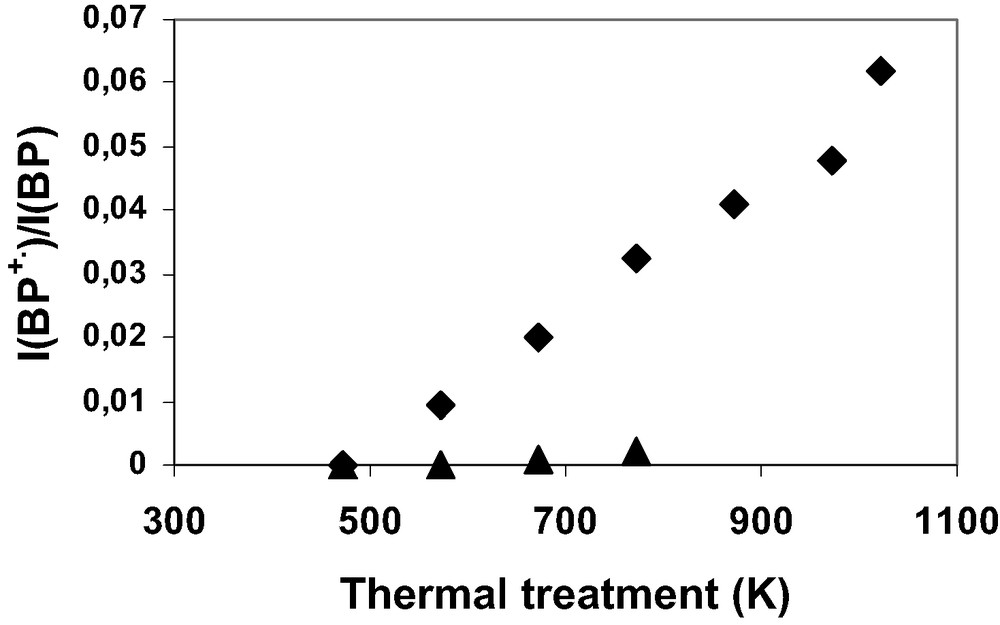
Initial BP+•/BP ionization yields as a function of the calcination temperature after the mixing of solid BP (1 BP per UC) and calcined H3.4ZSM-5. The H3.4(AlO2)3.4(SiO2)92.6 zeolite was calcined under Ar atmosphere (▲) or under O2 atmosphere (♦).
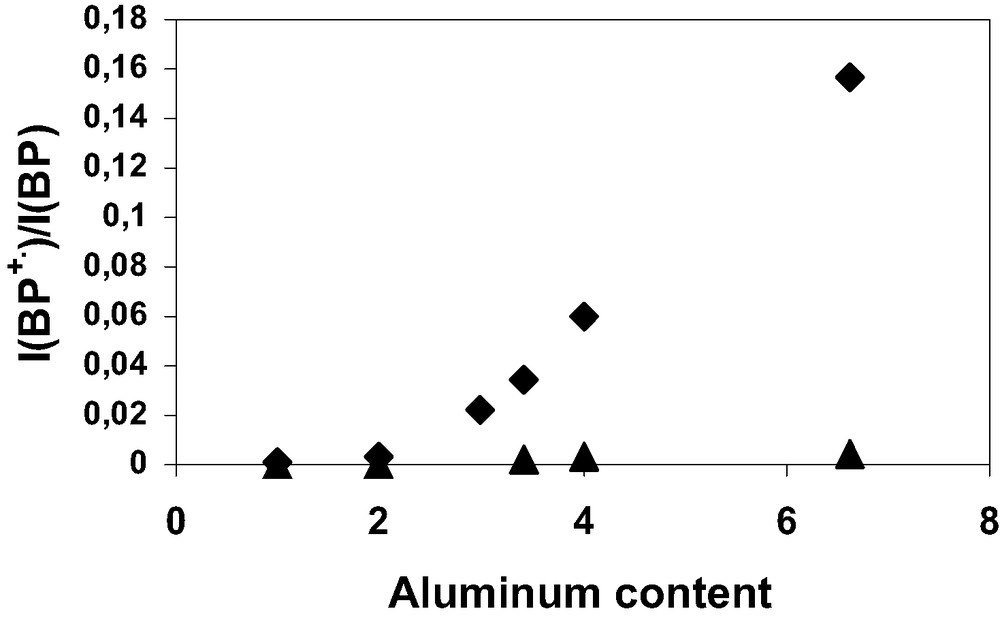
Initial BP+•/BP ionization yields as a function of Al content (n) after the mixing of solid BP and calcined HnZSM-5 (1 BP per UC loading). The Hn(AlO2)n(SiO2)96–n zeolite was calcined at 773 K under Ar atmosphere (▲) or under O2 atmosphere (♦).
3.3.2 Continuous wave electron paramagnetic resonance (CW-EPR) spectrometry
CW-EPR spectra of all zeolites Hn(AlO2)n(SiO2)96–n (n = 1, 2, 3, 3.4, 6.6) show rapidly intense signals after BP was adsorbed onto the ZSM-5 samples calcined under oxygen. The intensity pattern depends on the activation conditions of the zeolites, on the aluminum content and in lesser extent on the BP loading.
Particularly, a very weak signal appears after BP sorption into acidic HnZSM-5 zeolites dehydrated under argon at 773 K, whereas an intense spectrum appears after BP sorption into zeolites calcined under pure O2 at 773 K. The intensity of the signal is found to be very weak after activation at 473 K under O2 and increases with the activation temperature. EPR spectra (Fig. 18, curve a) recorded at room temperature immediately after the mixing of solid BP with H3(AlO2)3(SiO2)93 calcined at 773 K under O2 consist of seven resolved line signals which belong to the radical cation BP+•. In addition to the narrow seven lines, the EPR spectra contain broad overlapping features, which increase in intensity during the course of the sorption. Numerous spectra were recorded at different times, immediately after the exposure and until the reaction goes to completion over 1 month at 300 K. The processing data of all the spectra recorded during the BP sorption in each aluminated zeolite sample activated under O2 provide evidence of two independent spectra of paramagnetic species. Fig. 18 exhibits typical EPR spectra recorded during the BP sorption into H3(SiO2)93(AlO2)3 with a loading corresponding to 1 BP per UC. The extracted spectrum with well resolved seven line spectrum is readily assigned to radical cation BP•+ (Fig. 19 spectrum a) whereas the extracted broad signal is attributed to HnZSM-5•– and HnZSM-5+••– unpaired electrons (Fig. 19, spectrum b). The double integration of the extracted signal provides the intensity as a function of time of a and b signals, respectively (Fig. 20).
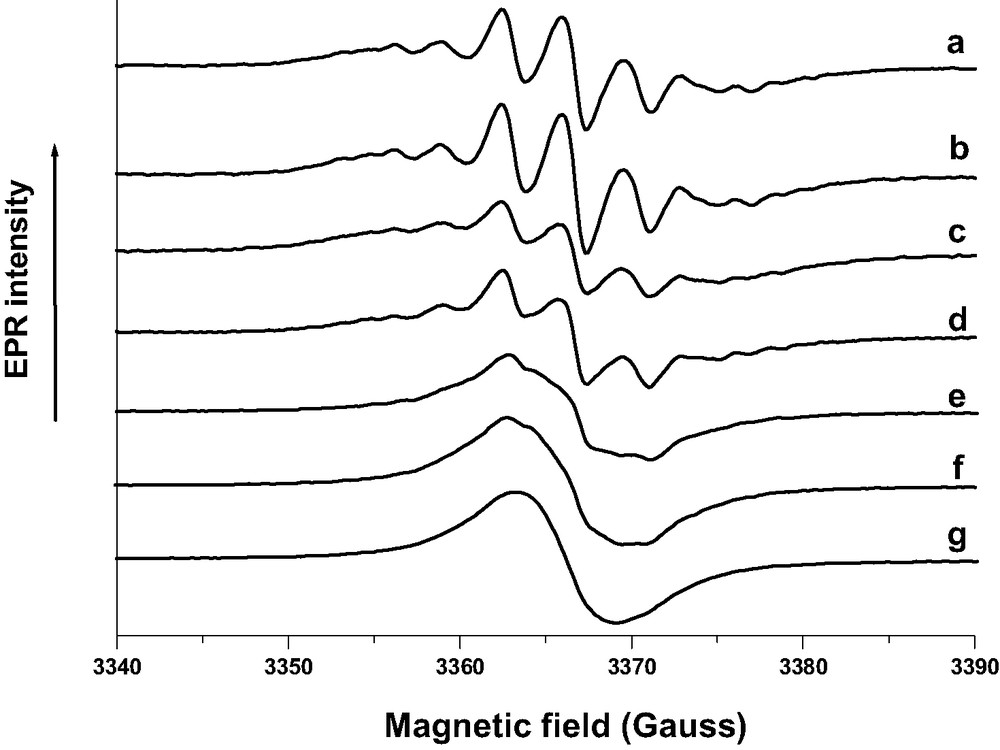
CW-EPR spectra recorded at room temperature during the course (1 month) of BP sorption into calcined acidic H3(AlO2)3(SiO2)93. Solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K were mixed under Ar. (a) 5 min after the mixture of the powders; (b) 45 min; (c) 8 h; (d) 16 h; (e) 24 h; (f) 50 h; (g) 1 month.
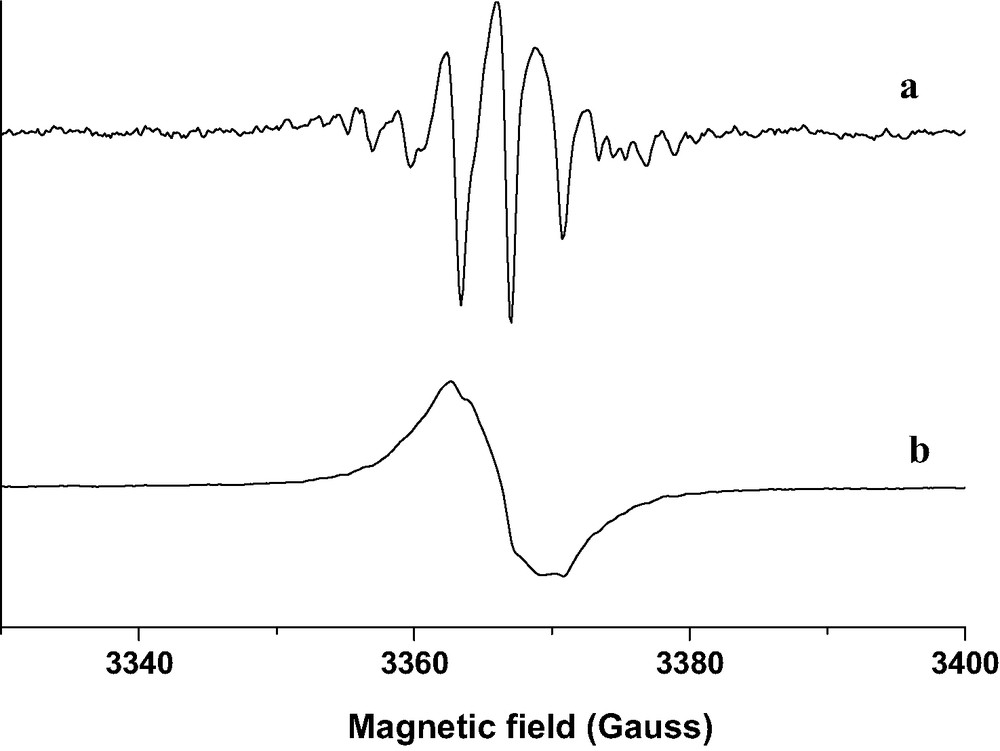
(a) Extracted seven line EPR spectrum assigned to BP+•; (b) Extracted broad EPR spectrum assigned to trapped electron in the framework during the course of BP sorption into calcined H3(AlO2)3(SiO2)93. Solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K were mixed under Ar.
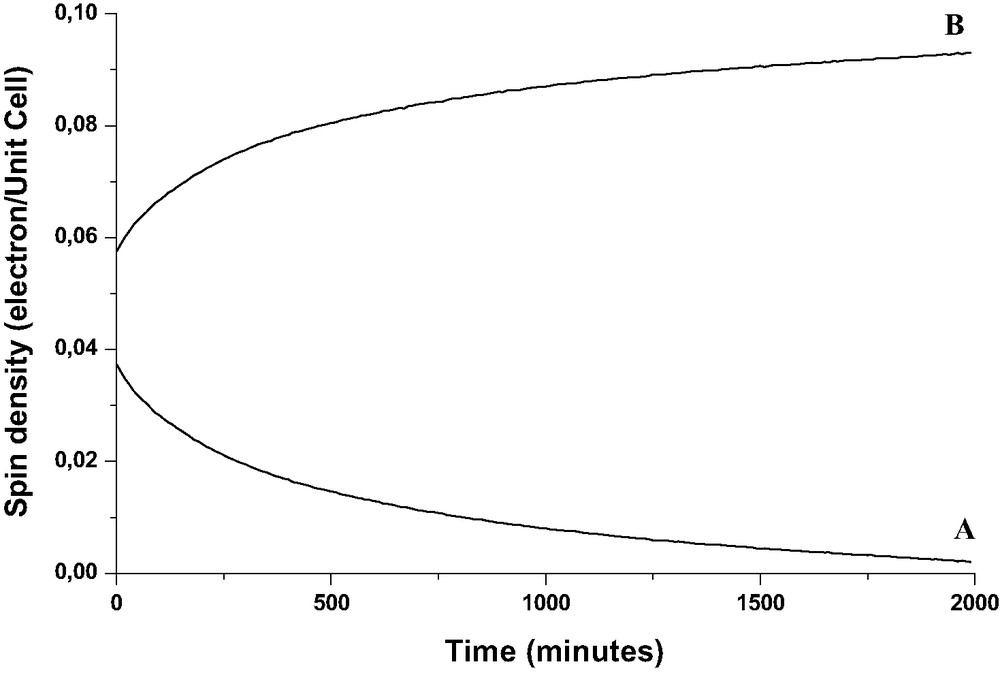
Variation as a function of time of the double integrated a and b EPR signals during the course of BP (1 BP per UC) sorption into calcined H3(SiO2)93(AlO2)3. Solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K were mixed under Ar.
The interaction of an unpaired electron with protons of BP•+ generates the seven line hyperfine structure of the A signal [22]. Through the data processing of the EPR spectra a broad structureless B signal was detected immediately after the mixing of the solids and was readily assigned to the transferred electron. From the double integration of the extracted A and B signals, the B signal carries about 50% of the overall paramagnetic signal at the beginning of the sorption (reaction [7]). The overall signal can reach 0.15 electron per UC. It is clear from the results shown in Fig. 20 that BP•+ amount decreases dramatically during the course of the BP sorption and practically disappears 2 days after the mixing of the solids in the experimental conditions. However, the overall EPR signal increases markedly with a concomitant change of the shape of the pattern to a quasi isotropic signal, (Fig. 19, spectrum b). The EPR signal width implies probably weak spin interaction through exchange and dipolar interactions. The B signal intensity increases as the A signal decreases and can reach 0.3 electron per UC. The disappearance of the BP•+ signals with the remaining of intense EPR signal does not indicate a geminal recombination BP•+-electron, but highly suggests that BP•+ captures one electron from the zeolite framework to restore BP and provides evidence of a durable electron–hole pair mediated through BP within the framework (reaction [8]).
The strong signal around g = 2 typical of BP@HnZSM-5•–•+ remains broad even at low temperature, (Fig. 21, spectrum b). The EPR signal was found to be persistent over at least 1 year. Applying CW-EPR as a function of temperature we were able to characterize the magnetic exchange coupling between unpaired electrons of BP@HnZSM-5•–•+. We can clearly see a weak signal around a g value of 4.22 that can be attributed to forbidden transition Δms = 2 resulting from electronic spin(1/2)–spin(1/2) coupling interaction. This results from population of the triplet state (S = 1) at low temperature and ferromagnetic behavior rather than the singlet state (S = 0) and antiferromagnetic behavior. There is no doubt that the occluded electron–hole pair acquires a ferromagnetic ground state (S = 1). The Boltzmann fitting of g = 2.0036 double integral signal of several spectra recorded from 10 to 300 K yields a weak ferromagnetic exchange value estimated to less than +0.1 cm–1 or +3000 MHz. The coupling constant estimates were not found to depend on the spin density and the ferromagnetic exchange value is representative of individual electron–hole pair in BP@HnZSM-5•–•+. The dipolar interaction was not estimated but was assumed to be in the same order of magnitude as the exchange coupling. These two interactions induce marked broadening of the CW-EPR signal.
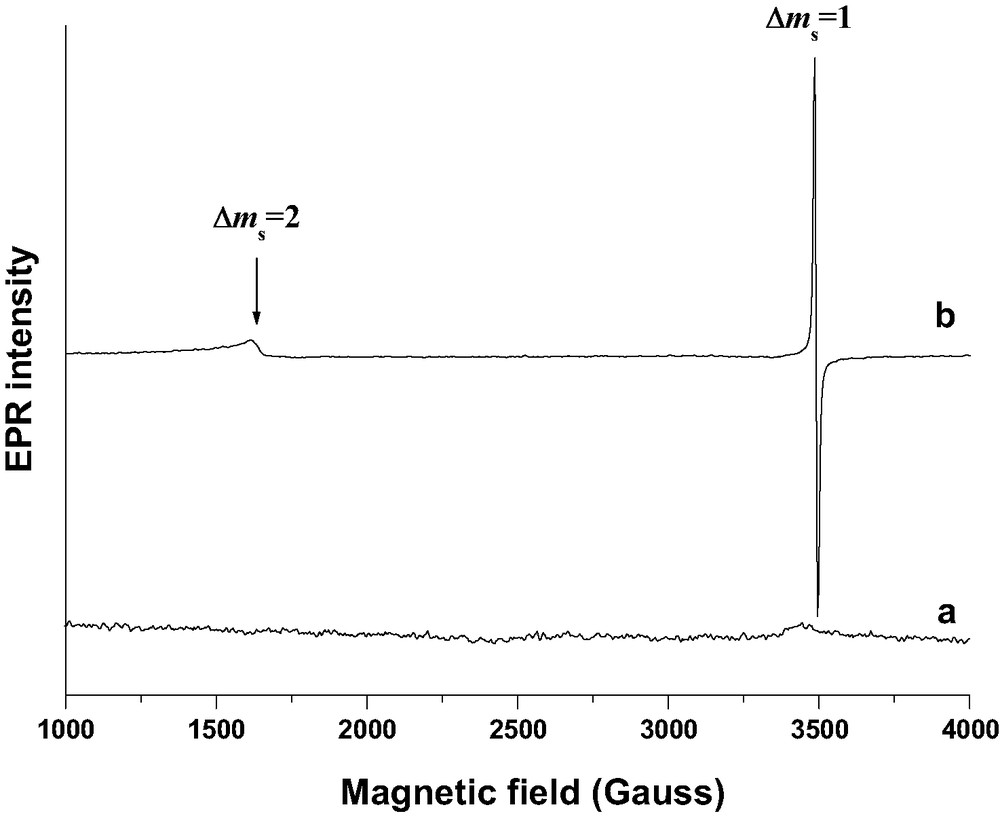
CW X-band EPR spectra (100 K) of: (a) H3ZSM-5 zeolite calcined at 773 K under O2 and evacuated under vacuum; (b) BP occluded in H3ZSM-5•–•+ zeolite (1 BP per UC) 6 months after the mixing under Ar of solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K.
3.3.3 SECSY and HYSCORE EPR measurements
No hyperfine interaction was detected in the broad persistent signal of the CW-EPR spectra even at low temperature. In order to precise the structural surroundings of unpaired electrons within BP@H3ZSM-5•–•+ we performed pulsed EPR experiments. The ESEEM spectroscopy measures the energy splittings of nuclei whose spins interact with the spin of unpaired electrons. Those energy splittings were used to determine the nature of the electron nucleus environments. Significantly higher resolution was achieved in a 2D four pulse HYSCORE experiment [10].
Such experiment is displayed on Fig. 22a and shows five ridges centered at the proton nuclear Larmor frequency of 14.5 MHz. We can directly measure an anisotropic hyperfine constant of 9 MHz. An additional peak centered at 2.9 MHz is also observed. The peak corresponds to the 29Si free nuclear Larmor frequency resulting from a 29Si coupling with electron. When the experiments were carried out at 4.2 K another peak centered at 3.8 MHz can be assigned to free Larmor nuclear frequency of 27Al nuclei, see the frame in Fig. 22a. No coupling of 13C with electron was detected. In order to raise the doubt of the origin of proton modulation, we perform the experiment with fully deuterated BP-d10. The substitution of 1H by 2H isotope in BP dramatically affects the 2D three pulse ESEEM spectra, (Fig. 22b). These findings indicate that the 1H or 2H isotope of BP are the main contributors to these lines. Effectively, the proton modulation has completely disappeared but a strong deuterium pattern is observed. The HYSCORE corresponding spectra (not shown) yield an anisotropic coupling constant of 1.6 MHz. In contrast, the substitution of 1H by 2H of bridging OH group of the H3ZSM-5 zeolite and subsequent BP-h10 sorption does not affect the CW-EPR spectra. However, recording two-pulse ESEEM spectrum yields a strong 1H-pattern modulation with an overlapped weak 2H one. Removing proton modulation by high pass filtering gives after Fourier transformation a single line centered at 2.2 MHz, assigned to 2H nuclear Larmor frequency. ESEEM results strongly suggest that the unpaired electrons are located near Al and Si atoms of the zeolite framework and in the vicinity of H atoms of occluded BP (Fig. 22b). There is no experimental evidence to assign the trapped electron and the positive hole, respectively. The experimental investigations of the present system of interest provide a reasonable picture of the electron–hole pairs stabilized in self-assembled array of BP@HnZSM-5•–•+.
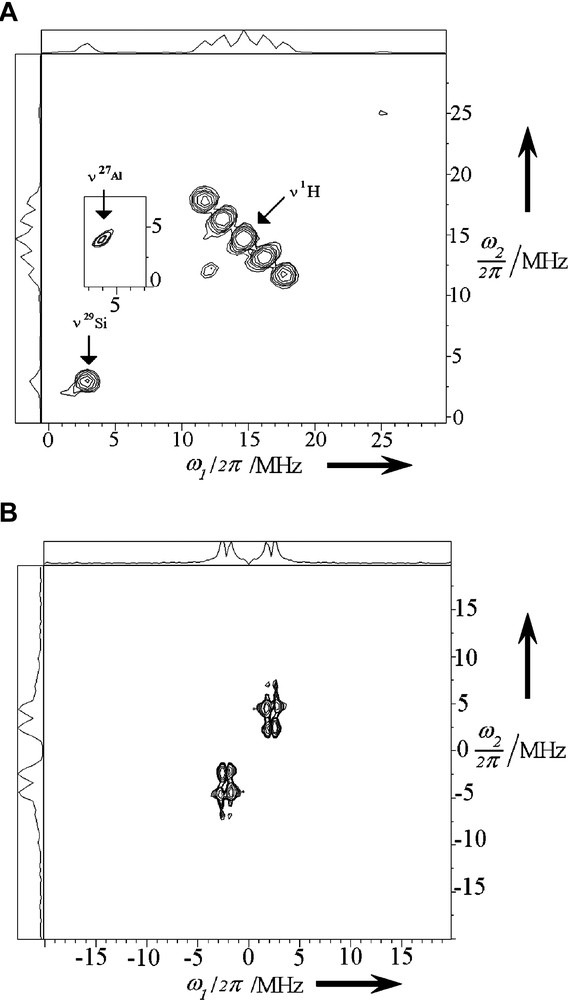
(a) HYSCORE spectrum of occluded BP-h10 1 BP@H3ZSM-5•–•+ 6 months after the mixing under Ar of solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K. The spectra were obtained at 300 K at the maximum of EPR signal at τ = 256 ns and 4.2 K (framed) at τ = 200 ns. Pulse lengths of, respectively, 12 ns for π/2 pulses and 24 ns for π pulse were used and a four-step phase cycling (π/2–τ–π/2–t1–π–t2–π/2–τ-echo) was applied to suppress unwanted echo. (b) 2D three-pulses ESEEM spectra of occluded BP-d10 1 BP@H3ZSM-5•–•+ 6 months after the mixing under Ar of solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K. Pulse lengths of 12 ns was set for π/2 pulses. (Reprinted with permission from Ref. [9]. Copyright (2002) Wiley-VCH Verlag). Masquer
(a) HYSCORE spectrum of occluded BP-h10 1 BP@H3ZSM-5•–•+ 6 months after the mixing under Ar of solid BP (1 BP per UC loading) and H3ZSM-5 calcined under O2 at 773 K. The spectra were obtained at 300 K at the maximum of EPR ... Lire la suite
4 Discussion
The preceding review of our recent results in direct relation with spontaneous ionization of polyaromatics upon sorption in ZSM-5 zeolites illustrates once again that this zeolite family with well known catalytic properties can behave as specific hosts for rod shape sorbates and exhibit versatile acceptor/donor electron properties. The unique properties of the inner surface of the ZSM-5 void space with respect to sorption and electron transfer behavior provide the ability of these microporous materials to generate and to stabilize species detected or assumed as transient species with short life time in solution. Many organic radical cations have been reported to be generated spontaneously in different zeolite types by mere sorption without additional activation. The rigid microporous solids serve as excellent matrices, stabilizing otherwise reactive or unstable radical cations, due to the combined contributions of the intense electrostatic fields inside the zeolite and from topological restrictions. The basement of the spontaneous ionization phenomena upon sorption in zeolites was recently reviewed [1]. This review goes back to the early incorporation studies and covers extensively the literature until mid-2000. The main structural and chemical parameters of zeolites governing the generation and stabilization of radical cations were briefly summarized here.
The topology of the zeolite host is crucial for the diffusion of the guest through the interior; it controls the maximum uptake achievable. Zeolites with tridirectional structures are more accessible and allow diffusion readily, whereas diffusion in monodirectional zeolites such as ZSM-5 is seriously restricted. The tight fit between the radical cation shape and the pore size was found to be an important factor for the stabilization in the pores of radical cations, which exhibit short life in solution [23,24]. A consensus appears to have been reached about the inability of purely siliceous zeolites to generate spontaneous ionization of organic molecules. It appears that the presence of heteroatoms such as aluminum in the siliceous framework and proton as counter balancing cation is a requirement for efficient stabilization of radical cations [1,23].
The necessary thermal treatment before any guest molecule can be accommodated in the zeolite pores affects the ionization efficiency and the molecular oxygen used during the calcination plays a central role in the ionization yield through the generation of Lewis acid sites. There is a continuing debate whether Brønsted or Lewis sites are responsible for the electron acceptor ability of zeolites [1].
In the previous and extensive review, several opened questions were pointed out in the perspective of future research. Among them, the insight into the nature of the electron acceptor sites and the fate of the ejected electron was of special interest [1].
In this short review of our works we have presented selected recent results about the spontaneous ionization of polyaromatic molecules by sorption in ZSM-5 zeolites. We provided a new insight into the fate of the ejected electron and subsequent electron transfer. Using pulsed EPR techniques [25] we have characterized the surroundings of unpaired electron species of unusual long-lived electron-radical cation pairs and subsequent electron–hole pairs. From all the results, we are also able to propose one comprehensive mechanism of the spontaneous ionization of polyaromatics upon sorption in ZSM-5 zeolites. The proposed mechanism includes size and ionization potential of polyaromatics, aluminum content of framework and extraframework cations of MFI hosts as well as Lewis acid sites generated during the calcination of the zeolites.
The ZSM-5 zeolites are extensively characterized porous solids; however, some key features in direct relation with the ionization efficiency of the used samples have to be summarized. After calcination procedure to evacuate template molecules and water, the framework structure of ZSM-5 zeolites contains two types of intersecting channels, both formed by rings of 10 oxygen atoms, characterizing them as a medium-pore zeolite [26–28]. The straight channels are sufficiently wide to allow rod shape molecules such as BP, NPH and ANTH to pass through openings of pores and to be occluded in the straight channels with restricted mobility [5,6,29].
No iron impurities can be invoked to explain the spontaneous ionization and paramagnetic behavior with the ZSM-5 samples used by us. In addition, no extraframework aluminum species was detected in Mn(AlO2)n(SiO2)96–n for n < 4 [6]. In contrast, the aluminum rich M6.6(AlO2)6.6(SiO2)89.4 sample was found to contain 0.3 extraframework Al2O3 per UC. However, no marked effect upon ionization of aromatics was observed as reported above [6]. Brønsted acid sites of acidic samples are assigned to bridging hydroxyl groups Al–OH–Si. So far, the problem of locating the Al atoms and associated proton or deuterium in H(D)-ZSM-5 by X-ray or neutron diffraction techniques was not resolved. However, numerous theoretical and experimental investigations provide significant findings about the distribution, structure and acid strength of Brönsted acid sites of H-ZSM-5 zeolites with Si/Al ratio corresponding to samples under study [30,31]. The location of the extraframework cations in bare non acidic Cs4ZSM-5 zeolites was resolved by X-ray diffraction [32]. The position of extraframework cations (Li+, Na+, K+) and Al atoms were deduced from theoretical calculations [33].
Lewis acid sites are essentially electron acceptor centers which are often invoked to explain the spontaneous ionization. The origins of Lewis acidity in zeolites are diverse and depend on the structure and chemical composition of the material under investigation. The structure of true Lewis sites is still controversial. Some authors associate Lewis sites with trigonal Al atoms formed as a result of zeolite dehydroxylation by thermal treatment. Other research groups have proposed Al3+ species or various other nonframework species (AlO+, Al(OH)2+, Al(OH)3, AlO(OH), Al2O3, etc.) leached from the zeolite framework during chemical or thermal treatment. The exact nature of these species is, however, still unknown [34]. It was previously demonstrated that the IR absorption in the OH stretching region decreases and broadens markedly after activation at higher temperature particularly under O2, such trends were observed with the present zeolite samples [35]. Numerous papers and reviews have been published about the IR spectroscopy measurements of adsorption of probe molecules such as pyridine, ammonia and acetonitrile to study the acidity of zeolites [36,37]. Strength of Lewis acidity generated after thermal treatment was probed by FT-IR diagnostic of sorption of pyridine through the intensity of the weak IR band at 1454 cm–1. Particularly, the calcination under Ar induced weaker band than calcination under O2. This finding indicates that Lewis acidity increases markedly after calcination under O2 but remains weak in our ZSM-5 samples. It was demonstrated previously by EPR diagnostic of NO sorption that the Lewis acidity of ZSM-5 zeolites increases markedly as a function of the calcination temperature under O2 between 600 and 1000 K whereas the nonframework Al content has a weak effect on the Lewis acidity. EPR of sorbed NO in zeolites is probing Lewis acidity strong enough to quench the orbital moment of NO molecule [34]. No such experiments were carried out on the zeolite samples used in our works. Molecular oxygen could play a central role in the spontaneous ionization process of polyaromatics with high IP values such as NPH and BP in acidic ZSM-5 [1]. No ionization was observed through sorption into non acidic ZSM-5 after calcination under O2 or Ar. Ionization in low yield was observed through sorption into acidic ZSM-5 calcined under Ar. In contrast, a dramatic increase in the ionization yield after calcination under O2 was noted in our [6,7] and previous cases [1]. The calcination treatment under O2 increases the Lewis acid sites with higher electron acceptor properties and initiates the ionization through a catalytic process. However, the fact that the sorption of ANTH with lower IP value generates ionization in high yield through sorption in acidic and non acidic ZSM-5 after activation under argon seems to indicate that O2 in the activation is not necessary in the process and the spontaneous ionization is an intrinsic property of the zeolite through sorption of compound with suitable IP. It should be noted that acidic zeolites loaded with metallic species and activated by appropriate treatment can generate radical cation through sorption of hardly electron donor molecule such as benzene with high ionization potential (IP = 9.25 eV) without any activation [38,39].
From the results concerning ANTH and 9,10-dimethyl antracene sorption, it is clear that the spontaneous ionization of ANTH is an intrinsic property of the inner surface of the ZSM-5 zeolites. The sorption process provokes the uptake of ANTH; NPH and BP at the pore openings of zeolite microcrystals and the slow diffusion in the straight channels. The sorption of ANTH in non acidic ZSM-5 zeolites MnZSM-5 (n > 2) with M = Li+, Na+, Mg2+, Ca2+ generates spontaneously the ionization. In contrast, the ANTH ionization does not occur upon sorption in MnZSM-5 with M = K+, Rb+, Cs+ as extraframework cation despite thermal treatment under Ar or O2 of the zeolites. No special calcination procedure except dehydration was necessary to induce the spontaneous ionization of ANTH in acidic and non acidic ZSM-5 zeolites MnZSM-5 (n > 2) with M = H+, Li+, Na+, Mg2+, Ca2+. The ionization yield was found to be 100% (2 electrons/1 ANTH per UC) with M = H+, but decreases as H+ > Li+ ~ Mg2+ ~ Ca2+ >> Na+. No such behavior was observed upon sorption of BP and NPH in identical zeolites and under identical experimental conditions.
In general, the ionization energy, Ie of molecules in condensed phase is represented by Ie = Ig + P+ + V0, where P+ is the polarization energy of the surrounding, Ig = 7.44 eV is the ANTH ionization potential in the gas phase, and V0 is the conduction band energy in the condensed phase [40]. The high polarization energy P+ of ANTH occluded in the straight channel in close proximity of H+, Li+, Mg2+ and Ca2+ induces probably spontaneous ionization by lowering the ionization energy. The ejected electron was found to be in close proximity of ANTH+• as ANTH+•-electron pair occluded in the straight channel (Fig. 23).
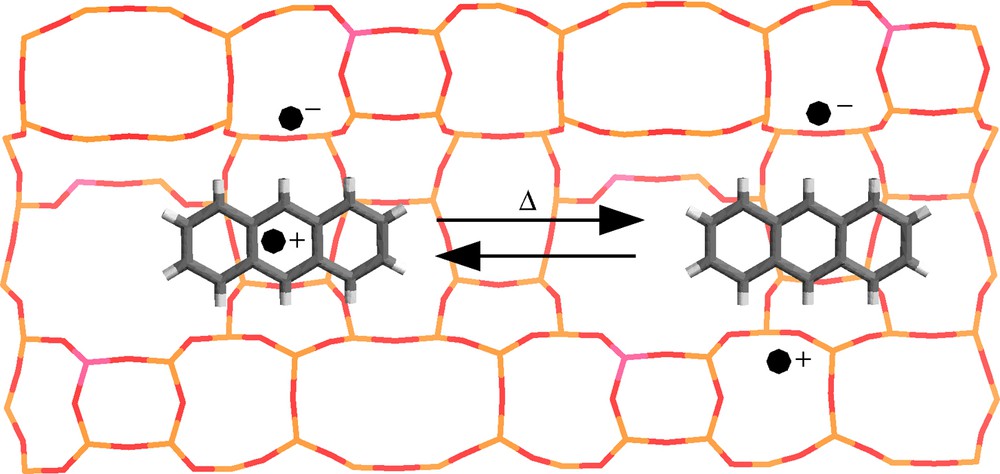
Schematic structures of 1 ANTH+• @H3ZSM-5–• ANTH radical cation-electron moiety and 1 ANTH@H3ZSM-5–•+• electron–hole pair occluded in the straight channel of H3ZSM-5 zeolite. Yellow, red and pink sticks represent Si, O and Al atoms of zeolite, respectively. Dark grey and white cylinders represent C and H atoms of ANTH. (•–) and (•+) represent unpaired electrons. (Reprinted with permission from Ref. [10]. Copyright (2003) Wiley-VCH Verlag).
However, the spin density of the trapped electron was probably distributed over several O nuclei in close proximity of Al–O(M)–Si moiety. The pulsed EPR results provide evidence of pairing effect and interaction with zeolite matrix nuclei through 27Al, 7Li and 13C couplings. No oxidizing effect of ANTH+• (Eox 1.09 eV vs. SCE as reference) with respect to electron donor ability of zeolite framework was observed except at high temperature (400 K). The weak mobility of ANTH•+ occluded in the pores of the ZSM-5 zeolites and the electron acceptor sites of the zeolite framework hinder efficiently the charge recombination.
The polarization energy P+ in the straight channel of MnZSM-5 is too low to generate spontaneous NPH (IP 8.14 eV) and BP (BP, IP 8.16 eV) ionization without chemical activation of the acidic ZSM-5 zeolites. After thermal treatment under molecular oxygen of acidic HnZSM-5 zeolites Lewis acid sites are created (see above). The higher polarization energy P+ of sorbate occluded in the straight channel in close proximity of Lewis sites induces spontaneous ionization by lowering the ionization energy of BP and NPH. The pulsed EPR results provide evidence of pairing effect and interaction with zeolite matrix through 27Al and 13C coupling. In contrast to ANTH•+ (Eox 1.09 V) strong oxidizing effect of BP•+ (Eox 1.96 V) and NPH•+ (Eox 1.54 V) with respect to electron donor properties of HnZSM-5 zeolite framework was observed at room temperature [41]. The experimental results supported the formation of positive holes within the zeolite framework and provided identification of electron-BP-hole and electron-NPH-hole as unique stable species after equilibration of the system before the final charge recombination. The occurrence of the electron transfer is not surprising considering results recently reported demonstrating electron transfer from zeolite to other powerful electron acceptors such as chloranil and cyano substituted benzene molecules [18,42]. A possible source of electron density for electron donation to BP•+ or NPH•+ is the lone pair of electrons on the bridging lattice oxygen atoms, particularly those in close proximity of Al. The tight fit between the shape of the BP, NPH and the pore size of straight channels of zeolites combined with efficient electron trapping sites appeared the most important factors responsible for the stabilization of the electron-sorbate-hole moiety and hinder efficiently the charge recombination. These electron-sorbate-hole moieties exhibit intense charge transfer bands in the visible region.
After the uptake of polyaromatic hydrocarbons (PAH) by pore opening of the microcrystals of ZSM-5 zeolites, the fast initial ionization occurs in the straight channel of suitable ZSM-5 according to:
| (9) |
With PAH0•+ radical cation, E0– electron trapping site and □ schemes the void space of the straight channel. The experimental results presented here suggest that both PAH0•+ and E0– migration can occur between electron acceptors spatially separated and concerted with the migration of PAH•+ within the straight channel of zeolite from one sorption site to another one.
| (10) |
The final charge recombination occurs in the straight channel after a very long time. The delay depends on the fitting between the radical cation size and the pore size of the zeolite.
| (11) |
For PAH molecules with high ionization potential such as BP and NPH, it is necessary to create Lewis acid sites by thermal treatment of acidic ZSM-5 zeolites to generate spontaneous ionization by sorption. However, the oxidizing power of the radical cation initiates the electron abstraction of the zeolite framework and generates positive hole D0+ according to:
| (12) |
The formation of long-lived electron–hole pair was due to the effective hole and electron migration concerted with the sorbate diffusion to more efficient trapping sites:
| (13) |
The deactivation of the long-lived electron–hole pair appears to be regulated by the long distance back electron transfer process to charge recombination:
| (14) |
5 Concluding remarks
The review of our recent works devoted to the spontaneous ionization of polyaromatic molecules upon sorption in ZSM-5 demonstrates once again that the tight fit between the size of rod shape polyaromatic molecules such as BP, NPH and ANTH and the diameter of the straight channel can stabilize radical cation-electron pairs or electron–hole pairs over long periods. Recent results provided a new insight into the mechanism of the ionization and subsequent electron transfer.
The spontaneous ionization is an intrinsic property of the inner surface of the porous materials, which depends both on the ionization potential of sorbate and the polarization energy of the host at the sorption site. For molecules with relatively low ionization potential such as ANTH, the ionization occurs in non acidic ZSM-5 zeolites. However, the polarization energy at the sorption site highly depends on the nature of the extraframework cation. It is probable that molecules with lower potential ionization can be ionized by sorption in purely siliceous silicalite-1.
It was established that the ejected electron is trapped as isolated electron in the oxygen framework in close proximity of Al atoms and extraframework cations with some pairing electronic effect with the radical cation.
The spontaneous ionization of molecules with relatively high ionization potential such as NPH and BP is effective upon sorption after generation of Lewis acid sites in the void space by thermal treatment of acidic ZSM-5 zeolites. The oxidizing power of radical cations initiates the electron abstraction of the framework and the generation of long-lived electron–hole pair before the final charge recombination. The electron and positive hole are trapped in the oxygen framework in close proximity of Al and extraframework cations with electronic interaction with the occluded sorbate.