

1 Introduction
Considered as the father of coordination chemistry, the Alsatian chemist, Alfred Werner, had an enormous influence upon much of twentieth century chemistry [1]. This is reflected in the words of another Alsatian chemist, and Nobel laureate, Jean-Marie Lehn, in describing supramolecular chemistry as “generalised coordination chemistry” [2]. Werner, working at the University of Zurich, conducted a remarkable variety of syntheses of cobalt(III) compounds in particular but did not live to see his rationalisations of their nature justified by another great scientific advance of the 20th century, X-ray crystallography [3]. Somewhat paradoxically, this advance has also revealed that Werner was fortunate in that the usual complexed form on Co(III) of a ligand he used extensively, 1,2-ethanediamine, ‘ethylenediamine’ = ‘en’, was that of a chelate. The characterisation of unidentate and bridging forms of coordinated ethylenediamine, however, is quite recent [4]1 and the development of syntheses specifically designed to produce ‘hypodentate’ ligand [6] complexes of Co(III) with polyamines more recent still [7].
One of the best known syntheses of a coordination compound is that of [Co(en)3]Cl3·3 H2O. The procedure first reported by Werner is one for which a yield of 95% is conventionally quoted [8]. Such a figure would be regarded as excellent, and it is of practical significance, but, as is usual with such information, it is not accompanied by any explanation of why the yield is not 100%. In general, any deficiency from 100% may be due to incomplete reaction, the diversion of some material along another reaction pathway, inefficiency in the actual isolation, an error in the assumed stoichiometry or some combination of all these factors. Having occasion to conduct the synthesis of [Co(en)3]Cl3·3 H2O on the scale of 10 kg, a natural frugality suggested to us that the amount of material represented by (100 – 95)% of 10 kg would be worthy of isolation and characterisation. A laborious separation using cation exchange chromatography was therefore conducted on the acidified reaction mixture obtained from the standard synthesis of [Co(en)3]Cl3·3 H2O after removal of the bulk of this material in close to the expected quantity. This proved to be extremely complicated, in part because its conduct in batches caused it to become apparent that slow reactions were still occurring in solution, as well as because the amounts of the numerous species present spanned a wide concentration range. In the end, not all species present could be isolated and characterised, but the nature of some of those that could provide an interesting tale in cobalt chemistry.
The relatively high (water) solubility of [Co(en)3]Cl3·3 H2O makes it unsurprising that it remained a significant component of the ‘wastes’. Given that its formation commences with the addition of ethylenediamine to Co(II), a labile aqua-ion [9], an equilibrium distribution at this point over many possible species but including mono-, bis- and tris-chelate species would be anticipated. It is therefore also unsurprising that ‘Wernerian’ bis(ethylenediamine)Co(III) species in sum proved to be a major component (as was indeed recognised by Werner himself [8]), and this explains at least part of the complexity of the attempted chromatographic separations, since relatively slow reactions interconvert the various forms shown below [10].
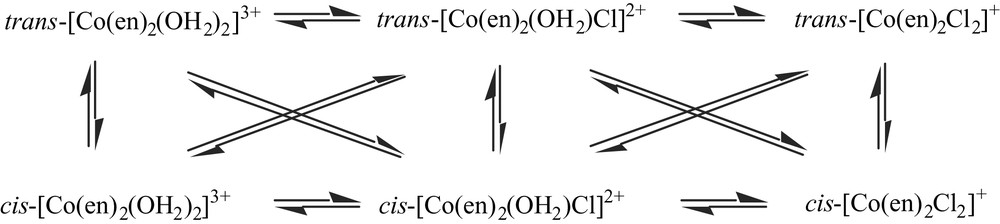
Whether mono-(ethylenediamine)Co(III) species were present initially is impossible to say, as their reduction to Co(II) would be expected under the conditions of the final acidified reaction mixture [11]. Both ‘free’ Co(II) and ethylenediamine (protonated) were isolated during the chromatography, but they could also have arisen because of incomplete oxidation or, for one or the other, because of inexactitude in the composition of the original mixture. Further, it is known that the peroxo-bridged dimers presumed to be intermediates in most oxidations of Co(II) to Co(III) using air (or O2) can undergo acid-catalysed decomposition to return to Co(II) and protonated ligand [12], so this reaction is another possible source of these two species in the wastes, presumably though as a minor pathway for any such peroxo dimers alongside the loss of H2O2 and the formation of Co(III) monomers.
The presence of ‘one-ended en’ complexes as side products in the formation of Co(III)-ethylenediamine chelates has been known for some time, though the structural characterisation of [Co(en)2(H2NCH2CH2NH3)Cl]Cl[ZnCl4], A, is quite recent [4]. This complex proved to be present in ~2% yield in our syntheses, its crystal structure (Fig. 1a), redetermined here at ‘low’ temperature (ca 150 K), being in complete accord with the earlier determination. The complex also is isomorphous with its Cr(III)/Hg(II) analogue [13] and was refined in a similar setting, the two systems being similar in that the range of M–N distances (M–N 1.968(6) (Co), 2.079(10) Å (Cr)) is narrow, with no indication of any significant trans effects, though the difference in M–N distances (0.11 Å) is considerably greater than that in M–Cl distances (2.302(3)–2.2621(9) ~ 0.040 Å), presumably reflecting differences in the Lewis acidity of Co(III) and Cr(III). There are significant differences between unidentate and chelate en in A, the Co–N–C bond angle for the unidentate ligand being 120.5(2)°, appreciably greater than the near-tetrahedral values of the other N-donor sites, and in the conformationally trans enH+ ligand, the central C–C bond (1.536(4) Å) is longer than those of the chelate ligands (1.503(5), 1.502(5) Å), while the two C–N bonds (N(31)–C(32) 1.476(4), C(33)-N(34) 1.483(4) Å) do not differ significantly in length from one another and are similar to those of the chelate ligands (1.483(4)–1.495(4) Å). We did not detect the en-bridged complex [(en)2CoCl(H2NCH2CH2NH2Co(en)2Cl)]4+, B (Fig. 1b), though this is known to be a side product in the synthesis of trans[Co(en)2Cl2]+ using H2O2 as an oxidant [4]. It is possible that the presence of [Co(en)2(H2NCH2CH2NH3)Cl]3+ results from the oxidation of the corresponding Co(II) species (perhaps with an aqua ligand in place of Cl), though it is tempting to attribute its presence to the expected intermediacy of a peroxo-bridged dimer [12] which, given that the maximum coordination number known for Co(III) is 6, could not contain three bidentate ethylenediamine ligands. This consideration leads to a dilemma in that, if it is assumed that all oxidation passes via a peroxo-dimer where each Co has two chelate and one ‘one-ended’ ethylenediamine ligands attached, and subsequently via [Co(en)2(H2NCH2CH2NH3)Cl]3+, the fact that the latter undergoes slow ring closure [14] makes the efficiency of [Co(en)3]3+ formation difficult to explain. It is well-known, however, that analogues of [Co(en)3]2+ involving cage amine ligands undergo rapid outer-sphere oxidation by O2 [15], so that the results of the Werner preparation are perhaps indicative of the operation of at least two oxidation mechanisms.
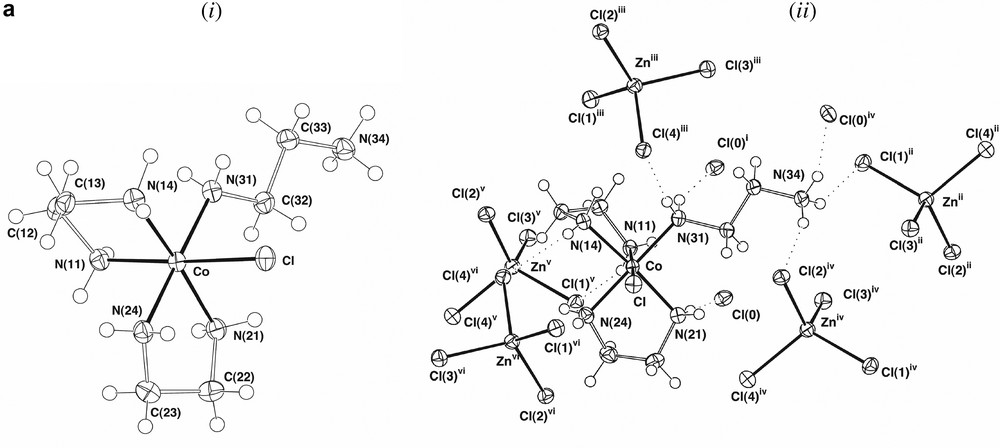
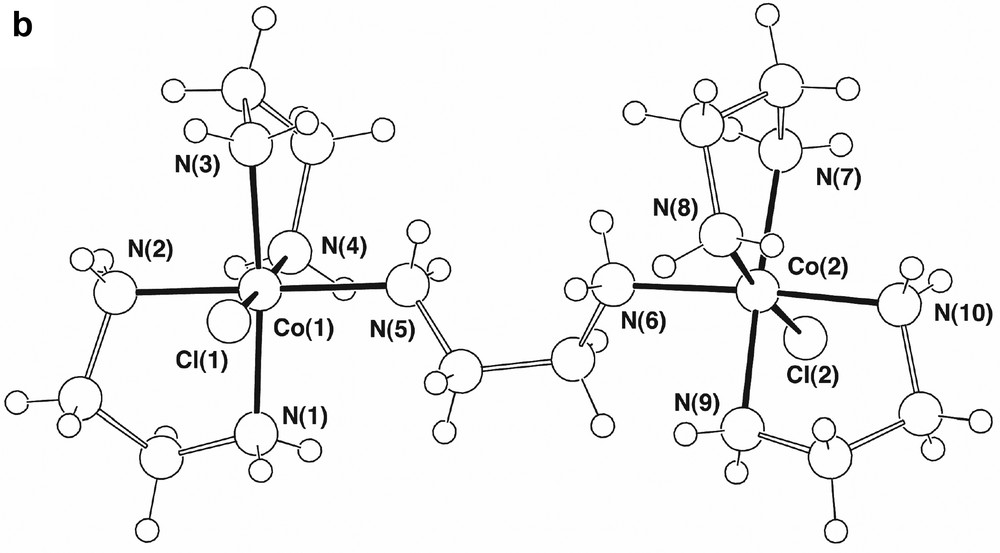
(a) Views of a single cation in the lattice of [Co(en)2(H2NCH2CH2NH3)Cl]Cl[ZnCl4], (A), showing (i) the isolated cation and (ii) the cation and its presumed hydrogen-bonding interactions with both Cl– and [ZnCl4]2– entities. (Symmetry transformations: (i) , 1 – y, ; (ii) , y – 0.5, 0.5 – z; (iii) x, y, z – 1; (iv) x, 0.5 – y, z – 0.5; (v) x, 1.5 – y, z – 0.5; (vi) 1 – x, 1 – y, 1 – z). (b) The binuclear cation present in [(en)2CoCl(H2NCH2CH2NH2Co(en)2Cl)][ZnCl4]2 [4], in which the two metal centres are of the same chirality.
A complex found to be present at approximately one-third of the level of [Co(en)2(H2NCH2CH2NH3)Cl]3+ was the well-known [16] species [Co(en)2(NH3)Cl]2+, C, presently structurally characterised as the dithionate (Fig. 2a), though structures are also known for both the (+)589-3-bromocamphor-9-sulfonate [17] and the chloride [18]. (Conformational differences between these species are discussed below.) Perhaps significantly, a third complex present in similar amounts to [Co(en)2(NH3)Cl]2+ was [Co(en)(H2NCH2CH2N=CHCH2NH2)Cl]2+, D (Fig. 2b), a known species [19] in which the tridentate diamine-imine ligand present may derive from intramolecular reaction of [Co(en)2(H2NCH2CH2NH3)Cl]3+ after the uncoordinated amino group has been oxidised to an imine and then hydrolysed to ammonia and the corresponding aldehyde. The released ammonia obviously could be the source of that in [Co(en)2(NH3)Cl]2+, the presence of this complex otherwise being difficult to explain, since free ammonia is not expected to be present in ethylenediamine.
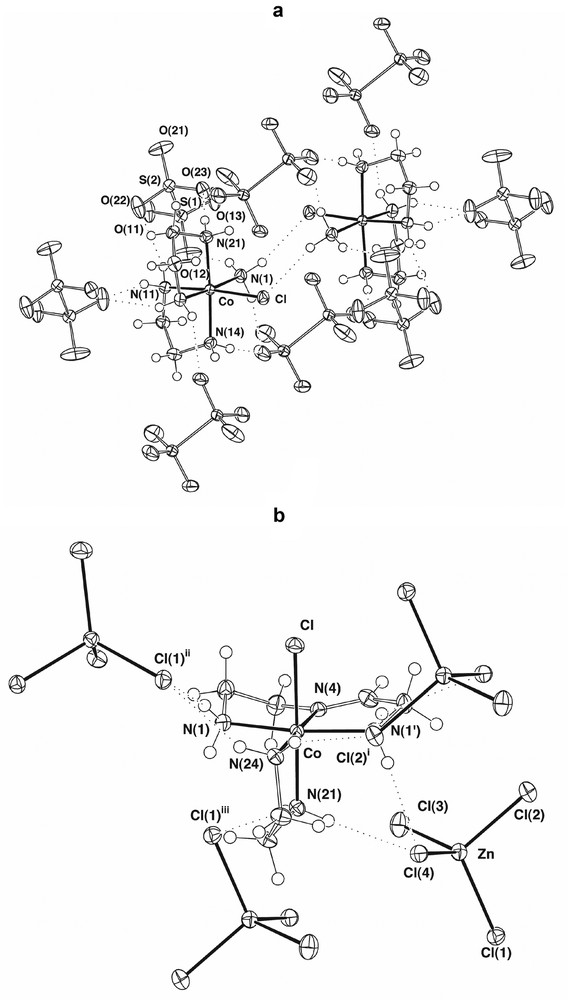
(a) Part of the lattice of cis-[Co(en)2(NH3)Cl][S2O6] showing the hydrogen-bonded centrosymmetric cation pair and its associated hydrogen-bonded anion capsule. (b) Part of the lattice of [Co(en)(H2NCH2CH2N=CHCH2NH2)Cl][ZnCl4], showing the cation and its neighbouring [ZnCl4]2– entities lying within hydrogen-bonding distances. (Symmetry transformations: (i) , , ; (ii) 1 + x, 0.5 – y, 0.5 + z; (iii) , 1 – y, ).
A yellow species (thus presumably one with a CoN6 chromophore) present in similar amounts to [Co(en)2(NH3)Cl]2+ proved difficult to purify chromatographically until 1H nuclear magnetic resonance spectroscopy was used to establish the presence of an NHCH3 entity at which slow (base-catalysed) inversion could occur [20], resulting in the slow interconversion of two isomers unless the chromatographic eluants were strongly acidic (e.g., 3 mol l–1 HCl). Crystallisation, as their [Co(CN)6]3– derivatives, of the two complexes ultimately separated showed them to be diastereomers of [Co(en)2(N-CH3en)]3+ (Fig. 3), E, F, only one of these (F) having been previously characterised by a structure determination [21]. While it is possible to write highly speculative mechanisms for the generation of such species from en, the real point of interest here is that the isomeric structures reveal an aspect of the complex ion stereochemistry not generally amenable to study at the time of Werner, though it is now well-recognised that each of the enantiomers of [Co(en)3]3+, which Werner could separate, actually consists of a mixture of rapidly converting conformers, usually designated as lel3, lel2ob, lelob2 and ob3 in reference to whether the C-C bond of the separate en units is oriented parallel (‘lel’) or obverse (‘ob’) to the C3 axis of the averaged complex ion form [22]. More precisely, using using upper- and lower-case Greek symbols to denote the helicities about the metal and of the ring conformations [23], the designation lel3 is equivalent to Δ(λλλ);Λ(δδδ), lel2ob equivalent to Δ(λλδ);Λ(δδλ), etc. In the case of N–methyl-en complexes, the assumption that the N-methyl group would prefer an equatorial orientation on and so ‘lock’ the ring formed by chelation, leads to the conclusion that two diastereomeric forms should exist, depending on whether the ring is λ or δ and the N-methyl centre of R or S configuration on a Co centre of one particular chirality (Fig. 4). This is as observed for the two isomers shown in Fig. 3, though of course these structures establish only the situation in the solid state and do not necessarily mean that in solution there cannot be an interconversion, not requiring inversion at N, with forms involving axial methyl substituents. In addition, the inversion of the N–methyl-en ring in passing from one diastereomer to the other is associated with inversion of one of the en rings, so that it happens that both these complexes can be described as lel2ob species. This is possibly in part a consequence of different hydrogen-bonding arrays in the two species, one of which is anhydrous while the other is a dihydrate. For simple en chelate rings, the energy difference between lel and ob orientations is expected to be small [22], and in the Cr(III) analogue of A, for example, crystallised as its chloride-tetrachloromercurate(II) rather than, as for the Co compound, as its chloride-tetrachlorozincate(II), the two rings have the same conformations, rather than the opposite as in A [13]. This sort of observation of subtle features of the complex ion stereochemistry is not uncommon [24] and a further example is the fact that in the present structure determination for C as its dithionate, a lel2 species was found, whereas in the (+)589-3-bromocamphor-9-sulfonate the species is a disordered mixture of ob2 and lelob , and in the chloride lelob. As well, the en-bridged species B crystallises in space group P212121, meaning that the individual crystals are resolved, and the dimers within them have Co(III) centres all of the same absolute configuration in the(lel2)(lel2) form, i.e., Δ(λλ);Δ(λλ) or Λ(δδ);Λ(δδ). All the present structures are of sufficient quality to define the complicated hydrogen-bonding networks present in all and that must play a role in influencing the solubility of the various species. (The reason for crystallising E and F as hexacyanocobaltates was that they were exceedingly difficult to precipitate from aqueous media in any other form.)

Diastereoisomeric species containing the [Co(en)2(N–CH3en)]3+ cation. (a) One of the cations of Λ chirality present in the lattice of [Co(en)2(N–CH3en)][Co(CN)6]·2 H2O; both en rings are of δ chirality, while the N–CH3en ring is of λ chirality, with its chiral-N centre of S absolute configuration. (b) One of the cations of Λ chirality present in the lattice of [Co(en)2(N–CH3en)][Co(CN)6]; the en rings are of opposite chirality (one δ, one λ), while the N–CH3en ring is of δ chirality, with its chiral-N centre of R absolute configuration.
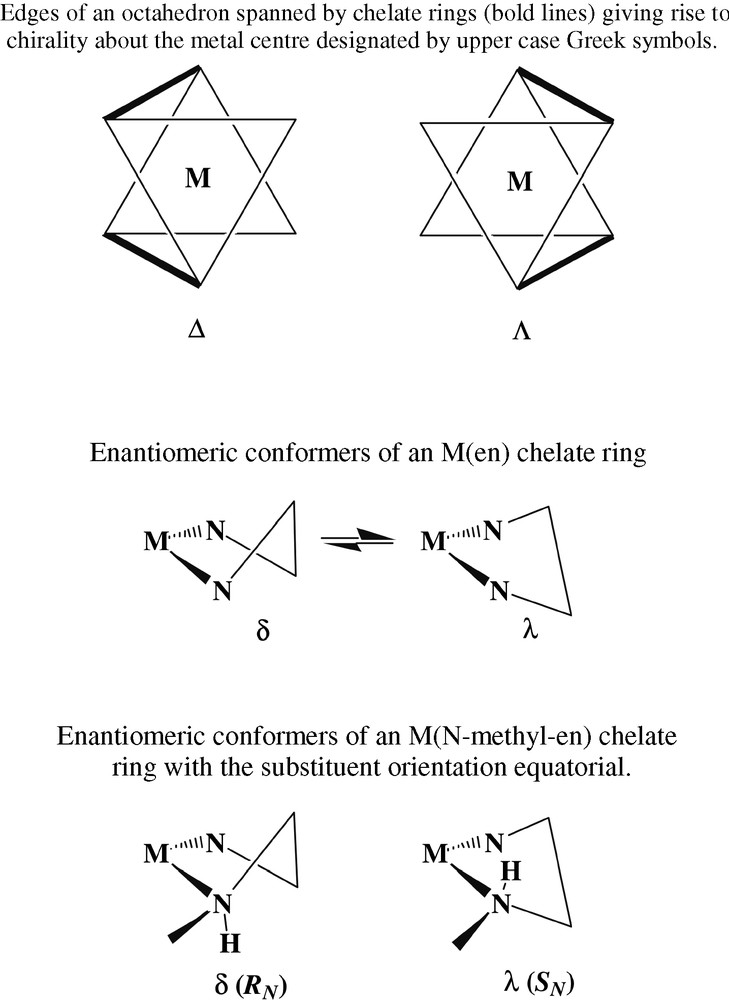
Chirality descriptors for saturated chelate-ring complexes.
Had Werner been confronted with [Co(en)2(H2NCH2CH2NH3)Cl]3+ as the principal product of his [Co(en)3]3+ synthesis, his rationalisation of the chemistry of Co(III)/en complexes might have been less convincing, though no doubt his understanding of the overall chemistry of Co(III) complexes would have allowed him to encompass this result. It is nonetheless worthy of note that a century of study following his work may not yet have revealed the full details of the reactions he so intelligently exploited. A similar remark may apply to one of the classical experiments in Pt(II) stereochemistry, the resolution of [Pt(H2NCH2C(CH3)2NH2)(H2NCH(C6H5)CH(C6H5)NH2)]2+ [25]. Were it to have been assumed that conversion between chiral forms of the chelate rings could be slow, the result may have been much more difficult to interpret. In an era of catchcries such as ‘atom economy’, ‘green chemistry’ and ‘molecular engineering’, it is a salutary lesson that an apparently efficient synthesis, such as that of [Co(en)3]Cl3·3 H2O may be a disguise for a remarkable range of different events. Clearly, no simple answer can be given to the question of how to improve the yield of [Co(en)3]Cl3·3 H2O from 95 to 100%.
2 Experimental
2.1 Structure determinations
Full spheres of CCD area-detector data were measured at ca 153 K (Bruker AXS instrument, ω scans; monochromatic Mo Kα radiation, λ = 0.71073 Å) yielding Nt(otal) reflections, reducing to N unique (Rint quoted) after ‘empirical’/multiscan absorption correction (proprietary software), No with F > 4 σ(F) being considered ‘observed’ and used in the full matrix least squares refinement (anisotropic displacement parameter forms for the non-hydrogen atoms; (x, y, z, Uiso)H were refined in all cases). Conventional residuals R, Rw (weights: (σ2(F) + 0.0004 F2)–1) are quoted on |F| at convergence. Neutral atom complex scattering factors were employed within the context of the Xtal 3.7 program system [26]. Pertinent results are given in the tables and figures, the latter showing 50% probability amplitude displacement ellipsoids for the non-hydrogen atoms, hydrogen atoms having arbitrary radii of 0.1 Å.
2.1.1 Crystal/ refinement data
All structures are monoclinic, space group P21/c (C2h5, No. 14), Z = 4.
A. cis-[Co(en)2(H2NCH2CH2NH3)Cl]Cl[ZnCl4] = C6H25Cl6CoN6Zn, M = 518.4. a = 13.332(2), b = 11.426(1), c = 13.872(2) Å, β = 118.863(2)°, V = 1850.6 Å3. Dc = 1.860 Mg m–3; specimen: 0.26 × 0.20 × 0.08 mm. μMo = 3.04 mm–1; ‘T’min/max = 0.80. 2θmax = 58°; Nt = 18246, N = 4684 (Rint = 0.031), No = 3642; R = 0.033, R′ = 0.037; nv = 281, |Δρmax| = 0.98(8) e Å–3.
C. cis-[Co(en)2(NH3)Cl]S2O6 = C4H19ClCoN5O6S2, M = 391.7. a = 7.8025(9), b = 13.783(2), c = 12.845(2) Å, β = 93.140(2)°, V = 1379.2 Å3. Dc = 1.886 Mg m–3; specimen: 0.45 × 0.20 × 0.08 mm. μMo = 1.77 mm–1; ‘T’min/max = 0.83. 2θmax = 58°; Nt = 13 510, N = 3491 (Rint = 0.021), No = 3016; R = 0.034, R′ = 0.042; nv = 248, |Δρmax| = 1.38(5) e Å–3.
D. [Co(en)(NH2CH2CH2N=CHCH2NH2))Cl][ZnCl4] = C6H19ClCoN5Zn, M = 462.8. a = 8.5593(8), b = 10.607(1), c = 18.112(2) Å, β = 103.646(1)°, V = 1597.9 Å3. Dc = 1.924 Mg m–3; specimen: 0.65 × 0.25 × 0.20 mm. μMo = 3.36 mm–1; ‘T’min/max = 0.65. 2θmax = 58°; Nt = 15 500, N = 4603 (Rint = 0.027), No = 3502; R = 0.027, R′ = 0.034; nv = 239, |Δρmax| = 0.77(5) e Å–3.
E. [Co(en)2(N-CH3en)][Co(CN)6]·2 H2O = C13H30Co2N12O2, M = 504.3. a = 13.872(2), b = 9.969(1), c = 15.423(2) Å, β = 95.258(2)°, V = 2123.9 Å3. Dc = 1.577 Mg m–3; specimen: 0.45 × 0.25 × 0.19 mm. μMo = 1.60 mm–1; ‘T’min/max = 0.73. 2θmax = 58 °; Nt = 20 615, N = 5310 (Rint = 0.026), No = 4221; R = 0.035, R′ = 0.043; nv = 373, |Δρmax| = 1.22(5) e Å–3.
F. [Co(en)2(N-CH3en)][Co(CN)6] = C13H26Co2N12, M = 468.3. a = 14.176(2), b = 9.462(1), c = 14.326(2) Å, β = 90.608(2)°, V = 1921.4 Å3. Dc = 1.619 Mg m–3; specimen: 0.25 × 0.24 × 0.22 mm. μMo = 1.75 mm–1; ‘T’min/max = 0.87. 2θmax = 58°; Nt = 18 614, N = 4860 (Rint = 0.029), No = 3869; R = 0.030, R′ = 0.035; nv = 351, |Δρmax| = 1.20(6) e Å–3.
The crystallographic data, in the form of .cif files, have been deposited with the CCDC, deposition numbers 246595–246599 . These data may be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax. (+44) 1223-336-033 or deposit@ccdc.cam.ac.uk.
1 Other metal ions also show various coordination modes with ethylenediamine, commonly with mixed modes within the one species. See, for example, [5]. Several other structurally characterised examples found in the CCDC are cited in reference [7] and early literature in the field is cited in reference [13].