

1 Introduction
In the field of second-order non-linear optics (NLO), two complementary approaches are currently actively studied. Beyond the ‘classical’ dipolar one, which has led to the elaboration of quasi-optimized electrooptic materials [1], the enlarged potential of the octupolar approach has been recognized since the early 1990s and have triggered the emergence of a new field of research [2]. In this latter case, the non-centrosymmetry can be obtained by a strict control of the molecular symmetry; and for molecules belonging to purely octupolar space groups, like D3, D3h, Td, D2d, the vectorial part of the hyperpolarizability tensor β(J = 1), corresponding to the dipolar contribution, is cancelled out, and only the octupolar contribution β(J = 3) remains [2]. Since the pioneering report on 1,3,5-triamino-2,4,6-trinitrobenzene, the concept of octupolar non-linearity has been widely demonstrated at the molecular level and numerous examples of octupolar molecules exhibiting large hyperpolarizability, in the same order of magnitude than that of the best dipoles, are reported in [3].
However, the extension of this concept at the macroscopic level still remains a challenge. For that purpose two different strategies can be envisaged for the elaboration of non-centrosymmetric materials, namely the orientation or the organization. The first one, of statistical nature, consists in the orientation of octupolar compounds possessing additional photoisomerization properties, dispersed or grafted in a polymeric matrix by means of the all-optical poling technique [4]. Here, we will focus on the second strategy that requires the non-centrosymmetric macroscopic organization of octupolar compounds by means of the control of the intermolecular interactions. Recently, Zyss [5] reported a general organization scheme study towards the optimization of 2D and 3D macroscopic arrangements of octupoles. The 2D organization, consisting in a non-centrosymmetric trigonal arrangement of trigonal octupoles (Fig. 1) [6], has been already illustrated by crystal engineering examples [7a] or by pre-organization of octupoles within a dendritic structure [7b]. The 3D generalization has been proposed in the form of a helicoidal hypercubic arrangement, by a stacking of the elementary cubic units along the threefold axis [5].
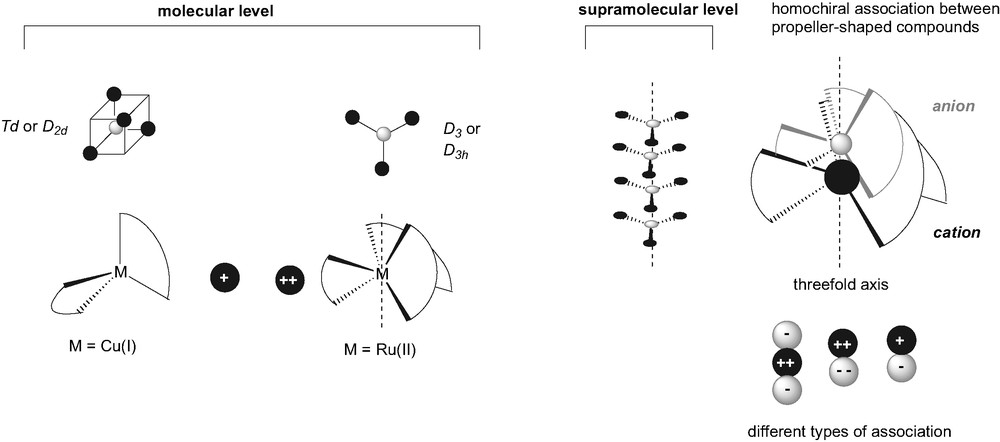
Schematic representation of octupolar symmetries and of the optimized supramolecular association of octupole (right).
In this communication, we would like to report on our first attempts to design such supramolecular architectures by means of the electrostratic shape recognition between D3 ruthenium(II) dication or D2d copper(I) cation and anions featuring the same symmetry.
The starting point of this study was the discovery of the homochiral interaction between (DEASbpy)3Ru2+ (DEASbpy = 4,4′-bis(diethylaminostyryl)-2,2′-bipyridine) and tris(tetrachlorobenzendiolato)phosphat anion also called TRISPHAT (Fig. 2) [8]. It is worth noting that these two ionic derivatives are propeller-shaped with a helicoidal axis of chirality (isomer Λ and Δ) and exhibit the D3 octupolar symmetry. A diastereoselective self-assembling process was evidenced at room temperature by 1H NMR in a dichloromethane solution containing a racemic mixture of anion and cation, resulting in the formation of the homochiral ion pairs (preferred association of propeller featuring identical handedness). This supramolecular association consists in the ‘stacking’ of the octupolar anion and cation along their threefold axis and corresponds exactly to the optimized macroscopic octupolar arrangement (Fig. 1) [6].
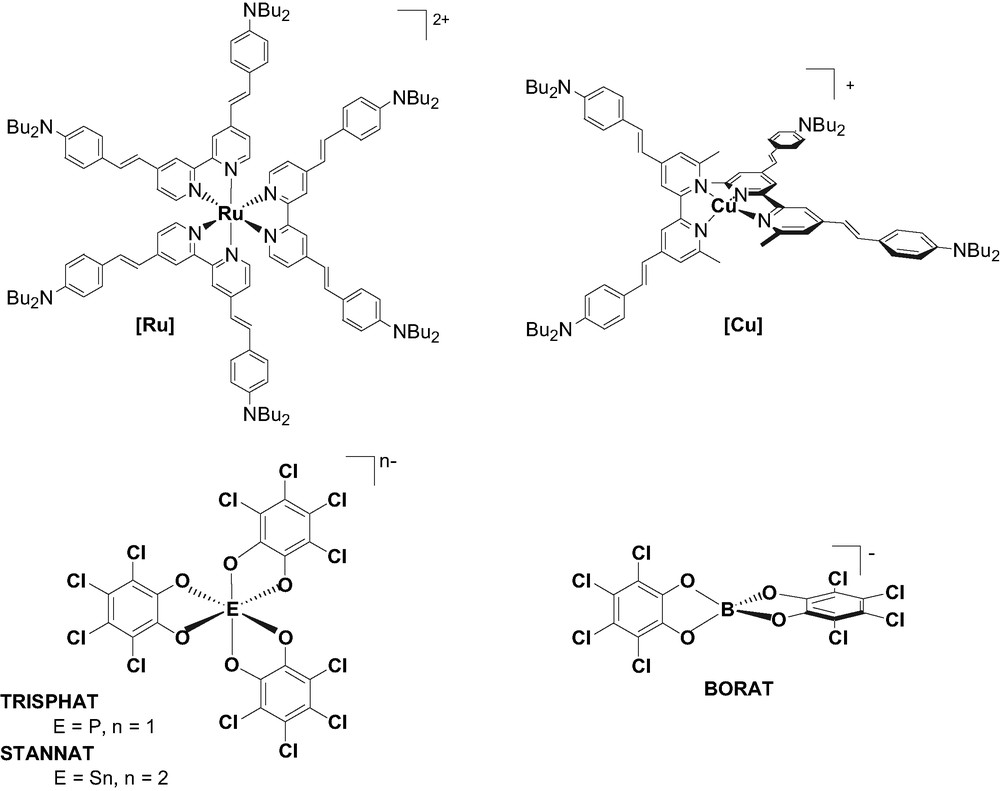
Cationic (top) and anionic (bottom) octupolar compounds.
This paper is devoted to the NLO study of such supramolecular assembly. To this end, we have synthesized mono and dianion based on P, Sn or B catecholate complexes presenting octupolar D3 or D2d symmetry and studied their interaction in solution with D3 tris{4,4′-bis(dibutylaminostyryl)-2,2′-bipyridine]}ruthenium(II) [Ru] or D2d bis{4,4′-bis(dibutylaminostyryl)-6,6′-dimethyl-2,2′-bipyridine}copper(I) [Cu] (Fig. 2).
2 Synthesis
The preparation of [Ru](PF6)2 and [Cu](PF6) has already been described [3]. The synthesis of [HNBu3][TRISPHAT] and [HNBu3]2[STANNAT] was achieved by mixing PCl5 or SnCl4 with three equivalents of tetrachlorocatechol in the presence of tributylamine according to procedures described by the groups of Lacour [9a] and Lambert et al. [9b] or Annan et al. [9c], respectively. The BORAT, [Na][B(O2C6Cl4)2] was simply prepared by treatment of NaBH4 with 2 equiv of catechol in tetrahydrofuran [10]. All these anions were characterized by heteronuclear NMR spectroscopy and exhibit a characteristic signal at –80.7, –554.5 and 14.5 ppm for TRISPHAT (31P NMR), STANNAT (119Sn NMR)1, and BORAT (11B NMR), respectively.
3 Supramolecular association
The association of the D3 symmetric anions and [Ru] dication was achieved by an anionic metathesis reaction. Since the STANNAT anion is not able to displace the PF6–, the [Ru]STANNAT complex can only be prepared from [Ru]Cl2 precursor. By contrast, the synthesis of [Ru](TRISPHAT)2 can be carried out either from the chloro, the hexafluorophosphate or even the STANNAT ruthenium complex and no other anion, even in large excess is able to displace the TRIPHAT. Finally from this competitive experiments, the following order of affinity of the tris(bipyridyl)ruthenium complex for the different anions can be proposed: Cl– < STANNAT2– < PF6– < TRISPHAT–. These three complexes were fully characterized by 1H, 13C NMR, UV–visible spectroscopy and microanalysis (see Section 6) and exhibit a completely different behavior in analytical thin layer chromatography (TLC) on silica plates with dichloromethane as eluent. Whereas the TRISPHAT salts are easily eluted (Rf = 1, Table 1), the hexafluorophosphate and STANNAT ruthenium salts does not migrate under the same conditions (Rf < 0.1). This different behavior indicates a strong difference of polarity dependent on the nature of the anion and is in agreement with the stronger ion pairing effect observed in the case of TRISPHAT. Indeed, TRISPHAT, as well as STANNAT is strongly lipophilic and a stronger anion/cation interaction will result in a decrease of the ion pair polarity.
1H NMR chemical shift (CD2Cl2, RT) and TLC behavior (silica plate, CH2Cl2) of various anion/cation combination
| Compound | H3–H5–H6 | H7–H8 | Rf |
| [Ru](PF6)2 | 8.36–7.33–7.52 | 6.92–7.43 | 0 |
| [Ru](TRISPHAT)2 | 8.34–7.28–7.98 | 6.57–7.22 | 1 |
| [Ru]STANNAT | 8.62–7.28–7.63 | 6.84–7.53 | 0 |
| [Cu](PF6) | 8.16–7.41 | 6.92–7.41 | 0 |
| [Cu](BORAT) | 8.22–7.5 | 6.96–7.47 | 0.58 |
In addition, 1H NMR spectroscopy (Fig. 3) gives significant information concerning the nature of the anion/cation interaction. All the spectra were recorded with the same concentration in ruthenium species. As already observed for TRISPHAT, the racemic mixture of [Ru] and STANNAT (isomer Λ and Δ) results only in the presence of a single set of signals for the bipyridyl ligand even at low temperature. This phenomenon is explained by the preferential co-threefold embrace of charged propeller-like derivatives [8]. In a non-dissociating solvent like dichloromethane, this strong ion pairing effect results in a modification of the 1H NMR spectra. The major effect is the deshielding of H6 proton in the case of TRISPHAT (ΔδH6 = 0.46 ppm vs. [Ru](PF6)2), whereas in the case of STANNAT, this deshielding effect occurs on the H3 proton (ΔδH3 = 0.26 ppm vs. [Ru](PF6)2). On the contrary, in a strongly polar solvent like deuterated dimethylsulfoxide, the charged species are completely dissociated and only the spectra of the solvated [Ru] species is observed (Fig. 3, bottom) [11]. It has to be pointed out that the shielding effect occurs on the proton closed to the metal (H6) in the case of TRISPHAT, and in the periphery of the ligand (H3) in the case of STANNAT. This should be explained by a more intimate interaction between [Ru] and TRISPHAT in agreement with the smaller size of the phosphorus anion when compared to that of the tin derivative (average d(P–O) = 1.71 Å [9a] vs. d(Sn–O) = 2.06 Å [[9b,c]).
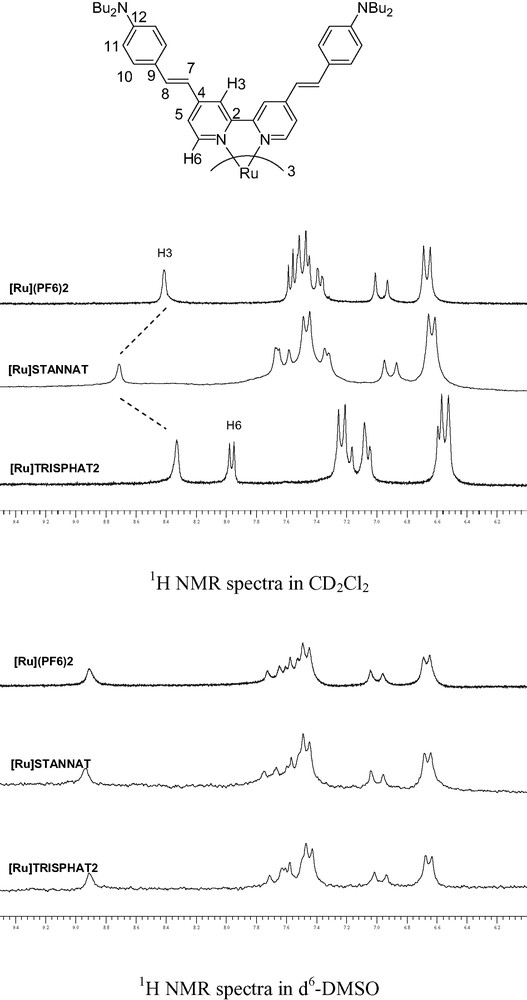
Influence of the anion on the 1H NMR spectra (conc. 10–2 mol l–1, room temperature).
In the case of the pseudo-tetrahedral derivatives, the anionic metathesis between [Cu]Cl and NaBORAT leads to the formation of [Cu]BORAT. This ion pair formation does not induced significant modification in the 1H NMR spectra but results in a completely different behavior in TLC (Table 1). Whereas the hexafluorophosphate salt is strongly retained, the BORAT one can be easily eluted with dichloromethane with a Rf value of 0.58.
4 Linear and non-linear properties
BORAT, TRISPHAT or STANNAT anions are completely transparent in the visible and exhibit only absorption in the UV (Table 2). Therefore, their association either with [Cu] or [Ru] does not modify the ILCT or MLCT transition located in the visible part of the spectrum (Table 2). The NLO activity of the D3 octupolar anions was estimated by means of the harmonic light scattering (HLS) technique using 1.06 μm as fundamental laser wavelength. Their hyperpolarizabilities are low (Table 2) in the same order of magnitude than that of trisisopropylaminotrinitrobenzene [12]. These low NLO activities are in marked contrast with the large β values of [Ru](PF6)2, β = 340 × 10–30 esu that must be measured at 1.91 μm, this fundamental wavelength precluding any perturbation of the experimental data by absorption effects at the second harmonic frequency or by two-photons induced fluorescence phenomena. Hence, changing the hexafluorophosphate by a TRISPHAT or a STANNAT anion does not induced any significant modification of the NLO activity (Table 2) and the observed variations remain within the experimental error of the measurement technique (±15%).
Linear and NLO data
| Compound | λmax (ɛ)a (nm l mol–1 cm–1) | βb (10–30 esu) |
| [HNBu3](TRISPHAT) | 300 (12,000) | 10 ± 2c |
| [HNBu3]2(STANNAT) | 306 (6000) | 30 ± 5c |
| [Ru](PF6)2 | 446 (142,000) | 340 ± 51d |
| 520 (150,000) | ||
| [Ru](TRISPHAT)2 | 449 (124,000) | 320 ± 48d |
| 524 (133,000) | ||
| [Ru]STANNAT | 447 (130,000) | 310 ± 46d |
| 517 (133,000) | ||
| NaBORAT | 301 (8000)e | – |
| [Cu](PF6) | 436 (106,000) | 113 ± 17d |
| 480 sh | ||
| [Cu](BORAT) | 437 (106,000) | – |
| 480 sh |
a In CH2Cl2.
b Precision ± 15%.
c Measured by HLS in CH2Cl2 at 1.064 μm.
d Measured by HLS in CH2Cl2 at 1.91 μm.
e In THF.
5 Concluding remarks
In this communication, we have described the supramolecular association between D3 or D2d symmetric octupolar cations and anions, evidenced by NMR or TLC. The too large difference between the NLO activity of the anion and the cation does not allow to measure the effect of this association on the NLO activity. This preliminary work opens interesting perspectives for the design of supramolecular assemblies of octupoles. The P, B, Sn catecholate derivatives seem to be well suited for the supramolecular association with tris(bipyridyl)ruthenium(II) or bis(bipyridyl)copper(I) complexes. Further functionalization of this catecholate ligands should increase the NLO activity of the corresponding octupolar derivatives enabling to measure the effect of supramolecular associations of octupoles on the NLO activity.
6 Experimental section
6.1 [Ru]STANNAT
In a Schlenk vessel RuCl2(DMSO)4 (131.32 mg, 0.27 mmol, 1 equiv) and 4,4′-[(N,N-dibutyl)aminostyryl]-2,2′-bipyridine (500 mg, 0.81 mmol, 3 equiv) were dissolved in ethanol (30 ml). The red mixture was heated at reflux under nitrogen for 7 h. After the solution was cooled to room temperature, (HNBu3)2STANNAT (0.515 g, 0.54 mmol, 2 equiv), was added and the solution was stirred for 3 h. The desired complex was precipitated by addition of water (300 ml), filtered off and washed several times with pentane and diethylether. The crude product was dissolved in CH2Cl2, dried with MgSO4; the solution was concentrated and the product was precipitated by addition of pentane (v/v = 1/10). Finally the red microcrystalline powder was dried under vacuum (400 mg, 55%). 1H NMR (200.13 MHz, CD2Cl2) δ ppm: 8.62 (s, 6H, H3); 7.63 (d, J = 5.8 Hz, 6H, H6); 7.53 (d, J = 16.0 Hz, 6H, H8); 7.40 (d, J = 8.5 Hz, 12H, H10); 7.28 (d br., J = 5.8 Hz, 6H, H5); 6.84 (d, J = 16.0 Hz, 6H, H7); 6.58 (d, J = 8.5 Hz, 12H, H11); 3.25 (t, J = 5.9 Hz, 24H, H13); 1.60–1.50 (m, 24H, H14); 1.40–1.22 (m, 24H, H15); 0.94 (t, J = 7.1 Hz, 36H, H16). 13C NMR (75.47 MHz, CD2Cl2) δ ppm: 157.0 (C2); 150.3 (C6); 149.4 (C12); 149.3 (Cstannat); 147.4 (C4); 137.0 (C8); 129.4 (C10); 123.0 (C5); 122.5 (C9); 120.2 (C3); 117.8 (C7); 117.5 (Cstannat); 116.1 (Cstannat); 111.5 (C11); 50.7 (C13); 29.4 (C14); 20.3 (C15); 13.7 (C16). Elemental analysis calculated for C144H162N12O6Cl12RuSn,3CH2Cl2: C 57.76; H 5.54; N 5.50. Found: C 57.51; H 6.01; N 6.14.
6.2 [Ru](TRISPHAT)2
Using similar procedure but with (HNBu3)TRISPHAT, the red complex was prepared in 65% yield. 1H NMR (200.13 MHz, CD2Cl2) δ ppm: 8.34 (s, 6H, H3); 7.98 (d, J = 6.2 Hz, 6H, H6); 7.25 (d, J = 8.9 Hz, 12H, H10); 7.22 (d, J = 16.3 Hz, 6H, H8); 7.08 (d. br., J = 6.2 Hz, 6H, H5); 6.57 (d, J = 16.3 Hz, 6H, H7); 6.56 (d, J = 8.9 Hz, 12H, H11); 3.26 (t, J = 6.4 Hz, 24H, H13); 1.60–1.50 (m, 24H, H14); 1.40–1.22 (m, 24H, H15); 0.94 (t, J = 6.9 Hz, 36H, H16). 13C NMR (75.47 MHz, CD2Cl2) δ ppm: 156.9 (C2); 150.9 (C6); 149.4 (C12); 147.0 (C4); 141.6 (d, 3J(P–C) = 6.6 Hz, 2C, C2trisphat); 136.3 (C8); 129.3 (C10); 122.6 (C5); 122.4 (d, 4J(P–C) = 2.2 Hz, 2C, C4trisphat); 122.1 (C9); 119.6 (C3); 117.2 (C7); 113.9 (d, 2J(P–C) = 19.8 Hz, 2C, C1trisphat); 111.4 (C11); 50.7 (C13); 29.4 (C14); 20.3 (C15); 13.7 (C16). 31P NMR (121.47 MHz, CD2Cl2) δ ppm: –80.71. Elemental analysis calculated for C162H162N12O12Cl24P2Ru: C 55.86; H 4.69; N 4.83. Found: C 55.61; H 4.83; N 4.60.
6.3 [Cu]BORAT
In a Schlenk vessel, the (4,4′-[(N,N-dibutyl)aminostyryl]-6,6′-dimethyl-2,2′-bipyridine)CuCl complex (100 mg, 7.22 mmol, 1 equiv), and one equivalent of NaBORAT (37.96 mg, 7.22 mmol, 1 equiv), were dissolved in dichloromethane (20 ml). The orange mixture was stirred for 3 h at room temperature. The crude mixture was filtered to remove NaCl. The solution was then dried with MgSO4; concentrated and the product was precipitated by addition of pentane (v/v = 1/10). Finally the brown microcrystalline powder was dried under vacuum (90 mg, 65%). 1H NMR (200.13 MHz, CD2Cl2) δ ppm: 8.22 (s, 4H, H3); 7.52 (d, J = 7.5 Hz, 8H, H10); 7,50–7.46 (m, 12H, H5, H8); 6.96 (d, J = 14.6 Hz, 4H, H7); 6.72 (d, J = 7.5 Hz, 8H, H11); 3.38 (t, J = 7.0 Hz, 16H, H13); 2.28 (s, 12H, H17); 1.64 (m, 16H, H14); 1.42 (m, 16H, H15); 1.02 (t, J = 7.2 Hz, 24H, H16). 13C NMR (75.47 MHz, CD2Cl2) δ ppm: 156.8 (C6); 152.2 (C4); 149.5 (C12); 147.9 (C2); 147.7 (Cborat); 135.2 (C8); 128.9 (C10); 122.5 (C9); 121.4 (C5); 122.5 (C9); 121.4 (C5); 120.3 (C3); 119.9 (Cborat); 115.7 (C7); 112.0 (C11); 111.5 (Cborat), 50.7 (C13); 29.4 (C14); 24.8 (C17); 20.3 (C15); 13.8 (C16); 11B NMR (96.29 MHz, CD2Cl2) δ ppm: 14.4.
1 STANNAT exhibits a singlet signal in 119Sn-{H}NMR (CD2Cl2, 111.9 MHz, SnMe4 as standard) δ = –554.5 vs. 353 ppm described in [9b] in the same conditions. This latter signal was also detected and attributed to a cavity resonance.