

1 Introduction
Ferrocene and ferrocenyl derivatives are well known for their ability to undergo reversible one-electron oxidation [1]. The redox potential depends on the electronic effect of the ring substituents on ferrocene. This change of the redox potential of the ferrocenyl center with the electronic effect of the substituent (chelating ligand) has been used to electrochemically sense a neutral or ionic guest molecule allowing its amperometric or potentiometric titration [1–5]. Phosphonic acids are known to react with metal salts to form organic–inorganic metal phosphonates, containing stable PO–metal bonds, with usually a two dimensional network [6]; they have also been used as grafting molecules [6]. For example, ferrocenylmethylphosphonic acid has been attached to a silicon surface as a charge storage molecule for memory applications [7].
In this paper we report the preparation and characterization of electro-active cadmium and manganese ferrocenylphosphonates.
2 Results and discussion
Ferrocenylphosphonic acid was prepared using tBuLi/tBuOK as a ‘super base’, according to Fig. 1 [8].
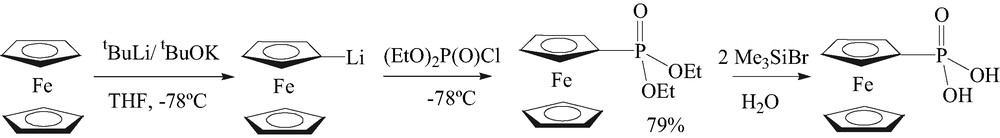
Preparation of ferrocenylphosphonic acid.
The pKa of the ferrocenylphosphonic acid determined by potentiometry, 31P NMR and by voltammetry are reported in Table 1 [9]. The pKa1 of the ferrocenylphosphonic acid cannot be determined by the potentiometric method due to its low solubility in acidic media. The low solubility of the ferrocenylphosophonic acid does not allow the control of the ionic strength of the solution (C = 0.01 mol l–1) in the potentiometric and NMR experiments. However, the values obtained for pKa2 by the potentiometric and 31P NMR methods are in good agreement. The higher values found by the voltammetric method should be attributed to the ionic strength of the medium which was maintained constant in this method. It is noteworthy that the oxidized forms of the ferrocenylphosphonic acid (pKa′) are more acidic than the reduced form, as previously observed for ferrocenyl pH-responsive compounds [10,11].
pK values of ferrocenylphosphonic acid
| Method | pKa1 | pKa2 | pK′a1 | pK′a2 |
| Potentiometric | < 3 | 8.3 | ||
| 31P NMR | 2.1 | 8.3 | ||
| Voltammetric | 2.4 | 9.1 | 1.0 | 6.2 |
Temperature 20 °C, in H2O, no control of the ionic strength except for the voltammetric method, KCl 0.1 N.
Fig. 2 shows the calculated mole fraction α of each ionic species of FcPO3H2 and of the oxidized form Fc+PO3H2.
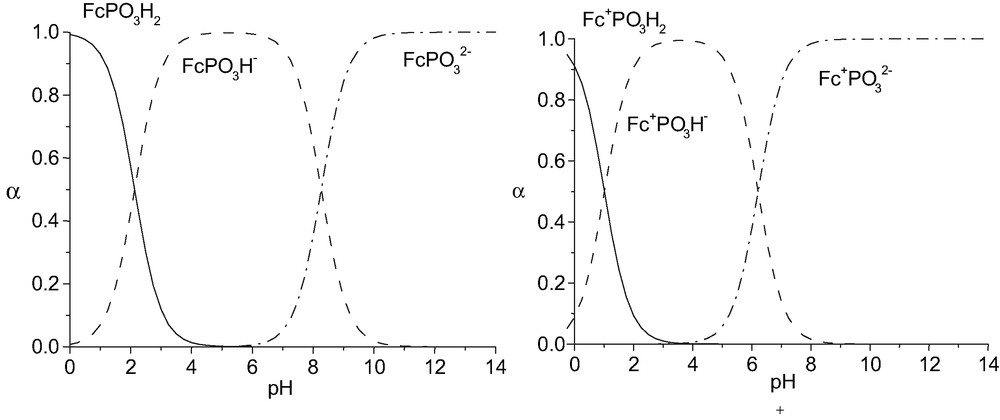
Mole fraction (α) of ionic species of FcPO3H2 and Fc+PO3H2.
2.1 Preparation of cadmium and manganese ferrocenylphosphonates at pKa1 < pH < pKa2
The reaction of ferrocenylphosphonic acid with cadmium and manganese nitrates was studied as a function of the initial pH of the solution.
For pKa1 < pH < pKa2 the general formula M(HO3PFc)2·x H2O was obtained (Fig. 3).
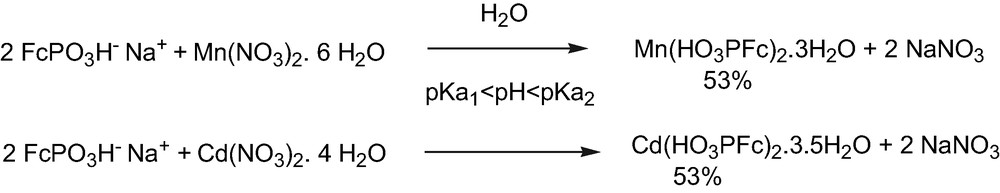
Preparation of metal phosphonates at pKa1 < pH < pKa2.
The compounds were characterized by IR spectroscopy, TGA, 31P and 113Cd MAS NMR, and X-ray analysis.
The IR spectra of the manganese and cadmium compounds are very similar (Fig. 4), the bands of OH from water appear at 3500–3100, and 1650 cm–1; the broad bands centered at 2800 and 2400 cm–1 correspond to POH, the bands between 1430 and 1350 cm–1 are related to PO3.
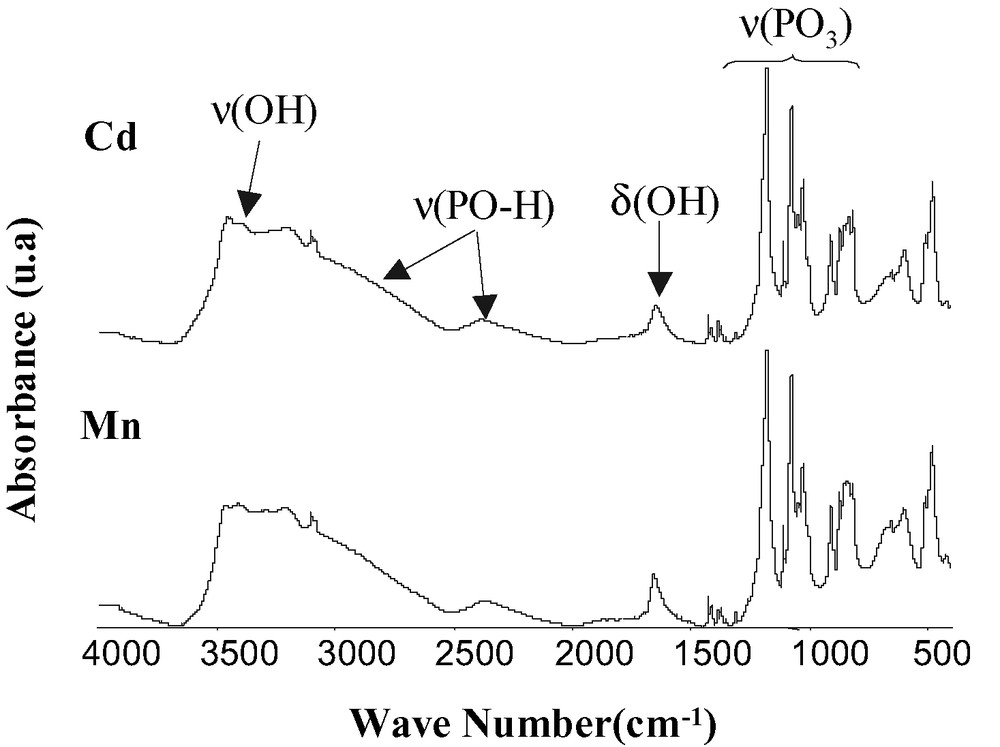
IR spectra of and Mn(HO3PFc)2·3 H2O and Cd(HO3PFc)2·3.5 H2O.
TGA allows the evaluation of the water content of the samples. The TGA trace of the Cd derivative (Fig. 5) reveals a first weight loss from 60 to 90 °C due to the loss of 3.5 equivalents of water. For the Mn compound the first weight loss occurs between 80 and 130 °C corresponding to the loss of 4 equiv of water, three are related to structural water and the last to the condensation of the POH groups. The degradation of the organic part occurs at about 200 °C.
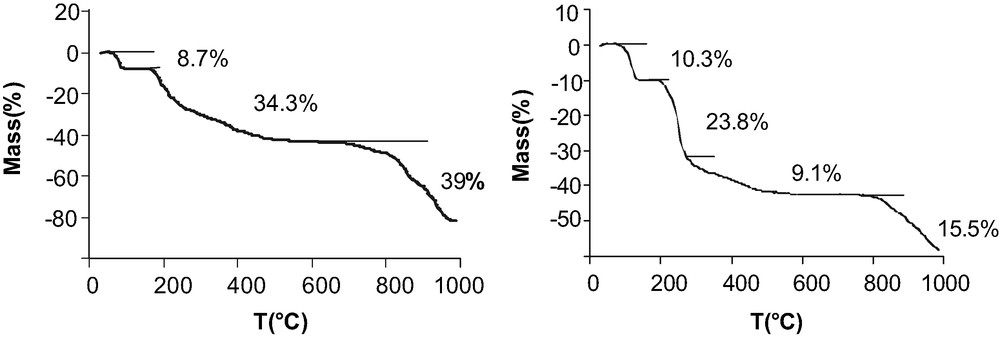
Thermal gravimetric analysis of Cd(HO3PFc)2·3.5 H2O and Mn(HO3PFc)2·3 H2O under nitrogen.
The 31P and 113Cd MAS NMR spectra of the diamagnetic compound Cd(HO3PFc)2·3.5H 2O show, respectively, one signal for the phosphorus atom at δ = 18.1 ppm and one environment for the cadmium atom with a chemical shift at –18.2 ppm vs. Cd(ClO4)2 1 M. This chemical shift corresponds to a hexacoordinated Cd atom [12,13].
The X-ray powder patterns shown in Fig. 6 are similar for both compounds. From the analytical data, the IR spectra and the X-ray diaffractograms, we can assume the iso-structurality of the two compounds.
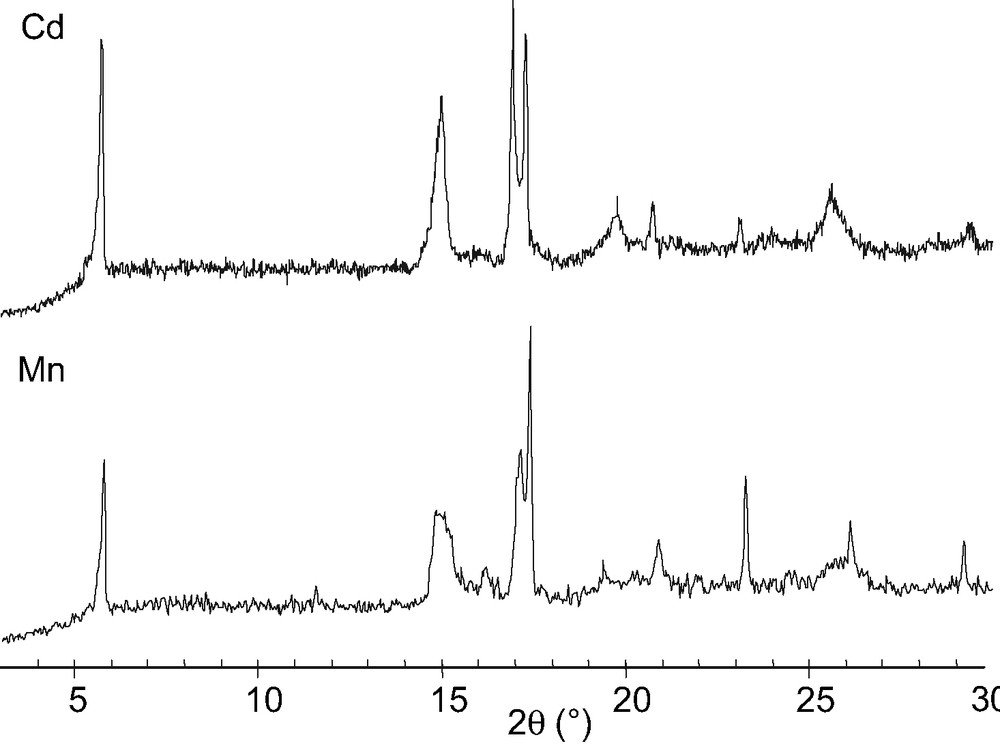
X-ray patterns of Cd(HO3PFc)2·3.5 H2O and Mn(HO3PFc)2·3 H2O.
2.2 Preparation of cadmium and manganese ferrocenylphosphonate at pH > pKa2
For pH > pKa2, the composition of the products were M(O3PFc)·2 H2O, M = Cd and Mn (Fig. 7).
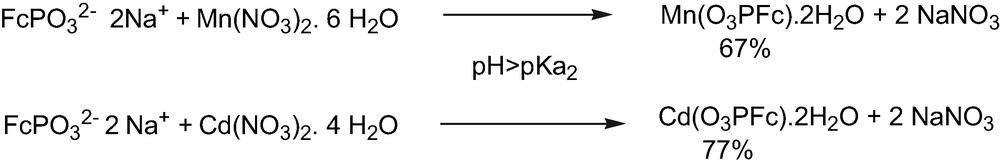
Preparation of metal ferrocenylphosphonates at pH > pKa2.
As shown in Fig. 8, the two compounds show similar IR spectra.
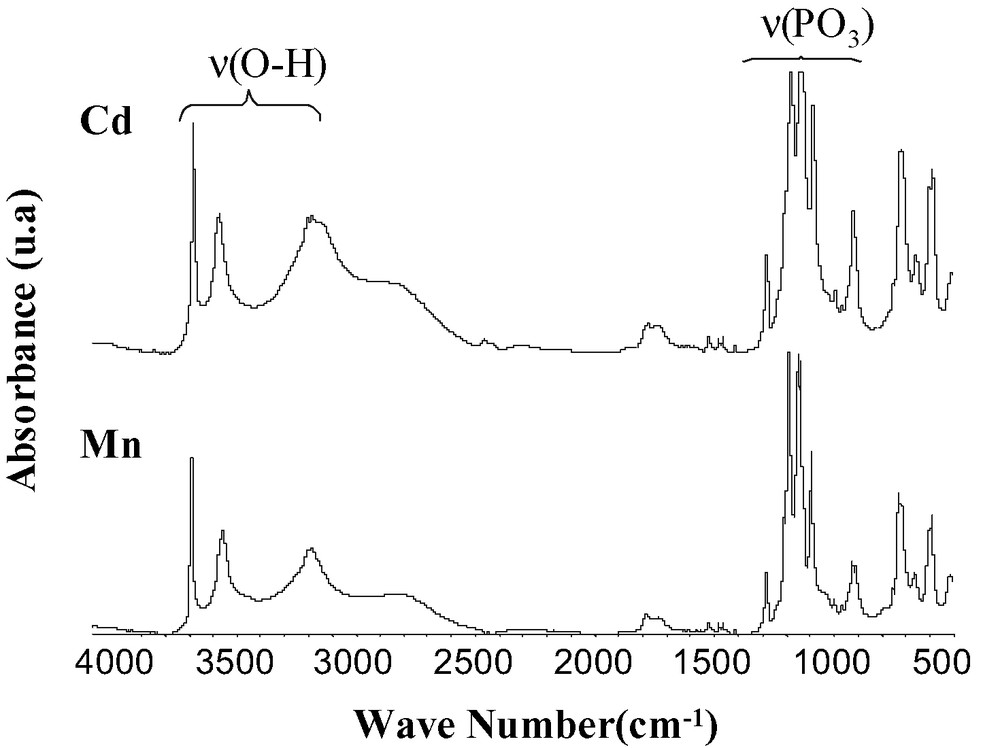
Ir spectra of Cd(O3PFc)·2 H2O and Mn(O3PFc)·2 H2O.
The TGA traces of the compounds reported in Fig. 9 show a first weight loss between 96 and 144 °C for Cd(O3PFc)·2 H2O and between 90 and 142 °C for Mn(O3PFc)·2 H2O corresponding to the dehydration of the compounds. The degradation of the organometallic part begins at 210 °C.
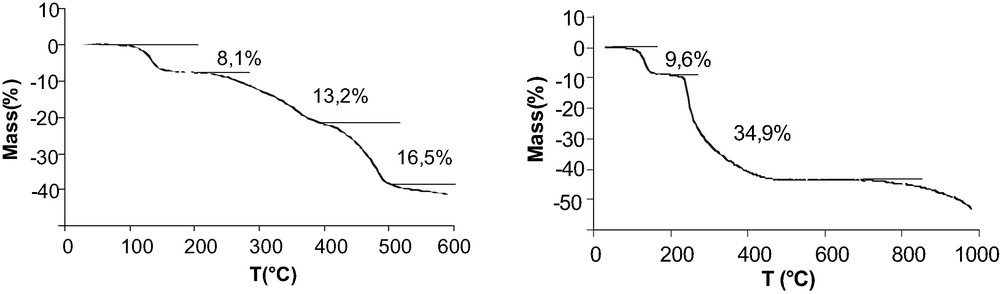
TGA traces of Cd(O3PFc)·2 H2O and Mn(O3PFc)·2 H2O under an inert atmosphere.
The diamagnetic compound, Cd(O3PFc)·2 H2O, presents a unique peak in the 31P NMR spectrum at δ = 23 ppm as well in the 113Cd MAS NMR spectrum, with a chemical shift at 15.2 ppm vs. Cd(ClO4)2 1 M corresponding to hexacoordinated Cd [12,13]. This implies that the oxygen atoms of the phosphonate groups act as bridges between two cadmium atoms (μ2O).
The X-ray diffraction patterns of the two compounds are similar as shown in Fig. 10.
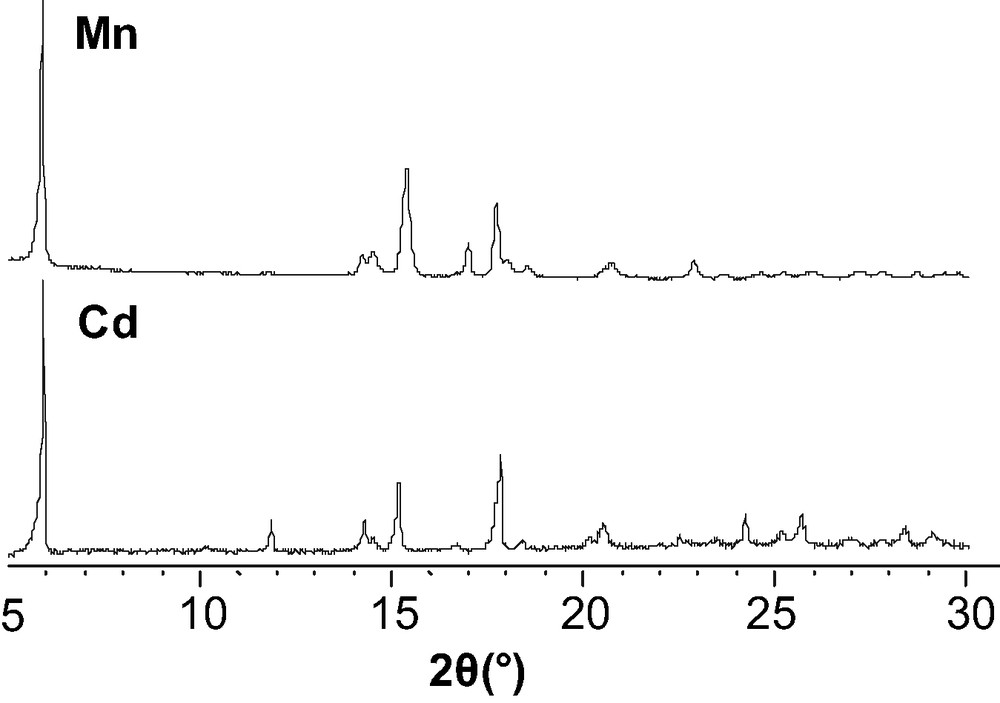
X-ray diffraction data of Cd(O3PFc)·2H2O and Mn(O3PFc)·2H2O.
From these results the iso-structurality of Cd(O3PFc)·2 H2O and Mn(O3PFc)·2 H2O can also be assumed.
2.3 Electrochemical study of the cadmium and manganese ferrocenylphosphonates
Due to the insolubility of the compounds in any solvent, the electrochemical study of these compounds by cyclic voltammetry was performed in the solid state using a cavity microelectrode provided by the ‘réseau micro electrode à cavité du CNRS’ (France) [14–18]. The results are shown in Table 2. The half wave potential of the ferrocenyl group for the same composition of the metal phosphonates does not depend on the nature of the metal (Zn, Cd, Mn). A maximum in ΔE1/2 of 8 mV is observed between Zn(HO3PFc)2·2H2O and Mn(HO3PFc)2·3 H2O and 3 mV between Cd(O3PFc)·2 H2O and Mn(O3PFc)·2 H2O. However the difference between the two compositions, M(HO3PFc)2·x H2O and M(O3PFc)·2 H2O is much more drastic with a ΔE1/2 up to 130 mV. It is noteworthy that the half wave potentials of the metal phosphonates depend on their composition. For M(HO3PFc)2 the half wave potentials are of the same order as the mono sodium salt of ferrocenylphosphonic acid FcPO3HNa (235–246 mV); while those of M(O3PFc) are the same for M = Cd or Mn (132–135 mV) but higher than that of the di sodium salt of ferrocenylphosphonic acid FcPO3Na2 (86 mV). The half wave potential of the metal ferrocenyl phosphonates does not seem to depend greatly on the nature of the metal but rather on the neutralization of the ferrocenylphosphonic acid.
Electrochemical data of metal ferrocenylphosphonates at 20 °C in water, electrolyte: KCl 1 M, saturated calomel as reference electrode, scan rate: 100 mV s–1
| Compounds | Epa (mV) | Epc (mV) | E1/2 (mV) | ΔE = Epa – Epc (mV) |
| Zn(HO3PFc)2·2 H2Oa | 260 | 210 | 235 | 50 |
| Cd(HO3PFc)2·3.5 H2O | 264 | 213 | 238 | 51 |
| Mn(HO3PFc)2·3 H2O | 270 | 217 | 243 | 53 |
| Cd(O3PFc)·2 H2O | 157 | 107 | 132 | 50 |
| Mn(O3PFc)·2 H2O | 155 | 115 | 135 | 40 |
| FcPO3H–Na+b | 281 | 211 | 246 | 70 |
| FcPO32–2Na+b | 119 | 54 | 86 | 65 |
a Prepared according to [19].
b Cyclic voltammogram in solution.
3 Conclusion
The composition of cadmium and manganese ferrocenylphosphonates, prepared from ferrocenylphosphonic acid and metal nitrate depends on the pH of the initial solution. The cadmium and manganese compounds are iso-structural. Their half wave potentials are the same for the same composition and do not depend on the nature of the metal. Ferrocenylphosphonic acid does not behave as a metal cation sensor but rather as a pH sensor.
4 Experimental section
IR spectra were performed on a Thermo Nicolet AVATAR 320 FT-IR apparatus as KBr pellets. MAS NMR spectra were taken on a Bruker DPX 300 operating at 121.5 MHz for 31P and 66.5 MHz for 113Cd. The reference used were H3PO4 30% for 31P and Cd(ClO4)2 1 M for 113Cd. Powder X-ray diffraction was performed on an X Pert Philips diffractometer. TGA were made on a Netzch 409 thermobalance. Cyclic voltammetry measurements were performed by means of a Radiometer Analytical PGZ 100 potentiostat in a three-electrode cell. A cavity microelectrode with a Pt working electrode, a platinum wire auxiliary electrode, and a saturated calomel reference electrode were used to record voltammograms. The electrolyte was KCl (1 M in water). The scan rate was 100 mV s–1. Elemental analyses were performed by the ‘Service central d'analyse’ of CNRS in Vernaison, France.
Ferrocenylphosphonic acid and zinc ferrocenylphosphonate Zn(HO3PFc)·2 H2O have been prepared as already reported [8,19].
4.1 Preparation of Cd(HO3PFc)2·3.5 H2O
Ferrocenylphosphonic acid (0.098 g, 0.36 mmol) was dissolved in 15 ml of water. The pH was adjusted to 3.7 by addition of NaOH 1 M. Cd(NO3)2·4 H2O (0.11 g, 0.36 mmol) was added to the solution without stirring. After 5 days at room temperature, the yellow precipitate was filtered, washed with methanol and ether, and dried under vacuum leading to 66 mg (0.09 mmol) of Cd(HO3PFc)2·3.5H2O (53% yield). 31P MAS NMR, 18.1 ppm; 113Cd MAS NMR, –18.2 ppm vs. Cd(ClO4)2 1 M. Anal. Calc. for CdP2Fe2C20H27O9.5: Cd, 15.93%; Fe, 15.83%; P, 8.78%; C, 34.02%; H 3.83%; found: Cd, 15.10%; Fe, 15.40%; P, 8.43%; C, 33.68%; H, 3.80%.
4.2 Preparation of Cd(O3PFc)·2 H2O
Ferrocenylphosphonic acid (0.1 g, 0.38 mmol) was dissolved in 28 ml of water. The pH was adjusted to 10.2 by addition of NaOH 1 M. Cd(NO3)2·4 H2O (0.12 g, 0.39 mmol) was added to the solution without stirring. After 5 days at room temperature, the yellow precipitate was filtered, washed with methanol and ether, and dried under vacuum leading to 120 mg (0.29 mmol) of Cd(O3PFc)·2H2O (77% yield). 31P MAS NMR, 23.4 ppm; 113Cd MAS NMR, 15.2 ppm vs. Cd(ClO4)2 1 M. Anal. Calc. for CdPFeC10H13O5: Cd, 27.26%; P, 7.44%; Fe, 13.24%; found: Cd, 26.61%; P, 7.52%; Fe, 13.50%.
4.3 Preparation of Mn(HO3PFc)2·3 H2O
Ferrocenylphosphonic acid (0.15 g, 0.56 mmol) was dissolved in 35 ml of water. The pH was adjusted to 3.5 by addition of NaOH 1 M. Mn(NO3)2·4 H2O (0.08 g, 0.32 mmol) was added to the solution without stirring. After 5 days at room temperature, the yellow precipitate was filtered, washed with methanol and ether, and dried under vacuum leading to 95 mg (0.15 mmol) of Mn(HO3PFc)2·3H2O (53% yield). Anal. Calc. for MnP2Fe2C20H26O9: Mn, 8.60%; P, 9.71%; Fe, 17.49%; C, 37.58%; H, 4.07%; found: Mn, 8.46%; P, 9.58%; Fe, 17.62%; C, 38.56%; H, 3.60%.
4.4 Preparation of Mn(O3PFc)·2 H2O
Ferrocenylphosphonic acid (0.10 g, 0.38 mmol) was dissolved in 28 ml of water. The pH was adjusted to 11 by addition of NaOH 1 M. Mn(NO3)2·4 H2O (0.09 g, 0.36 mmol) was added to the solution without stirring. After 5 days at room temperature, the yellow precipitate was filtered, washed with methanol and ether, and dried under vacuum leading to 86 mg (0.24 mmol) of Mn(O3PFc)·2 H2O (67% yield). Anal. Calc. for MnPFeC10H13O5: Mn, 15.48%; P, 8.73%; Fe, 15.74%; C, 33.88%; H, 3.66%; found: Mn, 15.11%; P, 8.68%; Fe, 15.57%; C, 33.90%; H, 3.83%.