

1 Introduction
Hydrogen-bonded systems have received much attention within the realm of supramolecular chemistry as well as in coordination polymer systems [1]. These directional, moderately strong inter- and intramolecular interactions have been used to engineer structural motifs capable of binding anions, cations and neutral species and are able to strongly support supramolecular architectures in 1-, 2-, and 3-D arrays [1]. Polyamines such as triethylenetetramine have been used extensively in coordination chemistry as multidentate ligands [2], as well as in hydrogen-bonded systems involving anion coordination [1,3].
As part of our study of some lanthanoid coordination chemistry involving multidentate ligands [4] and polyamines, we obtained a variety of by-products of interest with respect to their degree of protonation. These structures all have supramolecular interactions, particularly hydrogen bonding, but also, in those without classical hydrogen bond donors such as O–H and N–H, we observe C–H···I interactions in the solid state. Thus, we now report the structural characterisation of six new quaternary amine complexes, viz [TETA(H+)4][tos]4·4 H2O (1), [TETA(H+)2][NO3]2 (2) (TETA = triethylenetetramine), [Apip(H+)2][NO3]2, (3) and [Apip(H+)4][Cl]4 (4) (Apip = N,N'-bis(2-aminoethyl)piperazine), [dbm][I], (5A) and [dbm][I](C7H8), (5B) (dbm = N,N-dibenzylmorpholinium). In each of these complexes, supramolecular interactions dominate the structural packing.
2 Results and discussion
We have studied the interactions of multidentate ligands in coordination complexes of many metal salts. In particular, the polyethers have shown much structural diversity and second-sphere hydrogen-bonding interactions can dominate crystal packing [5]. It was of interest to us to extend this study to the polyamines, in particular triethylenetetramine (TETA), a potentially quadridentate ligand. Treatment of triethylenetetramine with four equivalents of p-toluenesulfonic acid (tosH) resulted in the deposition of large colourless crystals of [TETA(H+)4][tos]4·4 H2O while a reaction involving Sm(NO3)3·6 H2O and TETA deposited colourless crystals of [TETA(H+)2][NO3]2 (the low pH of the solution arising from hydrolysis involving the rare earth salt). In the latter, the lack of incorporation of Sm3+ in the final product is presumably due to the hard ionic character of the lanthanoid ion having a preference for water as a ligand and this dominates over the chelate effect of the polyamine. It is of interest to discuss compounds (1) and (2) together for a comparison of the structures obtained for the quadruply protonated (1) and its literature counterparts with the doubly protonated compound (2), being a rare example in the literature: a Cambridge Crystallographic Database search found seven hits for doubly-protonated triethylenetetramine [3,6]. In both of these compounds, hydrogen bonding dominates the crystal packing (see Figs. 1 and 2, Tables 1 and 2), and indeed in compound (1) all potential hydrogen bond donors (N–H) in the cations are involved in H-bonding, while in compound (2), 13 out of 16 N–H donors (of the two unique cations) are involved in the hydrogen bonding networks and the other three nitrogen atoms act as hydrogen bond acceptors. Thus, in each compound, all heteroatoms of the polyamine are involved in the hydrogen-bonding scheme. Interestingly, the configuration of the polyamine in compound (1) is significantly different to that in compound (2) (Fig. 3). In compound (1), the [TETA(H+)4] cation is in a linear-type configuration with the N atoms on alternating sides of the chain, whereas in compound (2) the [TETA(H+)2] cation has the nitrogens all displaced on the same side of the chain. The difference between the two therefore lies in the conformation of the –CH2CH2– linker units between the N atoms (see Fig. 3). Presumably this arises due to the different anions, where the nitrate anions of (2), having a small bite, position the hydrogen bond acceptors and donors in such a fashion. Additionally, compound (1) has four water molecules in the lattice, while interestingly, for compound (2), the species isolated was an anhydrous species, which is rather unusual in these hydrogen-bonded hydrophilic complexes, and it is possible that the hydrogen bonding capacity of the water molecules in compound (1) relieve the strain on the polyamine cation to give the less-strained conformation.

Hydrogen-bonding array in the X-ray crystal structure of the [TETA(H+)4][tos]4·4 H2O (1).
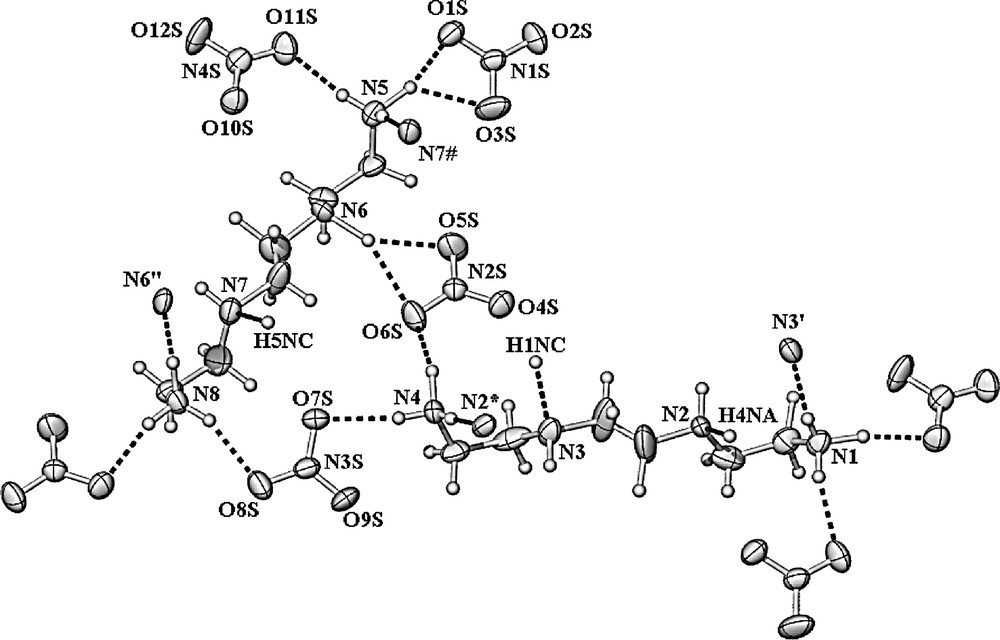
Hydrogen bonding array in the X-ray crystal structure of [TETA(H+)2][NO3]2 (2).
Hydrogen bonds for [TETA(H+)4][tos]4·4 H2O (1), [Å and deg.]
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
| N(1)–H(1NA)···O(13)#1 | 0.89 | 2.04 | 2.778(5) | 140.0 |
| N(1)–H(1NB)···O(3) | 0.89 | 1.88 | 2.744(5) | 164.7 |
| N(1)–H(1NC)···O(1S) | 0.89 | 1.92 | 2.751(5) | 154.5 |
| N(4)–H(4NA)···O(3S) | 0.89 | 2.00 | 2.809(6) | 151.1 |
| N(4)–H(4NB)···O(8)#2 | 0.89 | 2.00 | 2.884(5) | 173.3 |
| N(4)–H(4NC)···O(9) | 0.89 | 2.26 | 2.741(5) | 114.1 |
| N(4)–H(4NC)···O(7)#3 | 0.89 | 2·65 | 3.265(6) | 126.8 |
| N(4)–H(4NC)···O(8)#3 | 0.89 | 2·66 | 3.289(5) | 128.8 |
| O(1S)–H(1SA)···O(3)#3 | 0.991(10) | 2.29(5) | 2.958(8) | 124(5) |
| O(1S)–H(1SB)···O(4)#3 | 0.998(10) | 1.84(2) | 2.813(5) | 165(6) |
| N(2)–H(2NA)···O(5)#3 | 1.02(5) | 1.88(5) | 2.747(5) | 141(4) |
| N(2)–H(2NB)···O(4S) | 0.81(4) | 2.06(5) | 2.745(6) | 142(4) |
| O(2S)–H(2SA)···O(7) | 0.88(5) | 2.06(5) | 2.821(6) | 144(5) |
| O(2S)–H(2SB)···O(6) | 0.72(5) | 2.14(5) | 2.805(6) | 154(5) |
| N(3)–H(3NA)···O(11) | 0.90(4) | 1.98(4) | 2.741(5) | 141(3) |
| N(3)–H(3NB)···O(2S)#3 | 0.90(5) | 1.94(5) | 2.759(5) | 149(4) |
| O(3S)–H(3SA)···O(12) | 1.002(7) | 2.04(7) | 2.836(5) | 135(8) |
| O(3S)–H(3SB)···O(8)#4 | 0.83(5) | 2·42(8) | 2.910(6) | 119(8) |
| O(4S)–H(4SA)···O(10) | 0·61(4) | 2.22(4) | 2.804(6) | 162(5) |
| O(4S)–H(4SB)···O(1)#1 | 0.86(7) | 2.10(7) | 2.879(7) | 151(6) |
Hydrogen bonds for [TETA(H+)2][NO3]2, (2) [Å and deg.]
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
| N(1)–H(1NA)···O(10S)#1 | 1.14(6) | 1.82(6) | 2.939(6) | 165(4) |
| N(1)–H(1NA)···O(12S)#1 | 1.14(6) | 2.30(6) | 3.102(7) | 125(4) |
| N(1)–H(1NA)···N(4S)#1 | 1.14(6) | 2.39(6) | 3·453(8) | 154(4) |
| N(4)–H(4NA)···N(2)#2 | 0·67(4) | 2.32(4) | 2.991(8) | 173(5) |
| N(5)–H(5NA)···O(1S) | 1.03(6) | 1.99(6) | 2.957(7) | 156(4) |
| N(5)–H(5NA)···O(3S) | 1.03(6) | 2.27(6) | 3.098(7) | 136(4) |
| N(5)–H(5NA)···N(1S) | 1.03(6) | 2·46(6) | 3·457(8) | 161(4) |
| N(6)–H(6NA)···O(5S) | 1.18(9) | 2.28(9) | 3.308(6) | 145(6) |
| N(6)–H(6NA)···O(6S) | 1.18(9) | 2·41(8) | 3.502(6) | 153(6) |
| N(6)–H(6NA)···N(2S) | 1.18(9) | 2·66(9) | 3.814(7) | 168(6) |
| N(8)–H(8NA)···O(8S) | 0.85(4) | 2.08(4) | 2.905(7) | 164(4) |
| N(8)–H(8NA)···O(7S) | 0.85(4) | 2.71(5) | 3·432(7) | 144(4) |
| N(8)–H(8NA)···N(3S) | 0.85(4) | 2.75(4) | 3.587(7) | 170(4) |
| N(4)–H(4NB)···O(6S) | 1.12(6) | 1.79(6) | 2.870(7) | 159(5) |
| N(4)–H(4NB)···N(2S) | 1.12(6) | 2·49(6) | 3.572(8) | 161(4) |
| N(4)–H(4NB)···O(4S) | 1.12(6) | 2.54(6) | 3·430(7) | 136(4) |
| N(5)–H(5NB)···O(11S) | 0.89(4) | 2.03(4) | 2.882(7) | 159(4) |
| N(8)–H(8NB)···O(4S)#3 | 1.02(6) | 1.97(6) | 2.940(6) | 158(4) |
| N(8)–H(8NB)···O(5S)#3 | 1.02(6) | 2.25(5) | 3.113(7) | 142(4) |
| N(8)–H(8NB)···N(2S)#3 | 1.02(6) | 2·45(6) | 3·463(8) | 171(4) |
| N(4)–H(4NC)···O(7S) | 1.12(6) | 1.83(7) | 2.923(7) | 164(4) |
| N(4)–H(4NC)···O(9S) | 1.12(6) | 2.26(6) | 3.128(7) | 133(4) |
| N(4)–H(4NC)···N(3S) | 1.12(6) | 2.37(7) | 3·456(8) | 163(4) |
| N(5)–H(5NC)···N(7)#4 | 0.89(6) | 2.06(6) | 2.951(8) | 177(5) |
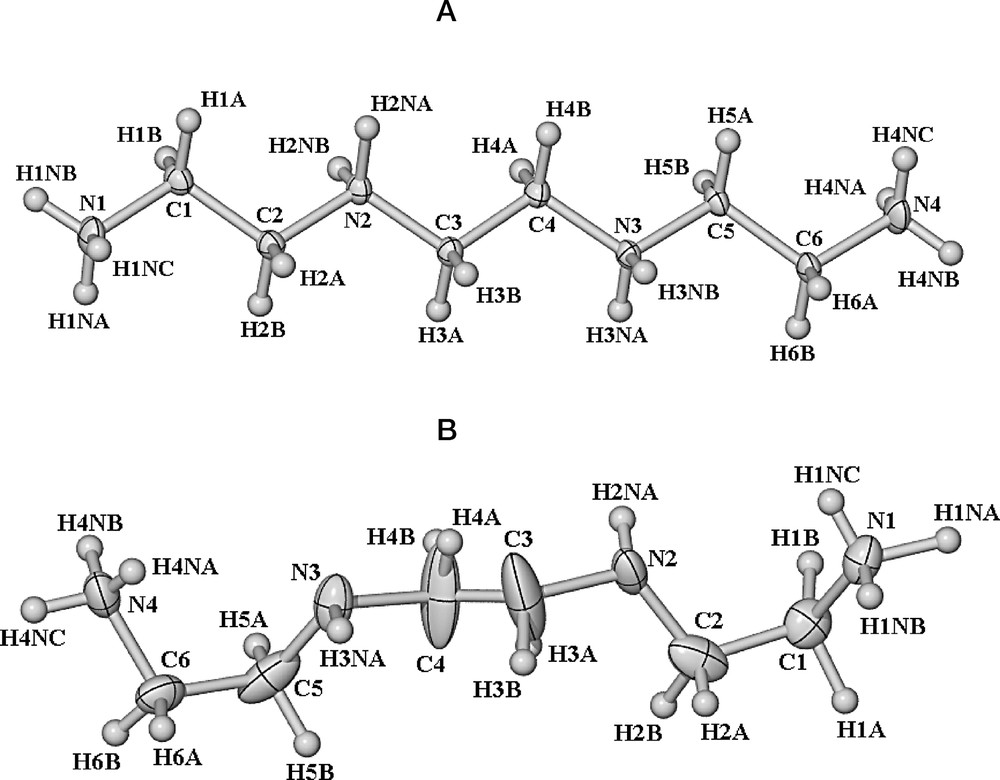
The two conformations of the (a) tetraprotonated TETA ligand in (1) and (b) the doubly protonated TETA ligand in (2).
Treatment of La(NO3)3·6 H2O or LaCl3·7 H2O with triethylenetetramine in aqueous media deposited crystals of compound [Apip(H+)2][NO3]2, (3) and [Apip(H+)4][Cl]4 (4) (Apip = N,N'-bis(2-aminoethyl)piperazine) respectively after slow evaporation. Several points of interest arise from this chemistry. Firstly, the lack of incorporation of the lanthanoid metal is apparent. Presumably under the aqueous conditions employed, water is a superior ligand for the hard lanthanum ion, and thus inhibits binding of the polyamine. Secondly, the polyamine ligand in both products is not the straight chain triethylenetetramine used in the synthetic procedure but rather the cyclic amine N,N'-bis(2-aminoethyl)piperazine (I) (Scheme 1). It has been previously noted in the literature [7,8] that similar ring-closed species have been obtained in reactions involving triethylenetetramine and this was accounted for by a ca 13% contamination of the commercially available polyamine by N,N′-bis(2-aminoethyl)piperazine [7,8]. The X-ray crystal structures of compounds (3) and (4) are dominated by hydrogen bonding (Figs. 4 and 5, Tables 3 and 4) and all hydrogen-bond donors on the cations are involved in the three-dimensional network solids. In compound (3), N,N′-bis(2-aminoethyl)piperazine is protonated at only the terminal primary amines, while the tertiary bridgehead nitrogen atoms are not protonated generating a 2+ charge on the cation. By contrast, in compound (4), all four nitrogen atoms of the amine are protonated, generating a 4+ overall charge. The overall structure in compound (3) is a three-dimensional motif with a two-dimensional framework obtained by direct hydrogen bonding between the doubly protonated N,N′-bis(2-aminoethyl)piperazine ligands and the nitrate counterions. The NO3– anions then link the sheets together to form the three-dimensional structure. On the other hand, in compound (4) there is a true three-dimensional network rather than sheets held together by anions in the hydrogen-bonded array. A further point to note is the anhydrous nature of both complexes.
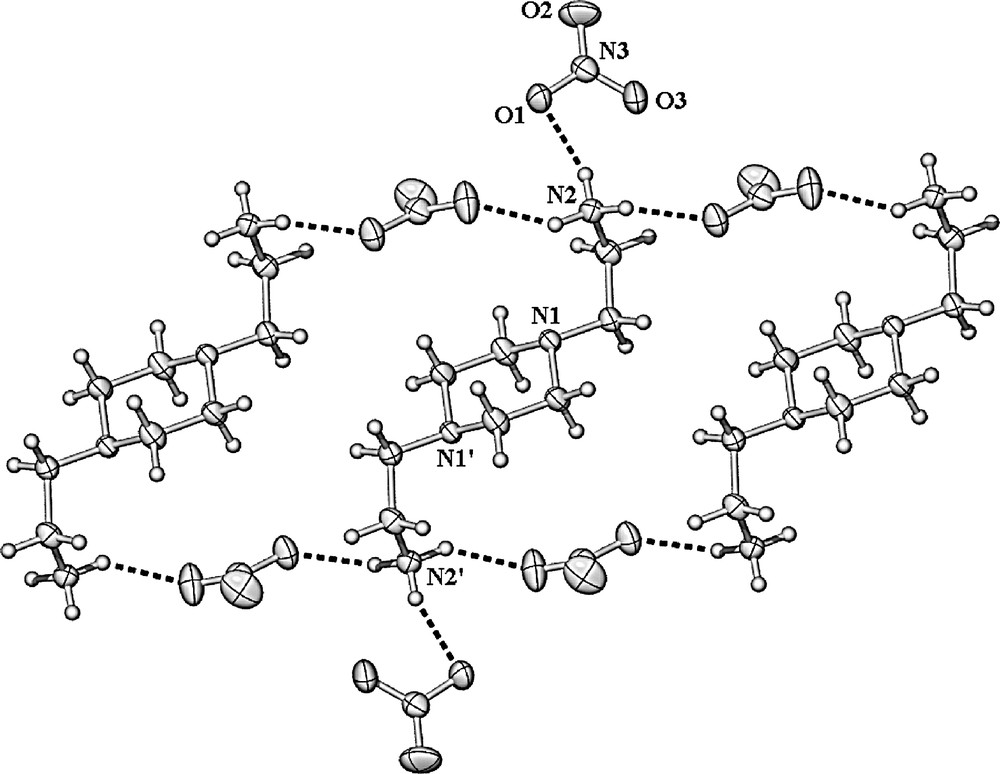
X-ray crystal structure of [Apip(H+)2][NO3]2 (3), showing the hydrogen-bonding pattern.
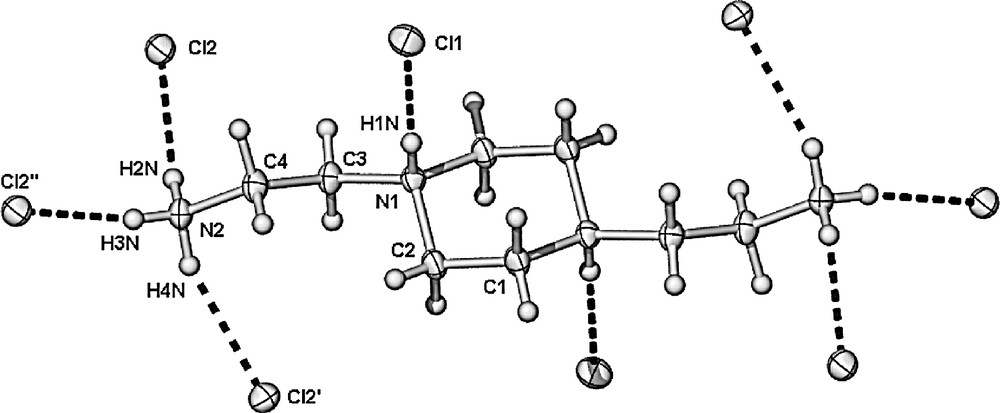
X-ray crystal structure of [Apip(H+)4][Cl]4 (4) showing the interactions between the protonated amines and chlorides ions.
Hydrogen bonds for [Apip(H+)2][NO3]2, (3) [Å and deg.]
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
| N(2)–H(2NA)···O(3)#1 | 0.95(3) | 2.05(3) | 2.889(3) | 147(3) |
| N(2)–H(2NB)···O(1) | 0.93(3) | 2.01(4) | 2.884(3) | 155(3) |
| N(2)–H(2NC)···O(1)#2 | 0.84(3) | 2.11(3) | 2.913(3) | 160(3) |
| N(2)–H(2NC)···O(3)#2 | 0.84(3) | 2·45(3) | 3.132(3) | 139(3) |
| N(2)–H(2NC)···N(3)#2 | 0.84(3) | 2·65(4) | 3·467(4) | 166(3) |
Hydrogen bonds for [Apip(H+)4][Cl]4, (4) [Å and deg.]
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
| N(2)–H(2N)···Cl(2)#1 | 0.88(3) | 2.30(3) | 3.131(3) | 156(2) |
| N(2)–H(3N)···Cl(2)#2 | 0.87(3) | 2.28(3) | 3.141(2) | 169(2) |
| N(2)–H(4N)···Cl(2) | 0.88(3) | 2.39(3) | 3.188(3) | 150(3) |
| N(1)–H(1N)···Cl(1) | 0.84(3) | 2.16(3) | 3.004(2) | 175(2) |
Compounds (5A) and (5B) were isolated from the reaction of diiodo ethylene glycol with benzyl amine after heating to reflux in acetonitrile in the presence of Na2(CO3). Removal of the acetonitrile and extraction with hexane gave [dbm][I], (5A) as large colourless crystals and in a likewise manner, [dbm][I](C7H8), (5B) (dbm = N,N-dibenzylmorpholinium) was isolated from toluene. X-ray crystal structures of both compounds revealed similar structures involving a ring closed heterocyclic tertiary amine with two benzylic groups bound to the amine. The counterions in both cases being iodide. The only difference in the structures of the two is there is a disordered molecule of toluene in the lattice of (5B). This does however, give rise to overall structural changes involving the interactions of the cations and anions. There are significant C–H···I interactions in both structures which are based on a maximum C–H···I distance of 3.35 Å, this distance being selected as the cut-off on the basis of ‘significant’ interactions being observed for N–H···I distances of 3.35 Å.3 Thus, in a system where the less polarised C–H bond has an H···I distance less than that in amine systems,3 these interactions should also be classified as ‘significant’. In compound (5A) I1 is hydrogen bound by two C–H(alkyl) groups and I2 is bound by five C–H(alkyl) and one C–H(aromatic) interactions whereas in compound (5B) I1 is hydrogen bound to three C–H(alkyl) one C–H(aromatic) and one C–H(toluene) (not shown in the figure) groups (see Fig. 6a and b and figure captions). The mechanism of formation of the cyclic system in compounds (5A) and (5B) presumably involves initial monosubstitution of the diiodo ethylene glycol by benzyl amine and, rather than the intended disubstitution, an intramolecular cyclisation takes place affording N-benzylmorpholine. The N-benzylmorpholine can then displace a morpholine group from another N-benzylmorpholine molecule giving the N,N-dibenzylmorpholinium quaternary amine (II) (Scheme 1).
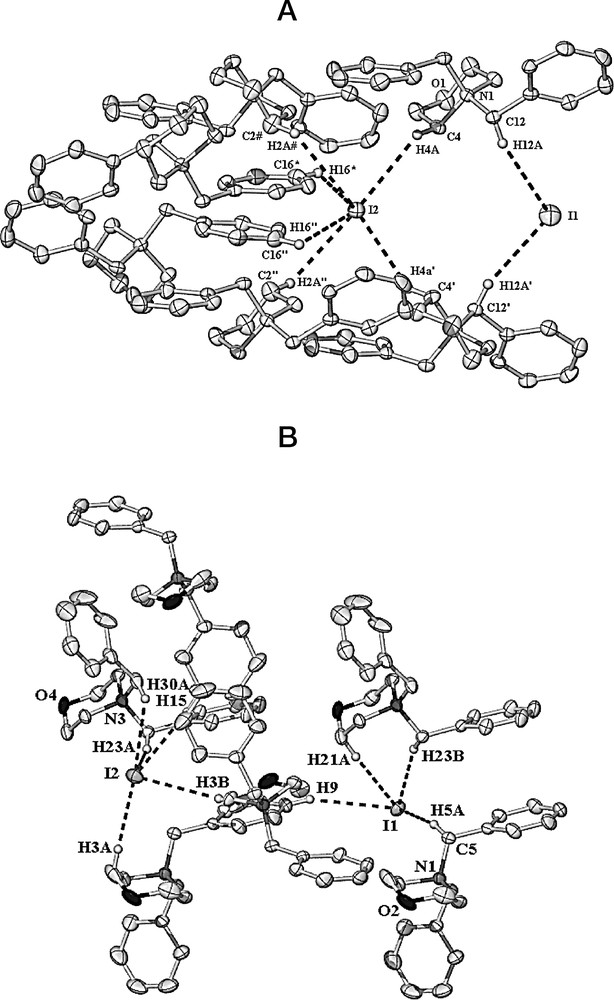
(a) X-ray crystal structure of [dbm][I] (5A), showing the C–H···I interactions (hydrogen atoms, except those involved in hydrogen bonding, have been omitted for clarity); selected distances: I1···H12A 3.334(5), I1···H12A' 3.334(5), I2···H4A 3.177(5), I2···H16” 3.302(5), I2···H2A” 3.188(5), I2···H2A# 3.188(5), I2···H16''' 3.302(5) Å. (b) X-ray crystal structure of [dbm][I](C7H8), (5B), showing the C–H···I interactions (the disordered toluene molecule of solvation and hydrogen atoms not involved in hydrogen bonding, have been omitted for clarity); selected distances: I1···H5A 3.074(8), I1···H21A 3.117(8), I1···H23B 3.076(8), I1···H9 3.260(8), I2···H3A 3.104(8), I2···H3B 3.280(8), I2···H23A 3.112(8), I2···H15 3.197(8), I2···H30A 3.246(8) Å. Masquer
(a) X-ray crystal structure of [dbm][I] (5A), showing the C–H···I interactions (hydrogen atoms, except those involved in hydrogen bonding, have been omitted for clarity); selected distances: I1···H12A 3.334(5), I1···H12A' 3.334(5), I2···H4A 3.177(5), I2···H16” 3.302(5), I2···H2A” 3.188(5), I2···H2A# 3.188(5), I2···H16''' 3.302(5) Å. ... Lire la suite
3 Conclusions
We have shown that protonated amines can coordinate anions through hydrogen bonding. Of particular interest is the degree of protonation of TETA can affect the configuration of the polyamine. In protonated N,N′-bis(2-aminoethyl)piperazine, the degree of protonation, and choice of anion can dictate the dimensionality of the overall structure. Furthermore, in systems where there is no water present in the lattice, significant C–H···I interactions prevail.
4 Experimental
All reagents were purchased from Aldrich Chemical Company and were used as received. Deionised water was used as solvent unless otherwise stated. In all syntheses, there were no attempts to optimise yields, rather, the pursuit of X-ray quality crystals was the overriding factor. However, in all cases, yields were moderate, ca 20–50%.
4.1 Synthesis
4.1.1 Compound (1)
Treatment of p-toluenesulfonic acid monohydrate (1.0 g, 5.25 mmol) with triethylenetetramine (0.19 g, 1.31 mmol) in deionised water (ca 20 cm3) resulted in a colourless solution that deposited crystals of compound (1) after evaporation.
4.1.2 Compound (2)
Treatment of Sm(NO3)3·6 H2O (1.0 g, 2.25 mmol) with triethylenetetramine (0.66g, 4.5 mmol) in deionised water (ca 20 cm3) resulted in a colourless solution which deposited crystals of compound (2) after evaporation.
4.1.3 Compound (3)
Treatment of La(NO3)3·6 H2O (1.0 g, 2.30 mmol) with triethylenetetramine (0.67 g, 4.60 mmol) in deionised water (ca 20 cm3) resulted in a colourless solution that deposited crystals of compound (3) after evaporation.
4.1.4 Compound (4)
Treatment of LaCl3·7 H2O (1.0 g, 2.69 mmol) with triethylenetetramine (0.79 g, 5.38 mmol) in deionised water (ca 20 cm3) resulted in a colourless solution that deposited crystals of compound (4) after evaporation.
4.1.5 Compounds (5A) and (5B)
Compounds (5A) and (5B) were isolated from the reaction of diiodo ethylene glycol (35 g, 0.11 mol) with benzyl amine (8.5 g, 0.08) and heating to reflux in acetonitrile (1 l) in the presence of Na2(CO3) (35 g, 0.33 mol). Removal of the acetonitrile and extraction with hexane gave (5A) as large colourless crystals. Alternatively, extraction with toluene gave colourless crystals of (5B).
4.2 X-ray crystallography
All X-ray-quality crystals were sealed and mounted in thin walled capillaries, with hemispheres of data collected at room temperature on a Bruker SMART CCD diffractometer using the omega scan mode with total reflections and unique data listed below. Datasets were corrected for absorption using the program SADABS [9]. Structures were solved using direct methods and refined on F2 using SHELXL97-2 [10] with X-SEED as the graphic interface [11]. All non-hydrogen atoms were located and were refined with anisotropic thermal parameters. For compound (1), hydrogen atoms were located on the nitrogen atoms and water molecules and were refined with isotropic thermal parameters, while all other hydrogen atoms were placed in calculated positions (riding model) and were not refined. In compounds (2), (3) and (4) all hydrogen atoms were located and refined isotropically. In compounds (5A) and (5B) all hydrogen atoms were placed in calculated positions (riding model) and were not refined. Details of the X-ray data collection and refinements appear below and hydrogen bond lengths appear in the tables and text and captions.
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 238869 for compound (1), CCDC 238870 for compound (2), CCDC 238871 for compound (3), CCDC 238872 for compound (4), CCDC 238873 for compound (5A) and CCDC 238874 for compound (5B). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223 336-033; email: deposit@ccdc.cam.ac.uk.
5 Crystal refinement data
5.1 Crystal data for compound (1)
C34H58N4O16S4, M = 907.08, 0.40 × 0.30 × 0.10 mm, triclinic, space group
5.2 Crystal data for compound (2)
C12H40N12O12, M = 544.56, 0.30 × 0.30 × 0.20 mm, monoclinic, space group P21/c (No. 14), a = 15.5974(18), b = 11.1993(14), c = 16.408(2) Å, β = 114.593(2)°, V = 2606.1(5) Å3, Z = 4, Dc = 1.388 g cm–3, F000 = 1168, Mo Kα radiation, λ = 0.71073 Å, T = 296 (2)K, 11 795 reflections collected, 3738 unique (Rint = 0.0974). Final GooF = 0.904, R1 = 0.0670, wR2 = 0.1570, R indices based on 1447 reflections with I > 2 σ(I) (refinement on F2), 486 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.121 mm–1.
5.3 Crystal data for compound (3)
C8H22N6O6, M = 298.32, 0.30 × 0.25 × 0.25 mm, monoclinic, space group P21/n (No. 14), a = 7.081(3), b = 13.647(6), c = 7.336(3) Å, β = 90.511(8)°, V = 708.9(5) Å3, Z = 2, Dc = 1.398 g cm–3, F000 = 320, Mo Kα radiation, λ = 0.71073 Å, T = 296(2) K, 2θmax = 46.5°, 3149 reflections collected, 1012 unique (Rint = 0.1065). Final GooF = 0.971, R1 = 0.0423, wR2 = 0.0955, R indices based on 723 reflections with I > 2 σ(I) (refinement on F2), 135 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.118 mm–1.
5.4 Crystal data for compound (4)
C8H24Cl4N4, M = 318.11, 0.20 × 0.20 × 0.15 mm, monoclinic, space group P21/c (No. 14), a = 8.3443(11), b = 8.6913(11), c = 10.5588(13) Å, β = 91.436(2)°, V = 765.51(17) Å3, Z = 2, Dc = 1.380 g cm–3, F000 = 336, Mo Kα radiation, λ = 0.71073 Å, T = 296(2) K, 3459 reflections collected, 1107 unique (Rint = 0.0449). Final GooF = 1.022, R1 = 0.0350, wR2 = 0.0803, R indices based on 926 reflections with I > 2 σ(I) (refinement on F2), 121 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.757 mm–1.
5.5 Crystal data for Compound (5A)
C36H44I2N2O2, M = 790.53, 0.30 × 0.30 × 0.20 mm, orthorhombic, space group Fdd2 (No. 43), a = 15.3347(11), b = 33.714(3), c = 12.9410(10) Å, V = 6690.3(9) Å3, Z = 8, Dc = 1.570 g cm–3, F000 = 3168, Mo Kα radiation, λ = 0.71073 Å, T = 296(2) K, 2θmax = 46.5°, 7475 reflections collected, 2376 unique (Rint = 0.0322). Final GooF = 0.991, R1 = 0.0254, wR2 = 0.0623, R indices based on 2148 reflections with I > 2 s(I) (refinement on F2), 192 parameters, 1 restraint. Lp and absorption corrections applied, μ = 1.914 mm–1. Absolute structure parameter = 0.18(3) [12].
5.6 Crystal data for Compound (5B)
C42.50H51I2N2O2, M = 875.65, 0.32 × 0.30 × 0.25 mm, monoclinic, space group P21/n (No. 14), a = 19.4096(18), b = 10.4350(10), c = 20.555(2) Å, β = 102.779(2)°, V = 4060.1(7) Å3, Z = 4, Dc = 1.433 g cm–3, F000 = 1768, Mo Kα radiation, λ = 0.71073 Å, T = 296(2)K, 18261 reflections collected, 5828 unique (Rint = 0.0550). Final GooF = 1.036, R1 = 0.0385, wR2 = 0.0884, R indices based on 4244 reflections with I > 2 σ(I) (refinement on F2), 497 parameters, 0 restraints. Lp and absorption corrections applied, μ = 1.585 mm–1.
Acknowledgements
We are grateful to the Australian Research Council for funding (RIEFP Grant) for the CCD X-ray diffractometer.