

1 Introduction
Although the mineralizing role of fluorine in hydrothermal synthesis was known by the ancient mineralogists and chemists [1], the first use of fluorine for the synthesis of microporous materials was only reported at the end of the seventies with the formation of silicalite-1 in slightly alkaline media by Flanigen and Patton [2]. From 1986 with the pioneering work by Guth, Kessler and Wey in Mulhouse, the fluoride route was then extensively developed by our group, first applied to silica-based zeolites, then to metallo- (alumino- and gallo-) phosphates microporous materials. Several other groups, in particular Corma's group (silica-based materials) in Valencia, Spain and Ferey's group (phosphate-based materials) in Versailles, France, also investigated the fluoride route. In this paper a special emphasis is made on the work performed by our group, with two successive parts corresponding to both kinds of materials just mentioned before. As it will be described, besides its mineralizing role, the fluoride ion is also able to play a templating role and thus allowed the discovery of a number of new porous materials.
2 Silica-based microporous materials
For the synthesis of silica-based zeolites, which are normally prepared in alkaline media, the replacement of the hydroxyde anions by fluoride anions as mineralizers makes it possible to obtain zeolites even in almost neutral media (pH 5–9). At such pH values the solubility of silica for example increases significantly in the presence of fluoride because of the formation of hexafluorocomplexes which were observed in particular in the mother liquor of fluoride silicalite-1 by 19F and 29Si NMR [3,4]. No other silicon-containing species could be observed but it can be assumed that the hydrolysis of fluorosilicate anions yields polycondensable hydroxylated species whose condensation leads to the crystalline material. In the synthesis of borosilicalite-1, the presence of the hydroxyfluoroborate anions (BF3OH)– and (BF2(OH)2)– besides BF4– and SiF62– was indeed evidenced by 19F NMR [3]. One important advantage of the fluoride route is to lead to materials with a much lower density of defects due to the charge balance role of the fluoride anions against the positive charge of the organic cations. Fluoride anions can also play a structure-directing role. Indeed, many discovered zeolites during the last decade contain double four membered ring (D4R) units, each of them occluding one fluoride.
Practically all the syntheses employing the fluoride route have been carried out in aqueous medium. Some exceptions correspond to the synthesis of large well-defined single crystals of all-silica materials from systems including pyridine, HF, reagent quantities of water, and, optionally, an organic template [5] or the crystallization of silicalite-1 by heating a dry powder of silica and NH4F (‘dry gel conversion method’), thus producing NH3 and water vapor [6].
Guth and coworkers developed in 1986 the use of F– ions as mineralizers with the synthesis of the MFI-type zeolite. Since then, the use of the fluoride route allowed the preparation of a large number of zeolites, some of them displaying a new framework topology. In what follows the main obtained results are reported in 3 sections. The first one is devoted to variously substituted MFI- and MEL-type zeolites (T = Al, Ga, B, Ti…), whereas the second section concerns the synthesis of silica-based zeolites with other framework topologies. The third section is specifically devoted to the recently discovered silicogermanate zeolites with new framework topologies.
2.1 Synthesis and characterization of MFI- and MEL-type zeolites
MFI-type zeolite with a large variety of framework composition [7–13] could be obtained in fluoride medium with PrxNH4–x+ templates (Pr = propyl, x = 1–4) according to the procedure developed in our laboratory [14]. The tetrapropylammonium cation seems to be the best template for the MFI-structure, other structure-types being obtained in some cases with the tri-, di- or mono-propylammonium ions. The efficiency of the Pr4NF species compared to the Pr3NHF and Pr2NH2F species was confirmed by the determination of the standard enthalpies of formation of the as-synthesized samples [15]. The use of other organic species such as ethylenediamine, diethylamine … was also reported for the synthesis of MFI-type zeolite in fluoride medium [16].
A remarkable and general characteristic of this fluoride route is the formation of large crystals. This is probably due to a smaller nucleation rate, since supersaturation is presumably smaller in fluoride medium than in alkaline medium. The largest crystals (800 μm in length) are obtained for pure silica MFI samples, Si substitution by Al or B leading to a decrease of the crystal size and the length/width ratio [14].
The unit cell formula of pure silica MFI samples is |4Pr4NF| [Si96O192] [17]. The symmetry of the as-synthesized samples is orthorhombic (space group Pnma) [18], whereas that of the calcined sample is monoclinic (space group P21/n) [19]. The question of the localization of the F– ions was a matter of debate. Whereas Price et al. [20] found from single-crystal X-ray diffraction data that the fluoride anions were located close to the Pr4N+ species (C–F distance of 2.11(9) Å) at the intersection of the 10-ring channels of the framework, Mentzen et al. [21] located, by Rietveld refinement, the F– ion far away from the Pr4N+ ion between the 2 five-membered rings of a [415262] cage near the intersection of the channels, with a Si–F distance of 2.20(4) Å. Fyfe et al. [22] determined the Si–F distances by multinuclear NMR spectroscopy and found for the F– ion a site similar to that proposed by Mentzen et al. [21]. Very recently Aubert et al. [23] confirmed from single-crystal X-ray diffraction the location of F– ion in a [415262] cage on a general position at an interacting distance from a silicon framework atom (Si–F = 1.915(3) Å), which is thus pentacoordinated by 4 oxygen framework atoms and one fluorine atom. The existence of SiO4/2F– units was also reported for a number of other as-made pure silica zeolites, as will be discussed in Section 2.2. Note also that, according to the latter study, the disordered Pr4N+ cations are qualitatively in the same locations than those found by Koningsfeld et al. [24]. Another characteristic of the as-made silica MFI-type samples prepared in fluoride media is their very low density or absence of structure defects, as shown first by Chézeau et al. by 29Si NMR spectroscopy [25]. This absence of structure defects was decisive in the study by Fyfe et al. [22] reported before about the location of the F– ion in the MFI-type zeolite. Finally, it should be emphasized here that, generally speaking, a high degree of perfection of the network is observed for high silica zeolites prepared in fluoride media [26,27]. The main reason for this effect is probably the occlusion of F–, which affords charge balance of the organic cation without recourse to Si–O– defects, and also avoids the stabilization of additional Si–OH defects as the extensive formation of strong Si–O–---HO–Si bonds is now prevented.
Due to this absence of connectivity defects, the corresponding calcined zeolite samples, especially of the MFI-type, are completely hydrophobic and found a new application, developed in our laboratory [28], in the field of energetics as ‘molecular springs or molecular bumpers’. This new application field is of course not limited to the MFI-type zeolite, and is therefore presented at the end of Section 2.2.
MFI-type zeolite samples, where Si is partly substituted by TIII elements [9–11,13] (T = B, Al, Fe, Ga) or TIV elements (T = Ge [12], Ti [11,29]) were also prepared according to this fluoride route. The Si/TIII ratio in the zeolite varies with the synthesis conditions and is always larger than 10. Nevertheless, for a ratio lower than 20, some extraframework TIII species are often present in the final material. This leads in the case of aluminum-rich H-MFI (Si/Al ≤ 20) zeolite samples prepared in fluoride medium to a much fewer number of acid sites in comparison with their OH counterparts [30]. Thanks to the large size of the obtained crystals, a study of the distribution of the TIII elements was performed by X-ray emission mapping. This study showed that the most homogeneous intra-and intercrystalline compositions are those with a Si/TIII ratio between 20 and 30 (corresponding nearly to 4 TIII per unit cell, i.e. four negative charges balanced by four Pr4N+ ions). For higher Si/TIII ratios, the homogeneity decreases with a core of the crystal richer in TIII than the outer shell. The concentration gradient varies with the nature of the TIII element and can be strongly reduced by using a combined source of SiIV and TIII elements [31]. (Si, Al) MFI-type zeolite samples were studied by thermal analyses, which allowed to determine whether the template is present in the form of Pr4N+F– ion-pairs or of Pr4N+ compensating cations and finally to derive the value of the SiIV/AlIII molar ratio of the framework [32]. The tetravalent element Ge was successfully substituted in the MFI-type framework [12] up to the very low ratio Si/Ge of 2/3. The incorporation of Ge in the framework was clearly established, for instance by the regular increase of the unit-cell parameters with the germanium content, the fourfold coordination of Ge being (both on as-made and calcined samples) proved by XANES and EXAFS analysis [33]. Titanium-containing MFI-type zeolite could also be synthesized in the presence of F– and Pr4N+ ions, but the substitution degree was low (Si/Ti ≥ 50). According to XANES and EXAFS analysis [29], titanium is incorporated into the framework by substitution for silicon, but a significant amount of Ti can also be present in the form of extracrystalline or intracrystalline inclusions of TiO2, depending on the F/Ti ratio in the synthesis mixture. The EXAFS technique revealed that, in the absence of water, Ti is essentially tetrahedrally coordinated, whereas in the presence of water its coordination becomes octahedral. As MFI-type titanium silicalite-1 (TS-1), first reported by Taramasso et al. [34] in 1983, is catalytically active for liquid-phase oxydation of organic substrates [35], the previous samples prepared in fluoride medium were tested in the hydroxylation reaction of phenol with H2O2 but proved to be rather inactive, due to a low amount of Ti in the framework and the rather large crystal size (about 10 µm). More recently [36,37], an original synthesis method based on the use of a SiO2/TiO2 xerogel (prepared by a sol-gel process) and its impregnation by a minimal volume of an aqueous solution of Pr4NBr and NH4F (‘wet impregnation method’) led to small crystals close to 1 µm, where the titanium is entirely incorporated in the framework (up to 1.8 Ti atoms per unit-cell). The corresponding samples exhibit high activity and selectivity for hydroxylation of phenol with H2O2, the catalytic results being close to the ones obtained with TS-1 samples prepared in alkaline fluoride-free media. The decisive advantage of the fluoride route is the use of cheap Pr4NBr and NH4F reactants instead of the much more expensive TPAOH reactant. The same wet impregnation method was also employed to prepare TS-2 [38], a Ti- substituted MEL-type zeolite in the presence of the very specific 3,5-dimethyl N,N-diethylpiperidinium cation [39]. The activity and the selectivity towards diphenols are comparable to the ones of the best TS-1 samples, an interesting result being the higher paraselectivity with a pyrocatechol/hydroquinone ratio close to 0.4 instead of 0.7 with MFI-type samples.
2.2 Synthesis and characterization of zeolites with other framework topologies
Up to now, a large number of zeolites could be prepared in fluoride media. However, only a few of them correspond to new framework topologies. In Table 1 the framework topologies of some zeolites synthesized in fluoride media are reported, with the corresponding name(s) of the organic template(s) used and the references. All the mentioned topologies in this Table have been previously discovered during syntheses performed in fluoride-free alkaline media.
Materials with already known framework topologies synthesized in fluoride media
| Type of topology | Nature of T atoms | Template used | References |
| FER, TON | Si, Al | CH3 (CH2)2-4NH2, NH2 (CH2)4-6 NH2 | [40,41] |
| MTT | Si, Al | di-n-propylamine, isopropylamine or pyrrolidine | [42] |
| ZSM-48 a | Si, Al | TBA+ + HM2+ + DADD | [43] |
| SOD | Si, Al | Na+ | [44] |
| MOR | Si, Al | Na+ | [45] |
| BEA | Si, Al | DABCO + CH3NH2 | [47] |
| Si; Si, Al; Si, Ti; Si, Ga | TEA+ | [54,56–58] | |
| Si, B | DABCO + CH3NH2 | [50] | |
| MTW | Si, Al | DABCO + (CH3NH2) | [51] |
| Si | M8BQ | [27] | |
| LEV | Si, Al | Q | [52] |
| GIS and MTN | Si, Al | TMA+ | [16] |
| RUT | Si, Al | TMA+ | [53] |
| CJS-2 a | Si | PY | [16] |
| CJS-3 a | Si | TMED | [16] |
| STT | Si | TMAda | [55] |
| STF | Si | DMASD | [60] |
| Si | DMATMBCO | [62] | |
| CFI | Si | MeSPA | [66] |
a No zeolite framework type defined.
FER and TON-type zeolites were prepared in the presence of linear aliphatic mono- or di-amines [40,41], respectively, from gels with low or high Si/Al ratios. MTT-type zeolite was prepared [42] under the same conditions as for the synthesis of TON-type zeolite. The occluded template is always present in its protonated form, its charge balancing the negative charges of the framework and of F– anions present in the channels. From gels containing TBA+, HM2+ and DADD organic species (see Table 1), and Na+ ions, the formation of ZSM-48 and silicalite-2 (MEL) was achieved, the formation of ZSM-48 being favored by high proportions of the diamine DADD; the amount of occluded fluoride is low, with a maximum of 0.4 F– ions per u.c. in the ZSM-48 samples [43]. Fluorosodalite samples (SOD structure-type) were obtained from gels with the following molar composition: 1 SiO2; 1 Al2O3; 1.4 Na2O; 1–5 NaF; 85 H2O, between 150 and 200 °C [44]. The unit-cell formula of the fluorosodalite is |(Na+)8 (F–)2|[Al6Si6O24], 4–6 H2O. Each sodalite cage contains one F– ion and 4 Na+ ions, the chemical shift value of the signal in the 19F MAS NMR spectra depending on the surrounding water molecules number. The signal at –178.7 ppm/CFCl3 in the 19F MAS NMR spectrum of the as-synthesized sample is assigned to fluorine located in the sodalite cage containing 2–3 water molecules. MOR-type zeolite was obtained under similar experimental conditions [45], with the exception of the SiO2/Al2O3 ratio of the starting gel, which is equal to 7. The mordenite samples display a SiO2/Al2O3 ratio close to 7, and do not contain any fluorine, which thus appear to play only a mineralizing role.
Zeolite beta, which is usually prepared from fluoride-free basic media in the presence of Na+ and (C2H5)4N+ cations [46], could be prepared from almost neutral fluoride silica–alumina gels with Si/Al ratio in the range 5–30 in the presence of DABCO [47] (diaza-1,4-bicyclo [2,2,2]octane) and methylamine. The obtained crystals are much larger than those synthesized in a high alkaline medium, with a size in the 5–10 µm range, and display a truncated square bipyramidal morphology. According to TEM and X-ray diffraction results, it is clear that disorder of the type described by Newsam et al. [48], the real structure being an intergrowth of at least two polytypes, is also present within the crystals of zeolite beta prepared in fluoride media. Upon dealumination by steaming, formation of mesopores occurs in a particular way [49]. Indeed the obtained mesopores are cylinders running along directions perpendicular to the c-axis, crossing the crystal and thus providing a direct connection to the exterior of the crystal, whereas, as it is commonly observed, randomly distributed spherical mesopores appear in the case of beta zeolite prepared in hydroxyde media. As suggested earlier, the mesopores probably form preferentially near structure defects, for instance near the interfaces between the domains corresponding to the two polytypes, and the different behavior shown by the zeolite crystals formed in fluoride media could be due to a much larger size of these domains, due to a lower crystallization rate [49]. The Si/Al molar ratio of the obtained samples lies between about 9 and 22, with an amount of organic species close to 25 wt.%. According to 13C MAS NMR spectroscopy, the occluded species is mainly a polyethylene piperazine polymer (partly protonated) resulting from an ‘in situ’ polymerization of the dabconium cation. Finally, the amount of occluded fluorine is low and corresponds to about 2 F– per u.c. (64 T atoms). Note that (Si, B) beta zeolite samples (Si/B molar ratio close to 14) were prepared in similar conditions [50] and also display only small amounts of fluorine, distributed over a large variety of non-characteristic species. Analogous silica–alumina reaction mixtures, with however higher Si/Al molar ratios between about 12 and 100, led to the formation of MTW-type zeolite crystals [51]. As for zeolite beta samples, the occluded organic species is mainly a polyethylene piperazine polymer and the amount of incorporated fluorine is low, corresponding to about 0.4–0.8 F– per u.c. In the latter case, the main fluorine-containing species identified by 19F MAS NMR spectroscopy is the SiF62– ion. LEV-type zeolite samples could be prepared [52] from gels with low Si/Al molar ratios (between about 5 and 10) in the presence of quinuclidine. Once again, F– plays essentially a mineralizing role, with a low amount of occluded fluorine (about 0.3–0.6 F– per u.c.) corresponding mainly to the SiF62– ion. As mentioned before for the syntheses performed in the presence of DABCO, an ‘in situ’ polymerization of quinuclidine occurs, the recovered samples containing beside the main quinuclidinium species some polyethylene piperidine. Zeolite P (GIS-type structure) and ZSM-39 (MTN-type structure) could be prepared [16] in the presence of tetramethylammonium cations from gels with low and high Si/Al molar ratios, respectively. The same cation also allowed the preparation of the Nu-1 zeolite (RUT structure-type) [53] but from more concentrated media. The 19F MAS NMR spectrum of the obtained Nu-1 sample (Si/Al molar ratio close to 15) displays several peaks at –58, –73 and –116.6 ppm, the first two being possibly characteristic of the RUT-type structure. Finally let us also mention the CJS-2 and CJS-3 clathrasils [16] which were synthesized in the presence of pyrrolidine and N,N,N′,N′-tetraethylethylenediamine, respectively.
Whereas in the examples given above the fluoride ion is incorporated in low amounts in the as-synthesized zeolites and seems to play essentially a mineralizing role, F– ion is, in some cases, clearly occluded in specific locations in order to compensate the positive charge of the cationic template as was already shown for the MFI-type zeolite (see Section 2.1.). Pure silica MTW-type [27], BEA-type [54], STT-type [55] zeolite samples could be prepared from gels with low H2O/SiO2 molar ratios (about 10 or less), tetraethoxysilane being used as silica source, in the presence of the bis(quinuclidinium) octane cation, the tetraethylammonium cation and the N,N-trimethyladamantammonium cation, respectively. Zeolite Beta could also be prepared in the (Si, Al) [56] (Ti, Si) [57] or (Si, Ga) [58] forms. The 19F MAS NMR spectra of these as-synthesized purely siliceous samples display well defined signals in the range between about –60 and –80 ppm/CFCl3 [27], which are characteristic of F– ions occluded in zeolite channels [59]. The cationic template/F– molar ratio equal to 1 in the purely siliceous samples decreases as the Si/TIII ratio decreases. The pure silica zeolite samples, as already mentioned for MFI-type zeolite, display no or very scarce connectivity defects according to 29Si MAS NMR spectroscopy and show thus a significant hydrophobic character.
Besides, the 29Si MAS NMR spectra of these pure silica samples [27] display a small component in the –115 to –150 ppm/TMS range, which is indicative of five-coordinate silicon units SiO4/2F–. Among the four zeolites mentioned just before, zeolites ITQ-4 (IFR) and ITQ-3 (ITE) correspond to new framework topologies which can only be obtained in fluoride media and which will be referred to later in the text. Actually the 29Si chemical shifts of five-coordinated silicon units without motion are between –140 and –150 ppm/TMS, whereas, when the fluoride ions exchange between several framework silicon sites, a broad averaged 29Si NMR signal is observed between –115 and –150 ppm. All signals in the –115 to –150 ppm/TMS range are logically somewhat enhanced on the 29Si{19F}CP MAS NMR spectra. The presence of pentacoordinated silicon atoms was also evidenced in the as-made fluorinated all-silica zeolite Mu-26 with the STF topology [60], prepared in the presence of a new organic template, the cis-6,10-dimethyl-5-azonia-spiro [4,5]decane. Previously the purely siliceous materials SSZ-35 [61] and ITQ-9 [62] with the same STF topology were also synthesized in alkaline or fluoride media, respectively, both as-made products displaying a monoclinic symmetry with eight non-equivalent-crystallographic silicon sites ; after calcination, the symmetry becomes triclinic with a unit cell volume twice smaller. The symmetry of the as-made Mu-26 sample proved to be triclinic with unit cell parameter values close to the ones of the calcinated Mu-26 and SSZ-35 materials [63]. The 29Si MAS NMR spectrum of the as-made Mu-26 shown in Fig. 1 displays, beside 12 components corresponding to (Si(OSi)4) sites with an area ratio of 1:1:3:1:1:1:1:1:1:2:1:1, two signals at –148 and –145.8 ppm relative to pentacoordinated silicon atoms [64]. The total number of non-equivalent crystallographic silicon sites in the as-made Mu-26 sample is of 16, instead of 8 in the as-made SSZ-35 and ITQ-9 materials. Finally let us note that all-silica zeolite CIT-5 (CFI-type topology) originally synthesized in alkaline medium [65], was prepared afterwards in fluoride medium [66] in the presence of the same template (N-methyl-sparteinium). As mentioned already before for other purely silicic zeolites, the sample obtained in fluoride medium present almost no connectivity defects, in contrast to that synthesized in the OH– medium [66].
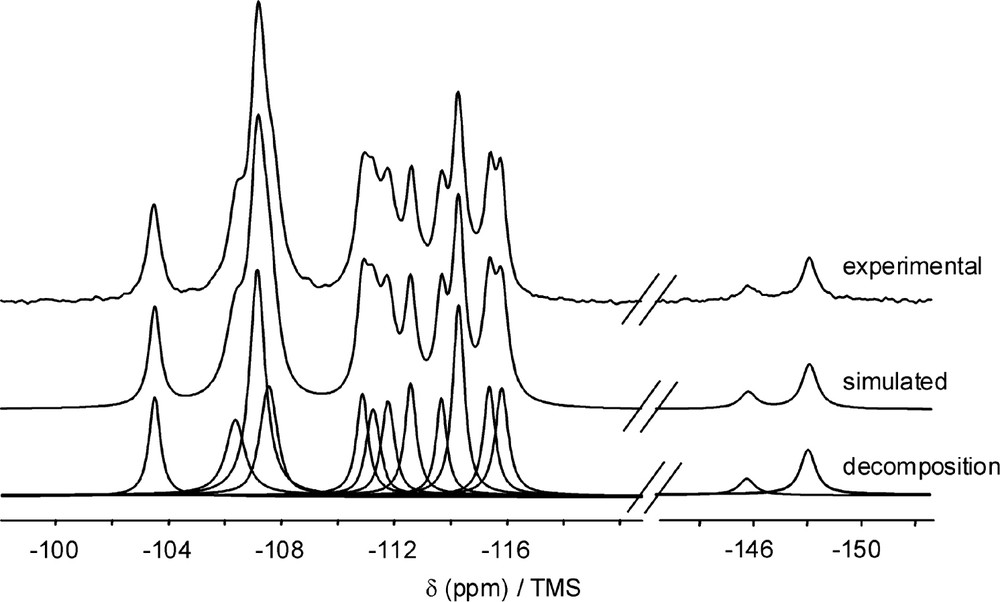
29Si MAS NMR spectra of Mu-26, (top) experimental, (middle) simulated, (bottom) decomposition. Only the spectral region of isotropic resonances is shown. The proton decoupled MAS NMR spectra were recorded on a Bruker DSX 400 spectrometer with a spinning frequency of 4 kHz and a π/2 pulse duration of 5.4 µs. A recycle delay of 60 s was used. The 29Si chemical shifts are referenced to TMS (adapted from [60]).
Up to now, the reported materials displayed topologies previously obtained during syntheses in fluoride-free alkaline media. However, a number of all-silica materials which present new framework topologies [67–73] were also obtained from fluorine-containing media. These topologies (or possibly these materials, when no structural code is defined) are indicated in Table 2, with the corresponding name(s) of the organic template(s) used and the references.
Purely siliceous zeolites with new framework topologies obtained via the fluoride route
| Framework code (material) | Template used | References |
| AST | Q | [67] |
| ITE (ITQ-3) | PMABCO | [68] |
| IFR (ITQ-4) | BQ | [69] |
| ISV (ITQ-7) | TMATCDD | [70] |
| ITW (ITQ-12) | TMI | [71] |
| ITH (ITQ-13) | HM | [72] |
| PREFER | ATMP | [73,74] |
Whereas, as already mentioned before, linear aliphatic amines were successfully used in synthesizing FER-type materials [40–42], the latter materials could also be prepared in fluoride media in the presence of pyridine [5] or other variously substituted heterocyclic amines [73,74], for instance dimethyl-substituted piperidines. Surprisingly, the use of a still bulkier organic template, the 4-amino-2,2,6,6-tetramethylpiperidine, led to the crystallization of a new material called PREFER (for ‘precursor of ferrierite’) [73]. This material crystallized only from fluorine-containing gels characterized by Si/Al molar ratios between 5 and ∞. The interesting point is that the calcination at about 500 °C of the as-made PREFER material leads to FER-type zeolite through the intermediate formation of poorly organized phases (Fig. 2). The powder XRD data and 29Si NMR spectra (not shown here) are consistent with a 2D → 3D transformation, the framework of PREFER material being made of non-interconnected (100) ‘ferrierite layers’, which link progressively during the calcination. The ferrierite layers bear Q3 (Si–O–) groups (8 per unit cell, with 36 T atoms), the charges of which being compensated by the positive charges of protonated (probably doubly) amine (4 per unit cell), which are located between the layers (Fig. 3). Whatever the value of the Si/Al molar ratio of the gel (5 to ∞), the PREFER samples are essentially siliceous with Si/Al molar ratios larger than 22. Solid-state 27Al and 19F MAS NMR experiments show that aluminum is mainly present as (AlFx) extra-framework species. Let us note finally that, according to a more general process also applied to other ‘lamellar zeolites’, such as the precursors of MCM-22 [76] (MWW), Nu-6 [77] and MCM-47 [78], the preparation of a delaminated material, called ITQ-6 [79] was later performed on (Al, Si) and (Ti, Si) PREFER samples.
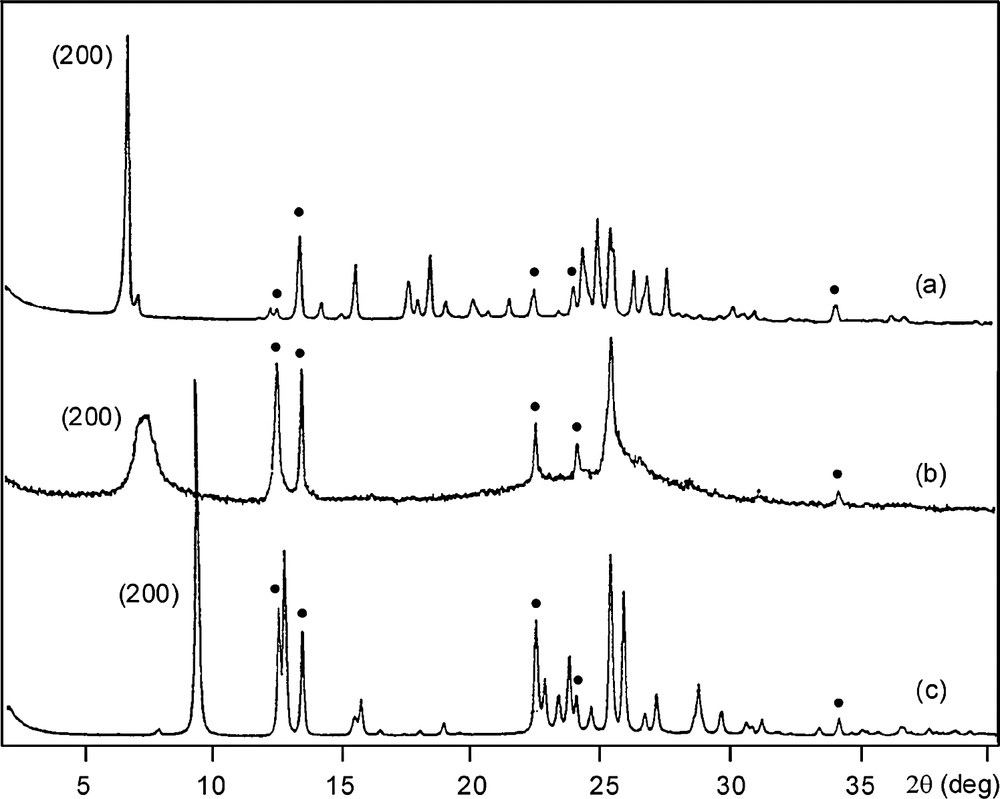
XRD patterns of a purely siliceous PREFER sample : (a) as-made, (b) calcined at 330 °C and (c) calcined at 550 °C (adapted from Ref. [74]). The peaks marked (•) correspond to the same (0kl) reflexions for both products.
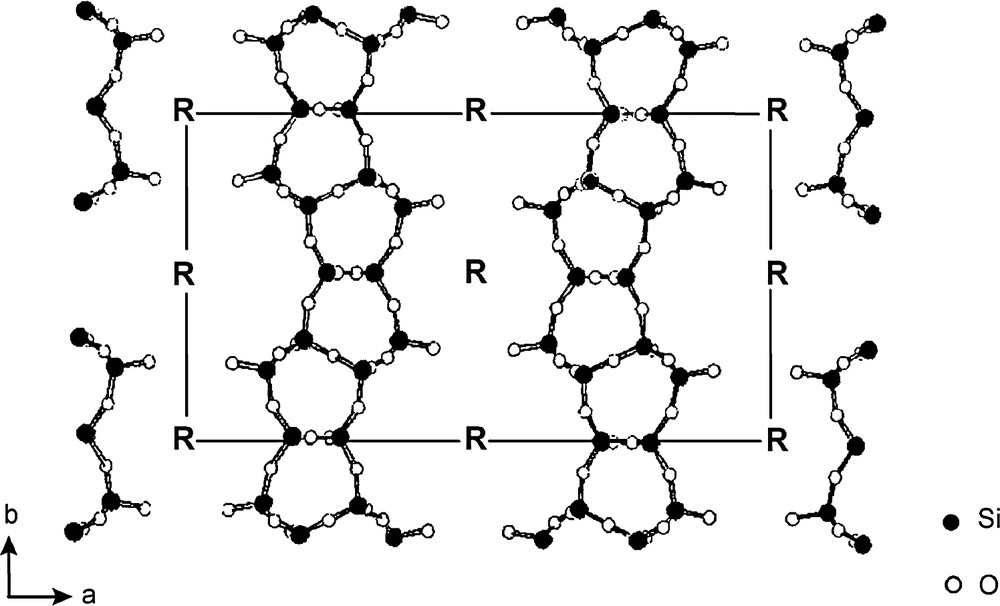
Proposed schematic structure of PREFER (R = 4-amino-2,2,6,6-tetramethyl-piperidine) (adapted from [74]).
Among the other topologies reported in Table 2, four of them, i.e. AST, ISV, ITW and ITH contain the small double four-membered ring cage (D4R), each cage being occupied by one fluorine, as revealed by the characteristic resonance at ca. –38 ppm/CFCl3 in the 19F MAS NMR spectrum (Fig. 4). It is important to note that, without the use of fluorine, no pure silica zeolite containing this small cavity could ever be prepared, which strongly suggests a structure directing and stabilizing effect [80] of the F– ion towards this secondary building block. More generally, fluorine tends to end-up trapped as the unique guest of small cages in zeolites prepared in fluoride medium. This tendency was very recently discussed in detail by Villaescusa et al. [81] in a well documented review.
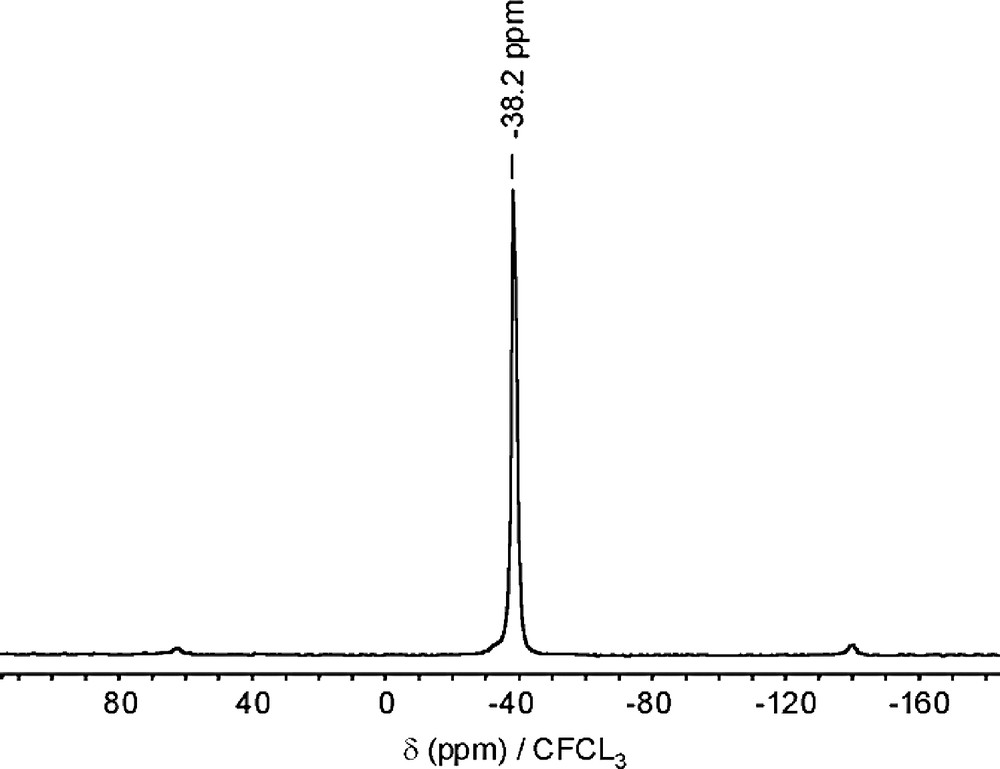
19F MAS NMR spectrum of as-made quinuclidinium-octadecasil (Bruker MSL 300; recycle delay: 6 s; chemical shift vs. CFCl3) (adapted from [67]).
We end this section by coming back to a new and promising field of application of pure silica zeolites as ‘molecular springs or molecular bumpers’, application which is only possible due to the absence of connectivity defects and to the resulting hydrophobic character of the samples prepared in fluoride media.
This original application in the field of energetics consists in transforming mechanical energy into interfacial energy, and vice versa. Thermodynamic systems consisting of a porous solid and a non-wetting liquid are known to have the property to accumulate, restore or dissipate energy [82,83]. By subjecting these systems to an increasing hydrostatic pressure, a strong decrease in volume is observed when the pressure reaches a certain value Pi, due to the intrusion of the liquid into the porous system. During this forced penetration or intrusion, the massive liquid is transformed into a multitude of molecular clusters which develops a large solid–liquid interface and which goes hand in hand with an increase in the free enthalpy of the system. Mechanical energy spent by the pressure forces is thus converted into interfacial energy. When the stress is released, for a certain pressure value Pe, closely related to Pi, the phenomenon may be reversible and spontaneous extrusion of liquid with a significant expansion of volume occurs. An entirely reversible system, able to accumulate superficial energy during the compression step and to restore mechanical energy during the pressure release, constitutes a real molecular spring. On the contrary, if the liquid contained in the pores of the material does not leach out, it is irreversibly trapped in the solid, and the system able to completely absorb mechanical energy acts as a bumper. Up to now, these thermodynamic systems consisted of amorphous mesoporous silica and a non-wetting liquid, such as metals – typically mercury – or alloys, melting at low temperatures. Their properties were used in various devices in order to yield important stresses and long displacements, for example the spreading of solar panels of satellites. The use of water as a non-wetting liquid would display obvious advantages, this polar liquid being non-polluting, inexpensive, easy to obtain as a pure phase, and characterized by a high interfacial energy. Moreover, the very small size of water molecules, which are comparable to spheres of 2.8 Å in diameter, can have access to very small micropores. Of course the choice of water implies that the solid phase is a very hydrophobic porous material.
Pressure-volume isotherms were thus determined for several ‘water–zeolite’ systems, using samples of silicalite-1 or purely siliceous zeolite β both prepared in fluoride media together with a more hydrophilic commercial sample of ZSM-5 zeolite [28,84]. The P–V curves are given in Fig. 5 (pressure is expressed in MPa and volume change in cm3 per gram of calcined zeolite). Although the highly hydrophobic silicalite-1 sample and the more hydrophilic material ZSM-5 sample are characterized by the same MFI structure, they display a completely different behavior. Indeed, the first sample acts as a molecular spring with a plateau of pressure corresponding to the intrusion and the extrusion of water (the reversibility of the phenomenon was confirmed on numerous cycles), whereas for the second sample a linear variation is observed on the P–V isotherm due to the fact that the corresponding porous system is already saturated with water at the beginning of the compression. The behavior of the zeolite beta sample differs from the one of silicalite-1, the P–V diagram showing a plateau only during the compression step. In this latter case, the system is not reversible and the material behaves as a molecular bumper. This absence of reversibility might be due to the presence of silanol defects formed at the interfaces of the polytypes the framework is made of.
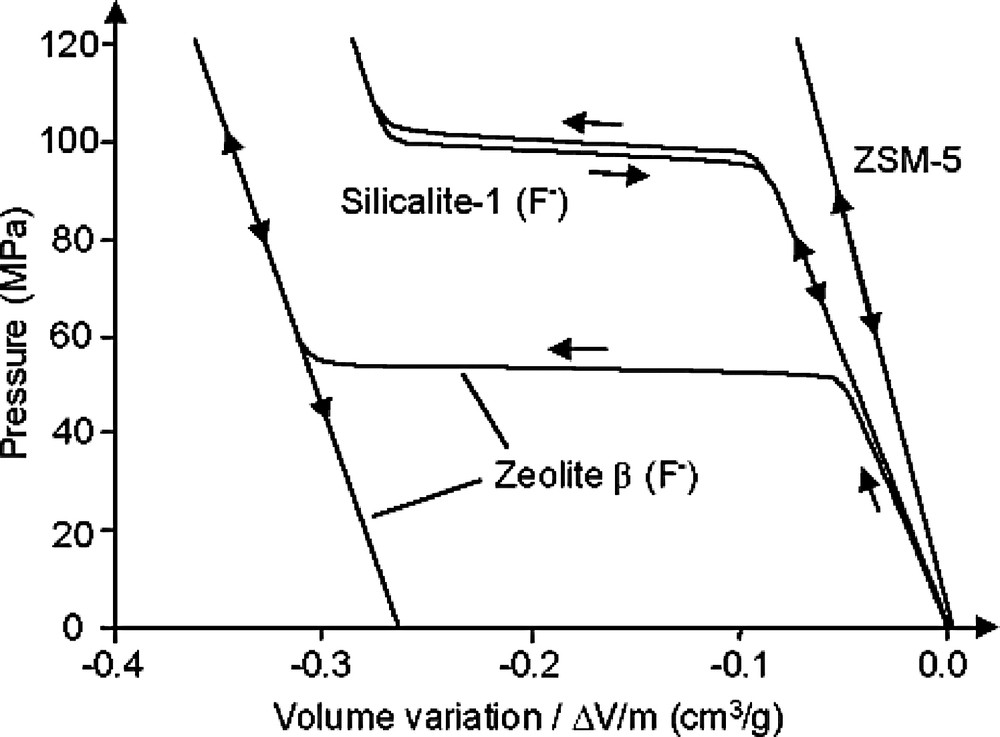
Pressure-volume isotherm of various ‘water–zeolite’ systems (silicalite-1 and zeolite β prepared in fluoride media; commercial sample of zeolite Na-ZSM-5) (adapted from [28]).
These studies open new application perspectives in the field of energetics for very hydrophobic zeolites in contact with water and are currently extended to other porous materials, such as clathrasils or grafted organized mesoporous silicas.
In addition to the influence of the nature of the organic template and of the F– ion, it appears quite logical to assume that the nature of the framework atoms, which allows T–O–T bonds with different distances and angles to be formed, can also exert a structure-directing effect. In particular, the partial or complete substitution of Si by Ge in the synthesis mixtures led to several new zeolite topologies [85], which are described in the next section.
2.3 Silicogermanate zeolites with new framework topologies
Due to its position in the periodic table of the elements, the germanium presents properties and reactivity similar to the silicon ones, with for example isostructural SiO2 and GeO2 quartz forms [86]. In spite of this similarity, germanates with framework structures containing four-, five- and six-coordinated Ge are known, whereas framework silicon atoms are exclusively tetracoordinated in zeolites.
The first porous germanates were synthesized in aqueous medium in 1991 with an amine as the structure directing agent [87] but proved to be unstable upon calcination. In 1998, solvothermal syntheses in ethylene glycol media led to a new germanate with an open structure of formula |(NH4+)2| [Ge7O15] [88] and also to the family of the non-thermally stable porous products ICMM-n (Instituto de Ciencia de Materiales de Madrid) [89]. The use of the fluoride route allowed the preparation of the ASU- n family (Arizona State University-n), with four-, five- and six-coordinated Ge atoms. Among these materials, ASU-7 and ASU-9 display all-tetrahedral Ge atoms. ASU-7 of topology ASV is formed by an assembling of D4R units [90] and is characterized by a monodimensional 12-ring channel system which remains intact upon calcination, whereas ASU-9 [90] is of topology AST [67]. FOS-5 of chemical formula |(CH3)3N)6 (H2O)4.5| [Ge32O64] was prepared in the presence of DABCO [91]. The structure code is BEC and corresponds to the hypothetical polymorph C of zeolite Beta [48]. Interestingly, the three previously mentioned materials display F–-containing D4R units in their structures.
One other important family of zeolitic materials, also synthesized via the fluoride route, is represented by the silicogermanates of the ITQ-n series. The silicogermanates ITQ-7 (topology ISV) [70], ITQ-12 (ITW) [92], ITQ-21 (no structure code assigned) [93] and ITQ-22 (IWW) [94] present new topologies and possess also F–-containing D4R units in their structures. Among the new topologies just mentioned, topology ISV is the only one that can crystallize under a purely siliceous form. One may also mention the case of zeolite ITQ-14, an intergrowth of several Beta polymorphs, in which a small amount of pure Si-form of BEC has been observed from high resolution electron microscopy [95]. The other topologies cannot be obtained from germanium-free fluoride-containing mixtures, which is related to the fact that the smaller values of the Ge–O–Ge angles in comparison to the Si–O–Si angles diminish the geometric constraints in the D4R units thus stabilizing the resulting structure [95]. Theoretical calculations confirm this interpretation, together with the reported preferential occupation of the T sites of the D4R by Ge in the zeolites formed from (Si, Ge) systems [96,97] and, also, with the observed faster crystallization rate of the ITQ-7 zeolite in its (Si, Ge) form [97]. It is worthy to note that the (Si, Ge) ITQ-17 [98] (BEC) and ITQ-21 [99] zeolites can also be prepared from fluoride-free mixtures but with a slower crystallization rate, thus confirming the stabilizing role of the F– ion towards the formation of the D4R unit.
The organic structure-directing agent hexamethonium [(CH3)3N–(CH2)6–N(CH3)3]2+ is of a great interest. Indeed, a systematic study on the synthesis of microporous materials from (Si, Ge) fluoride-containing gels with hexamethonium as the template allowed us, with Ge/Si molar ratios ≥ 1, to obtain IM-10 (UOZ) [103] (‘Institut français du pétrole–Mulhouse-10’), a new member of the clathrasil family.
Actually, when Si is progressively substituted by Ge in the synthesis mixtures, one observes successively the formation of, ITQ-13 (ITH) from a purely siliceous system [102,103], for a low Ge/(Si + Ge) ratio (0.2) a partial transformation of the gel into ITQ-17 [98] of topology BEC and, for Ge/(Si + Ge) ratios ≥ 0.5, a full transformation of the gel into the new germanium-containing IM-10. The Si/Ge molar ratio of the latter material is much lower than the corresponding ratio in the starting gel (0.32 instead of 1), showing that Ge is preferentially incorporated in the framework of IM-10. It is worthy to note that IM-10 can be obtained as a pure germanate form. The 19F MAS NMR spectra of the samples show a single resonance at –14.6 or –13 ppm for (Ge) IM-10 and (Si, Ge) IM-10, respectively. In both cases, this resonance corresponds to a unique crystallographic site and is characteristic of F– ions occluded in Ge-containing D4R units, with an overall Si/Ge molar ratio of the framework lower than 2. Indeed, according to the literature, the chemical shift values of F– trapped in Ge-containing D4R units lie between about –6 and –16 ppm [93,96,101].
As in ASU-7 (ASV) and ASU-9 (AST) [90], the topology of IM-10 may be described by an assembling of cubic D4R units bridged by GeO4 tetrahedra. In IM-10, each D4R unit is bonded to 13 others via eight GeO4 tetrahedra that connect corners as drawn on Fig. 6. Such an arrangement leads to [410620] supercages each of them occluding one hexamethonium dication. Every D4R unit or [46] cage is common to five [410620] supercages, the sixth face being one of a small [4264] cage (Figs. 6 and 7c).
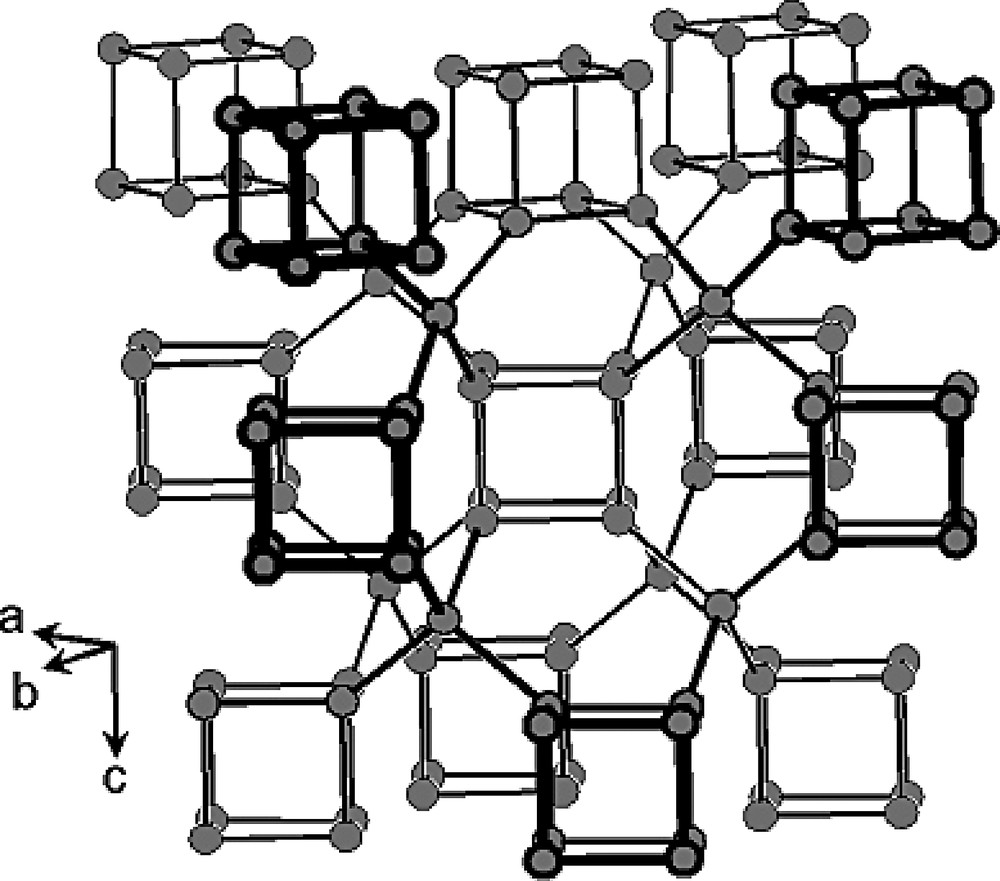
Connectivity between the F-containing D4R units in IM-10 (the oxygen and fluorine atoms have been omitted for clarity). Grey circles are Ge atoms (adapted from [103]).
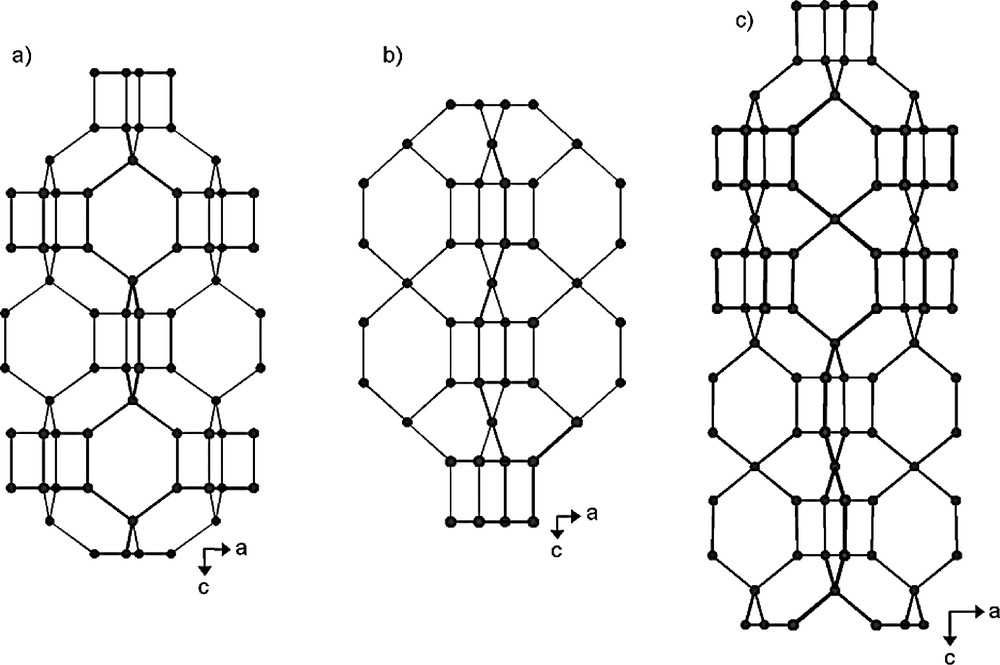
Projection along [010] of the a) ASU-9 (AST) b) ASU-7 (ASV) and c) IM-10 (UOZ) structures showing the different stacking of the D4R units (the oxygen atoms have been omitted for clarity) (adapted from [103]).
For comparison, Fig. 7 illustrates the different orientation of connecting tetrahedra in ASU-9 [90], ASU-7 [90], and IM-10. In fact, these three germanium oxides display a tetragonal symmetry with similar unit cell parameters, IM-10 having a double c unit cell parameter compared to ASU-9 and ASU-7. In ASU-9 (S.G.: I4/m, AST), the simplest structure, every D4R unit is surrounded by six AST cages (Fig. 7a). The framework topology ASV (S.G.: P4/mcc) (Fig. 7b) can be built from interconnected chains being formed by a stacking of D4R or [46] cage and [6442] along the c-axis, the stacking sequence being …–[46]–[6442]–[46]–[6442]–…. Each chain is directly linked to four identical chains via Ge–O4 tetrahedra, thus generating 12-ring channels along the c-axis [90]. In IM-10 (S.G.: Pn2), the same [46]–[6442]–[46] units are also present but the orientation of the Ge–O–Ge bonds of ASU-7 is changed in IM-10 and involved a lowering of the symmetry. Consequently the stacking mode in IM-10 becomes …–[46]–[6442]–[46]–[620410]–… along the c-axis (Fig. 7c). It is interesting to note that the new large cage [620410] seems to be a double AST or [61246] cage.
The structure-directing effect of Ge towards the D4R unit is strong enough to allow, as mentioned before, the obtention of ITQ-17 and ITQ-21 even in the absence of fluoride. This effect of Ge is also clear if one considers the synthesis results in the presence of the organic agent (6R,10S)-6,10-dimethyl-5-azoniaspiro [4,5]decane. Whereas, as presented before in Section 2.2., fluoride-containing purely siliceous mixtures led to Mu-26 (STF), (Si, Ge) systems led to two new phases, IM-9 [104] and IM-12 [105] (Institut Français du Pétrole/Mulhouse-9 and 12), in the presence or the absence of fluoride, respectively, both frameworks displaying D4R units [104,105]. Only IM-12 proved to be stable upon calcination. As illustrated in Fig. 9a, the 3-D structure of IM-12 may be described as layers stacked in the [100] direction, connected to each other by their four-membered rings (4MR) and thereby forming D4R units. From the structure determination and the Rietveld refinement, the germanium atoms can only be localized at T-sites of the D4R unit. Such a distribution is in agreement with those previously reported for the microporous silicogermanates with the topologies ISV, ITH and AST [97,106,107]. Therefore, each layer is essentially siliceous and constructed of fused small [415462] and [5262] cages. The new porous material IM-12 possesses a novel topology (submitted to the IZA Structure Commission for evaluation; following the rules, the code should be UTL [102]) with a two-dimensional channel system formed by 14- and 12-rings intersecting channels parallel to the c and b-axes, respectively (Fig. 8a, b).
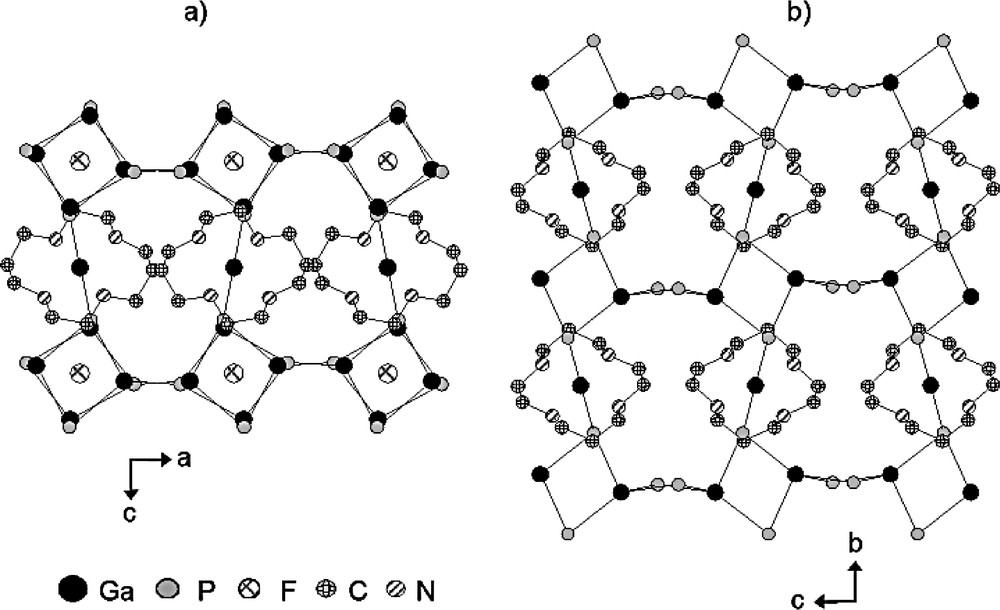
Projection of the structure of: a) Mu-5 [119] along [010] and b) Mu-6 [124] along [100] showing the presence and the absence of D4R-F units, respectively (for clarity, the hydrogen atoms, oxygen atoms and terminal Ga–F and Ga–OH groups are omitted).
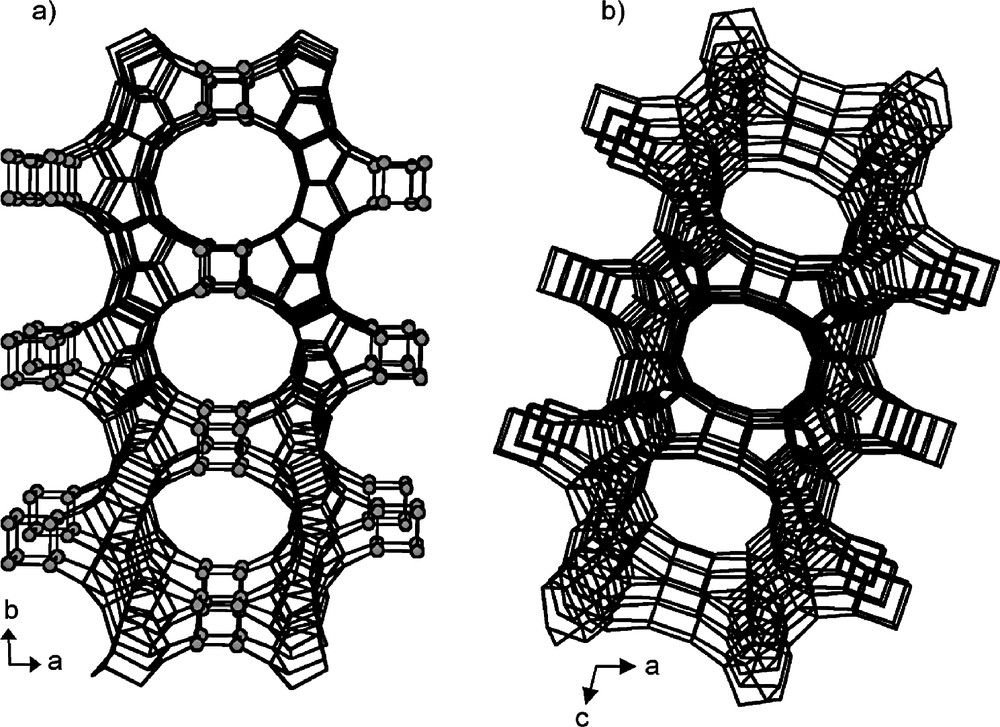
The 3D structure and the 2D channel system of the IM-12 zeolite: (a) connection between the silicogermanate layers lying in the (100) plane showing the 14-ring channel along [001] and (b) view down the 12-ring channel along [010]. The dark gray circles are the Ge atoms. There are no openings down [100] (adapted from [105]).
3 Phosphate-based microporous materials
The fluoride route was initiated and extensively investigated by our group for the synthesis of alumino- and gallophosphates. The first results were exciting with the synthesis of AlPO4-5 (AFI-structure-type) [108], the tetragonal variant of AlPO4-16 (AST structure-type) [109], the triclinic form of AlPO4-34 (CHA structure-type) [110], the discovery of the LTA-type gallophosphate [111] and the gallophosphate cloverite (CLO structure-type) [112] which is still then the molecular sieve with the largest 3-D pore channel system. Since then, several other research groups in the world developed this route for preparing new microporous phosphate-based materials [113]. Compared to the silicates and aluminophosphates, the gallophosphates possess more complicated framework structures, which result in the variety in the coordination of the gallium atom. Gallium can easily adopt four-, five- or six-coordination, which allows a greater flexibility in the building of inorganic frameworks.
Therefore, in our group, the synthesis of gallophosphates is mainly developed and particularly in fluoride medium. In these latter materials fluorine is generally part of the framework bonded to Ga atoms as terminal or bridging species, or even trapped in small structural units, the so-called double-four ring units, as it was mentioned above and firstly observed in the clathrasil octadecasil [67]. In the latter case it could act a templating role by stabilizing these units.
3.1 Templating role of F–
One characteristic of a large number of phosphate-based microporous solids prepared in fluoride medium is that F– is found located inside the small cubic building units, the so-called D4R units (double-four ring units). Such a situation is mainly observed for gallophosphates but also in a few aluminophosphates, most of these materials belonging to the Mu-n family (Mu standing for Mulhouse). Molecular anion (0-D)- , chain (1-D)-, layered (2-D)- and 3-D framework gallophosphates are thus formed (Table 3).
Phosphate-based materials with a framework showing D4R units hosting a fluoride anion (D4R-F units)
| Framework atoms | Structure code (dimensionality of the structure) | Size of the largest pore opening (number of T atoms) | References |
| Al, P | AST (3D) | 6 | [109] |
| Al, P | LTA (3D) | 8 | [114] |
| Ga, P | LTA (3D) | 8 | [111] |
| Ga, P | –CLO (3D) | 20 | [115] |
| Ga, P a | Mu-1 (molecular anion) (0D) b | [116] | |
| Ga, P a | Mu-2 (3D) b | 8 | [117] |
| Ga, P a | Mu-3 (1D) b | [118] | |
| Ga, P a | Mu-5 (3D) b | 12 | [119] |
| Ga, P a | Mu-15 (3D) b | 10 | [120] |
| Ga, P a | ULM-5 (3D) b | 16 | [121] |
| Ga, P a | ULM-18, GaPO4-TMED-1 (2D) b | [122] |
a In these materials Ga–F terminal groups and/or bridging fluorine (Ga–F–Ga) are also present.
b No zeolite framework code.
It is worthy to note that in the absence of fluoride in the starting mixture, with the exception of the molecular anion Mu-1 which can also be prepared in a fluorine-free medium [123], none of these materials is obtained. For instance, in the absence of F–, GaPO4-a crystallizes instead of cloverite [115]. Therefore, beside its mineralizing role, F– probably plays a structure-directing role stabilizing these small building units. The templating role of F– can be illustrated with the synthesis of the gallophosphates Mu-5 [119] and Mu-6 [124] but also with the synthesis of gallophosphates GaPO4-TMED-1 (ULM-18 [122]) and GaPO4-TMED-2 (Mu-8 [125]).
3.1.1 The gallophosphates Mu-5 [119] and Mu-6 [124]
The gallophosphates Mu-5 and Mu-6 were prepared under very similar conditions with the macrocycle 1,4,8,11-tetraazacyclotetradecane (cyclam) as organic structure-directing species. A summary of the most representative results is given in Table 4.
Syntheses performed in the system Ga2O3–P2O5–HF–Cyclam–H2O. Starting molar composition 1 Ga2O3/1 P2O5/x HF/0.5 Cyclam/80 H2O. The pH value was adjusted to 4–4.5 with tripropylamine (TRIPA) (0.8 TRIPA per 1 P2O5)
| Sample | HF (x) | T (°C) | pH | Crystallization time (days) | XRD results |
| A | 2 | 130 | 4.5 | 1 | Mu-5 |
| B | 2 | 90 | 4.5 | 5 | Mu-5 |
| C | 1 | 150 | 5.5 | 1 | Mu-5a + Mu-6 |
| D | 0.3 | 150 | 6.5 | 1 | Mu-5 + Mu-6 |
| E | 0 | 130 | 5 | 1 | Mu-6 |
| F b | 0 | 100 | 5 | 5 | Mu-6 |
a The underlined phase corresponds to the major phase.
b Amount of water in the starting mixture: 200 H2O.
As it can be seen from this table, when no fluorine is introduced in the starting mixture (samples E and F), pure Mu-6 crystallizes, whereas Mu-5 is obtained as a major phase when the HF/Ga2O3 is higher than 0.3 (samples C and D) and as a pure material for a ratio of 2 (samples A and B). On the basis of the chemical and thermal analyses and according to the structure determination, the unit cell formula of these two products are Ga20P16O64F8(OH)4(C10H24N4)4 for Mu-5 and Ga12P16O52OH12(C10H24N4)4 for Mu-6. The structures of the as-synthesized solids were determined for powder and single XRD data for Mu-5 [119] and Mu-6 [124], respectively. A view of these structures along the [010] (Mu-5) and [100] (Mu-6) directions is given in Fig. 9.
The 3-D structure of Mu-6 can be described as gallophosphate chains of gallium-corner-sharing Ga2P2O4 single-four rings (SARs), whereas in Mu-5, the 3-D gallophosphate framework consists of layers of D4R units hosting F– (D4R-F units). In both materials the chains or the layers are connected to each other via a Ga-cyclam complex to form the 3-dimensional framework. It has to be noticed that in Mu-5, fluorine is also present as Ga–F terminal groups sharing this position with an OH group [119]. These two fluorine positions are clearly evidenced by 19F MAS NMR spectroscopy (Fig. 10).
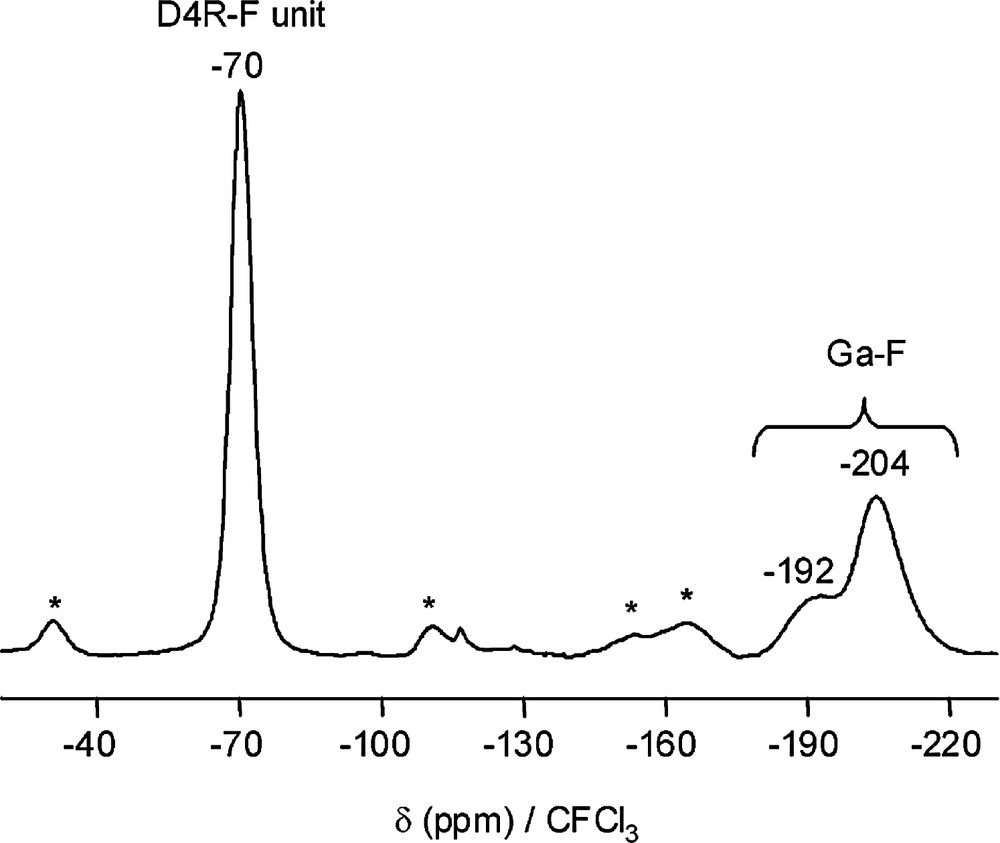
19F MAS NMR spectrum of the fluorogallophosphate Mu-5 (* spinning sidebands) (adapted from [119]).
Indeed, this technique is an efficient tool for characterizing the environment of fluorine in these materials. Thus, for phosphate-based microporous solids whose structures display D4R-F units, a NMR component with a chemical shift value (reference CFCl3) ranging from –60 to –70 ppm and –87 to –97 ppm is observed for fluorogallophosphates and fluoroaluminophosphates, respectively (Table 5). For the terminal and bridging species, the chemical shift is observed in a larger scale. Such differences could be explained by the different coordinations that gallium atoms can adopt (four, five or six). Furthermore, as it will be seen below the coordination of the fluorine atoms can change. It can be twofold or threefold coordinated to gallium atoms or water molecules via hydrogen bonds. A summary of the different fluorine chemical shift values observed in fluorine-containing phosphate-based microporous solids is reported in Table 5.
Fluorine chemical shift for some gallophosphates and aluminophosphates (reference CFCl3)
| Material | D4R-F | Terminal species | Bridging species |
| Mu-1 | –68.5 | ||
| Mu-2 | –72 | ||
| Mu-3 | –73 | –166 | |
| Mu-5 | –70 | –192, –204 | |
| Mu-12 | –82.3 | ||
| Mu-15 | –69 | –181.8 | |
| Mu-16 | –83.8 | ||
| Mu-17 | –102.1, –105.1 | ||
| Mu-20 | –110, –130 | ||
| Mu-23 | –139 | –130.7 | |
| Mu-28 | –78.8 | –84.3 | |
| AlPO4-16 | –90 | ||
| AlPO4-CHA | –126.8 | ||
| AlPO4-LTA | –94 |
3.1.2 The gallophosphates GaPO4-TMED-1 (ULM-18) [122]) and GaPO4-TMED-2 (Mu-8 [125])
The templating role of F– can also be illustrated with the synthesis of the gallophosphates GaPO4-TMED-1 (ULM-18) [122]) and GaPO4-TMED-2 (Mu-8) [125]. Both solids are prepared from similar starting mixtures in the presence of N,N,N′,N′-tetramethylethylenediamine (TMED) as organic template; GaPO4-TMED-1 (Ga8P10O40HF2(C6H18N2)3·2 H2O) in a fluorine medium, and Mu-8 (Ga12P12O48(OH)4(C6H18N2)2·4H2O) in the absence of F–. The structure of the former was determined by Taulelle and co-workers. The 2-D framework of the fluorogallophosphate ULM-18 is built from four-, six- and eight-membered openings forming a double sheet of D4Rs units containing fluorine (Fig. 11a). However, from 1H-31P CP MAS NMR spectroscopy, it was shown by these authors that HF instead of F– is occluded in these building units. It is interesting to note that the aluminophosphate Mu-4 [126] which is isostructural with ULM-18 was obtained from a fluorine-free mixture containing diethylformamide as main solvent. The structure of the gallophosphate Mu-8 is more complex. It consists of gallophosphate chains of edge-sharing four rings (two PO4, one GaO4 tetrahedra and one GaO4(OH)(H2O) octahedron). These ‘rope ladder’ chains are parallel to the [100] direction (Fig. 11b) and are connected to each other by a PO4 tetrahedron and a GaO4OH pentahedron leading to the 3-dimensional framework. Such an arrangement generates a 8-membered ring channel system along [100]. In that case, there are no D4R units.
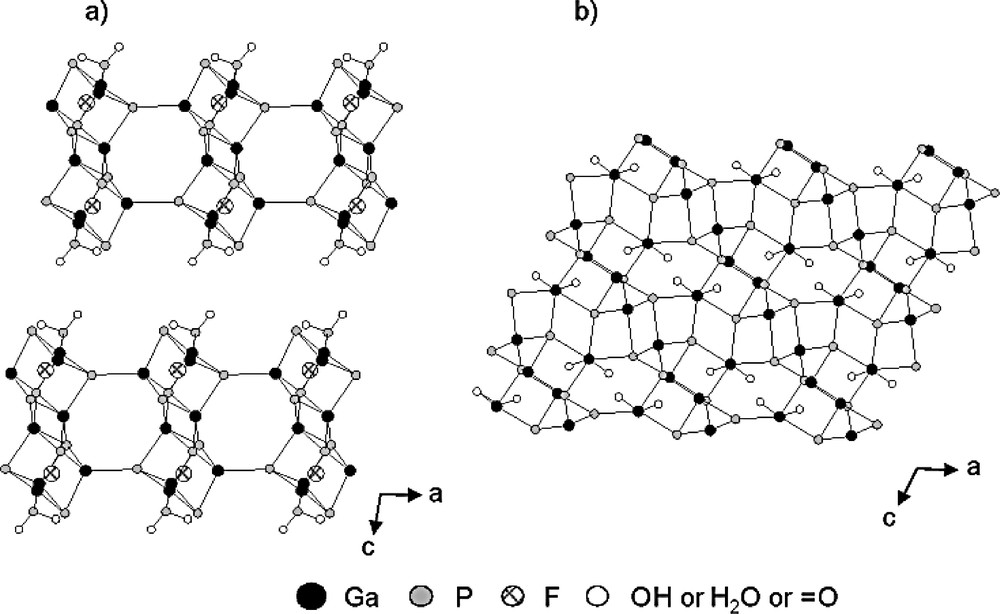
Projection of the structure along [010] of: a) ULM-18 [122] (GaPO4-TMED-1) showing the double sheets of D4R and b) Mu-8 [125] (GaPO4-TMED-2) showing the disposition of the inorganic chains of S4Rs parallel to the [100] direction (for clarity, the organic template, the hydrogen atoms and the framework oxygen atoms (Ga–O–P) are omitted).
These two series of materials illustrate well the templating role of fluorine for stabilizing small building units. However, at the present time there is no real proof of the existence of the D4R-F units in the precursor solutions. From this point of view, the synthesis of the two fluorogallophosphates Mu-2 [117] and Mu-3 [118] was very promising. Indeed, both materials present the same D4R-F building units and Mu-2 can be obtained using Mu-3 as reactant.
3.1.3 Synthesis of the fluorogallophosphates Mu-2 [117] and Mu-3 [118]
The fluorogallophosphates Mu-2 (Ga32P32O120(OH)16F6(C9H21N2)6·12 H2O) and Mu-3 (Ga12P12O48F12(C9H21N2)6) are both prepared in the presence of 4-amino-2,2,6,6-tetramethylpiperidine as organic template. Whereas Mu-3 crystallizes in the presence of ethylene glycol as main solvent, Mu-2 is obtained from an aqueous medium.
The structure of Mu-3 [118] consists of anionic chains of D4R units, hosting F– and presenting terminal Ga–F groups (Fig. 12a). The inorganic framework of Mu-2 [117] is also built from D4R-F units, the latter being, in this case, interconnected, forming a three-dimensional interrupted (presence of terminal OH groups) framework, with a three-dimensional pore system delimited by 8MR openings (Fig. 12b). As already observed for the other fluorogallophosphates presenting such D4R-F units (Table 3), the 19F MAS NMR spectrum of these two compounds displays a similar and characteristic signal at about –70 ppm. For Mu-3, the richest fluorine compound (F/Ga = 1), another 19F NMR signal, corresponding to Ga–F terminal groups, is also observed at –166 ppm (Table 5).
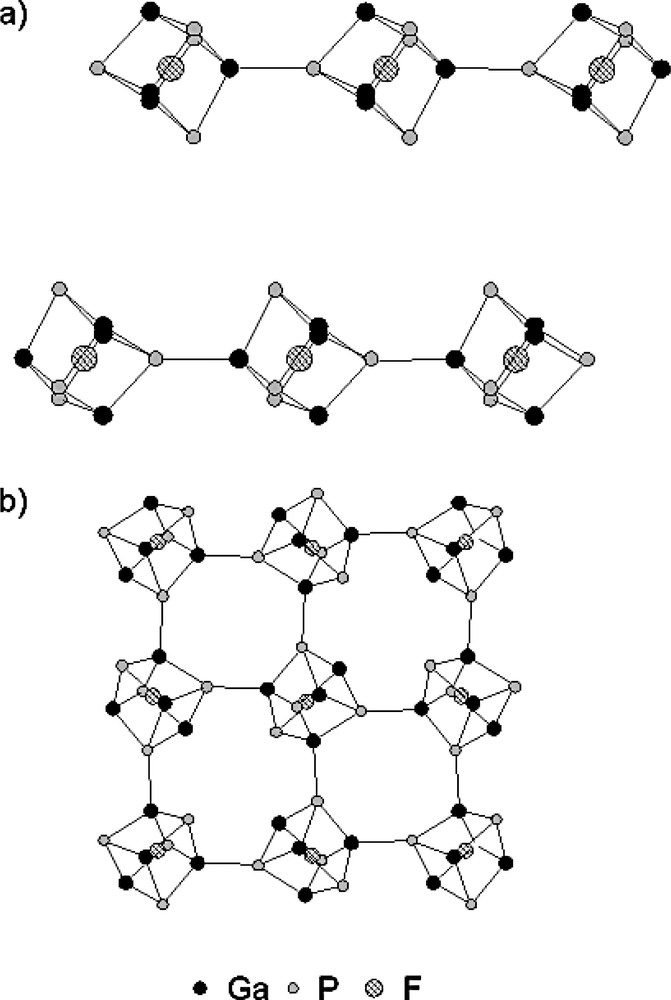
Projection of the structure of (a) Mu-3 [118] along [110] showing the chains of D4R-F units and (b) Mu-2 [117] along [100] showing the 3-D framework built from D4R-F units (for clarity, the organic template, the water molecules, the terminal F or OH groups, the hydrogen atoms and the framework oxygen atoms (Ga–O–P) are omitted).
3.1.4 Study of the transformation of Mu-3 into Mu-2 [127]
The different results obtained using Mu-3 as reactant in the presence of ethylene glycol (EG) and variable amounts of water are summarized in Table 6.
Results of the syntheses performed with Mu-3 as reactant for a reaction time of 5 days
| Sample | H2O/EG a molar ratio | T (°C) | Products |
| A | 0 | 170 | Mu-3 |
| B | 0.1 | 170 | Mu-2 + phase X + ɛMu-3 |
| C | 2 | 170 | Phase X b |
| D | 0.45 | 130 | Mu-2 + Mu-3 |
| E | 2 | 130 | Mu-2 |
a EG : ethyleneglycol.
b Unidentified phase.
In the absence of water, the fluorogallophosphate Mu-3 remains intact after heating at 170 °C during 5 days (sample A), whereas a slight increase in the H2O/EG molar ratio (= 0.1) leads to the formation of Mu-2 and another unidentified fluorinated phase (Phase X, sample B). It has to be noticed that the fluorogallophosphate Mu-2 is particularly stable in fluoride medium. Indeed, all attempts to crystallize the phase X, from Mu-2 as starting material, were unsuccessful (Fig. 13). This is not the case when Mu-3 is used as reactant. The phase X is obtained as a pure phase at this temperature (170 °C) in a mainly aqueous medium (H2O/EG = 2, sample C). As confirmed by 19F MAS NMR spectroscopy, no fluorinated D4R units are present in this material (no signal at –70 ppm). Therefore, we can conclude that in that case, this phase results from a complete dissolution of Mu-3. At a lower temperature (130 °C) and whatever the water content, phase X is never obtained. Under such mild conditions (low temperature), Mu-3 is partly (H2O/EG = 0.45, sample D) or completely (H2O/EG = 2, sample E) transformed into Mu-2. Since both gallophosphates are built from D4R-F units, one might assume that the transformation of Mu-3 into Mu-2 does not occur from a complete dissolution but results from a rearrangement of the fluorinated building blocs. A scheme of the different results obtained is given in Fig. 13.
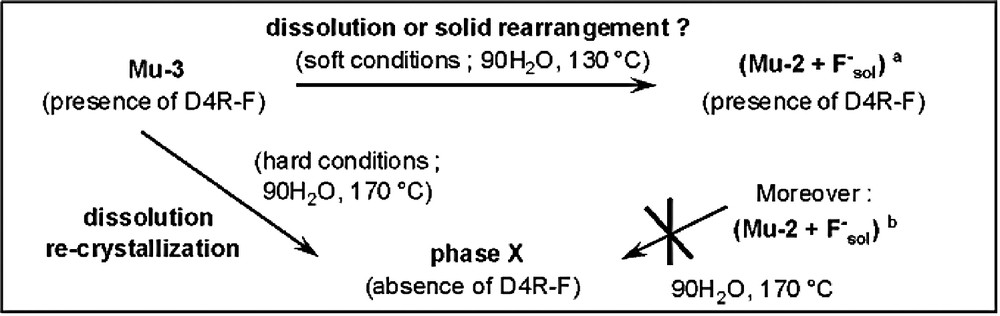
Scheme illustrating the different results obtained using Mu-3 (or Mu-2) as reactant. a As the fluorine content is larger in Mu-3 compared to Mu-2, the transformation of Mu-3 into Mu-2 should release F– anions in the solution, b F– anions were introduced in the starting gel in order to have the same fluorine amount as for Mu-3 (adapted from [127]).
The transformation of Mu-3 into Mu-2 was followed by XRD and 19F and 31P liquid state NMR spectroscopy [127]. The 19F NMR study gave interesting information. Two main components were observed on the NMR spectrum, one at –128 ppm (reference CFCl3) assigned to free F– anions in solution, and the second one at –134 ppm. The latter which is related to the transformation rate (vanishing of the NMR signal after 4 days of reaction) probably reveals fluorine in interaction with gallophosphate species. The latter is consumed during the reaction and takes therefore directly part in the transformation of Mu-3 into Mu-2. Nevertheless, it is difficult to unambiguously assign this 19F NMR signal to D4R-F units. Although the 19F chemical shift of such species in solution is not known, the value of the latter should not be too far from that observed in the solid phase, i.e. –70 ppm/CFCl3.
3.2 Incorporation of fluorine in the framework
By calcination, fluorine-containing species can be removed together with the organic template. The resulting solid is therefore essentially fluorine free. But in some cases, fluorine is part of the framework as bridging and/or terminal species. As it is the case for instance for the triclinic CHA-type aluminophosphate [110] and the GIS-type aluminophosphate [128]. Such a situation is mainly observed in fluorogallophosphates.
3.2.1 Fluorine as terminal group
As previously reported in Section 3.1., the fluorine atoms introduced in the starting mixture for the synthesis of Mu-3 and Mu-5 play not only a templating role but are also incorporated into the framework in terminal position. In the fluorogallophosphate Mu-3, the d(Ga–Fterminal) are in the range 1.821–1.862 Å, whereas for fluorine trapped inside the D4R units, the corresponding Ga–F distances are much longer, ranging from 2.328 to 2.508 Å (Fig. 14a). In Mu-5 the d(Ga–Fterminal) is a little bit longer (1.96(2) Å) since F– or OH– ions are assumed to be randomly distributed over the corresponding crystallographic site (Fig. 14b).
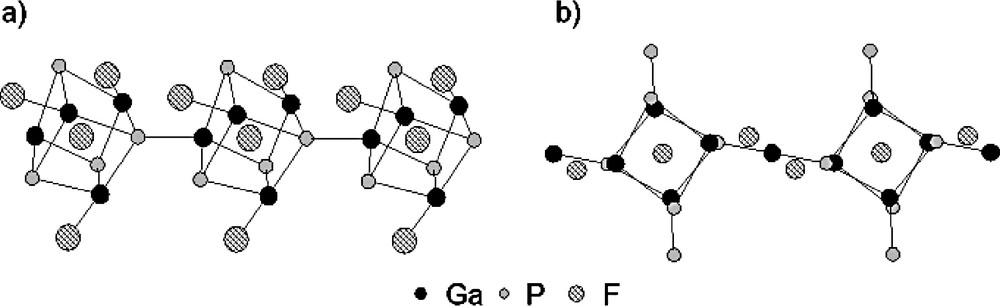
View of the D4R building unit showing the fluoride ion trapped inside the unit and the terminal Ga–F group for the gallophosphates (a) Mu-3 and (b) Mu-5 (in Mu-5, this latter position is shared with an OH group).
Such a terminal position has been recently observed in two other fluorogallophosphates named Mu-23 [129] and Mu-28 [130].
The fluorogallophophate Mu-23 is the first layered fluorinated gallophosphate with a Ga/P molar ratio of 5:4. It crystallizes in a narrow range of synthesis conditions and the amounts of Ga and amine are critical to obtain a pure phase. Syntheses are performed with the following molar ratio 1 Ga2O3/4 P2O5/8 HF/640 H2O/8 1,4-dimethylpiperazine (DMPIP) at 130 °C for 6 days [129]. The layers consist of GaO2F3(H2O), GaO4F2 ocahedra, and GaO4 and PO4 tetrahedra. One part of the fluorine is present as bridging atom (see below). The other part corresponds to terminal Ga–F group (Fig. 15) with Ga–F bond lengths of 1.854 and 1.885 Å. Such distances have previously been observed in other fluorogallophosphates: ULM-9 (1.800(2) Å) [131], GaPO4-CJ2 (1.903(2) Å) [132], TMP-GaPO (1.79(2) Å) [133], pseudo-KTP-type gallophosphate (1.900(3) Å) [134] and Mu-15 (1.86(1) Å) [120]. In the fluorogallophosphate Mu-23 the GaO4F2, GaO4 and PO4 tetrahedra are quite regular. The GaO2F3(H2O) octahedron is more distorted because of the presence of terminal groups.
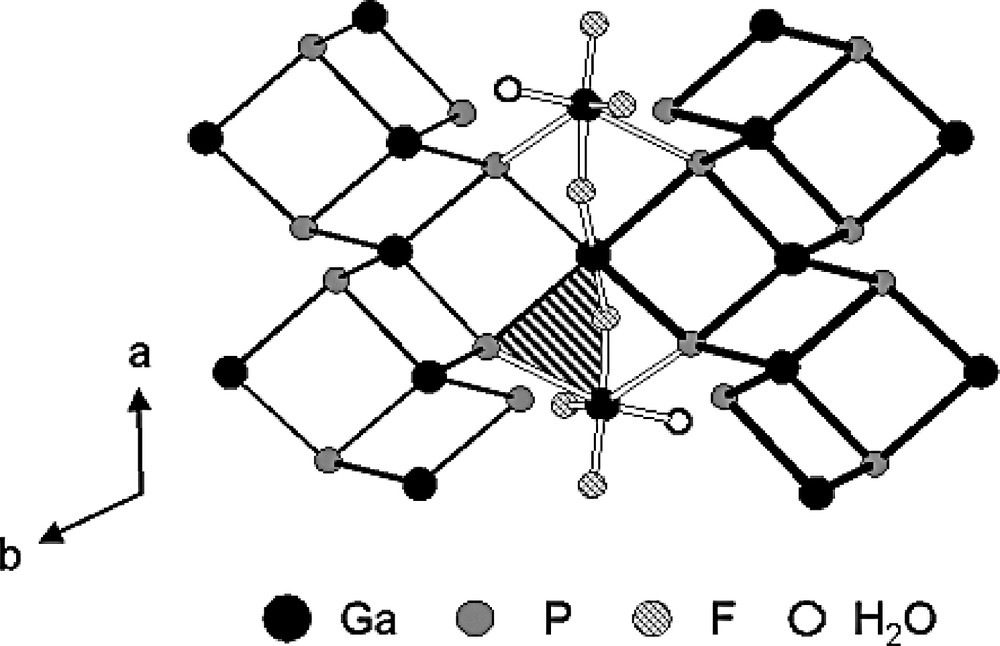
Projection of the structure of the fluorogallophosphate Mu-23 [129] in the (001) plane showing the terminal Ga–F groups and the bridging fluorine atoms (Ga–F–Ga) (for clarity, the framework oxygen atoms are omitted). The hatched area exhibits a three-membered-ring (Ga2P).
The fluorogallophosphate Mu-28 was obtained from synthesis conditions close to those of Mu-23 (molar composition of the starting mixture 1 Ga2O3/1 P2O5/2 HF/160 H2O/2 DMPIP, crystallization temperature: 130 °C, crystallization time: 6 days [130,135]). It displays a complex three-dimensional framework built from GaO3(OH,F)(H2O)2, GaO4(OH)F and GaO4(OH)2 octahedra, GaO4F trigonal bipyramid, GaO3(OH,F) and PO4 tetrahedra. The structure consists of 4-, 6-, 12-membered rings and an interesting secondary building unit (the [2134] pentacyclic unit) composed of 8T atoms (T = Ga, P) with 2 threefold coordinated oxygen atoms (O(18) and O(18′)) (Fig. 16a). Short distances Ga–X (d = 1.86(1) Å) and Ga–X′ (d = 1.80(2) Å) correspond to the presence of a 50/50 statistic repartition of hydroxyl groups and fluorine atoms over the two positions (Fig. 16a). Mu-28 structure can also be described as an arrangement of [4662] cages (called G unit) (Fig. 16b). The connection of those G units via 4-MR leads to the 3-D structure which displays a 3-D channel system delimited by 12-MR openings.
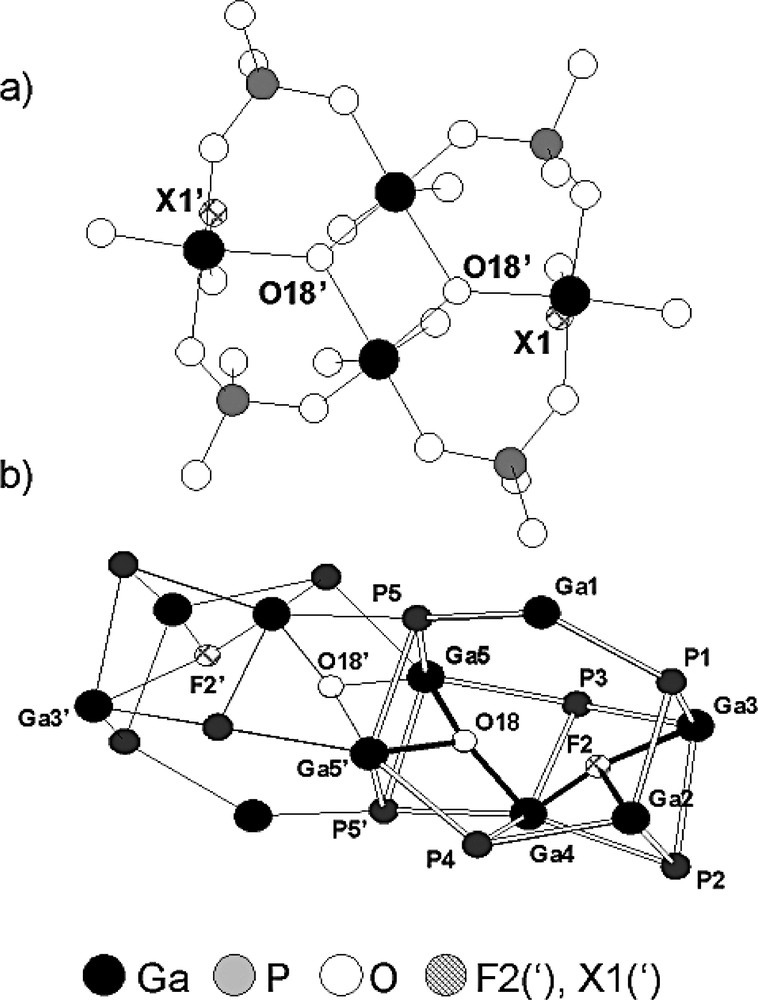
View of the building units observed in the fluorogallophosphate Mu-28 [130] a) the [2134] pentacyclic building unit (21: 1 ring with 2 T atoms (Ga2O2) and 34: 4 rings with 3 T atoms (Ga2PO3); T = Ga, P; X1, X1′ = F(1)(50%), OH(1)(50%)), b) the G unit showing the coordination of the F2 and F2′ fluorine atoms (adapted from [130,135]).
3.2.2 Fluorine as bridging species
As previously mentioned above, the fluorogallophosphates Mu-23 and Mu-28 have the particularity to exhibit both terminal and bridging fluorine atoms (Ga–F–Ga). For Mu-23 (Fig. 15), the presence of such a bridging atom leads to a 3-MR (Ga2P). The Ga–F bond length is about 1.95 Å.
In Mu-28 and Mu-16 [135] (see below) the fluorine is threefold coordinated to gallium atoms with one weak bond (d(Ga(3′)–(F2′)) = 2.77(3) Å; d(Ga(2)–(F2)) = 2.47(2) Å) and two stronger bonds (2.02(1) < d(Ga–(F2) or Ga–F(2′)) < 2.29(2) Å) (Fig. 16b). Such bond lengths are longer than those previously found in other fluorogallophosphates (typically 1.94 < d(Ga–F) < 2.04 Å [136,137] but the presence of the third weak interaction could explain the gap to ideal distances.
The fluorogallophosphate Mu-16 [135] was prepared from a starting mixture with the following molar composition 1 Ga2O3/1 P2O5/2 HF/160 H2O/2 N-methylpiperazine (MPIP) for 2 days at 180 °C [135]. It displays a three-dimensional framework with a three-dimensional channel system delimited by 12-ring opening. As observed for Mu-28 the same [2134] pentacyclic unit is present. However, the main differences between these two structures are situated in the 4-ring connecting the pentacyclic unit (Fig. 17). In Mu-16, a bridging F atom is observed whereas in Mu-28 terminal Ga–X groups (X = OH/F) are present.
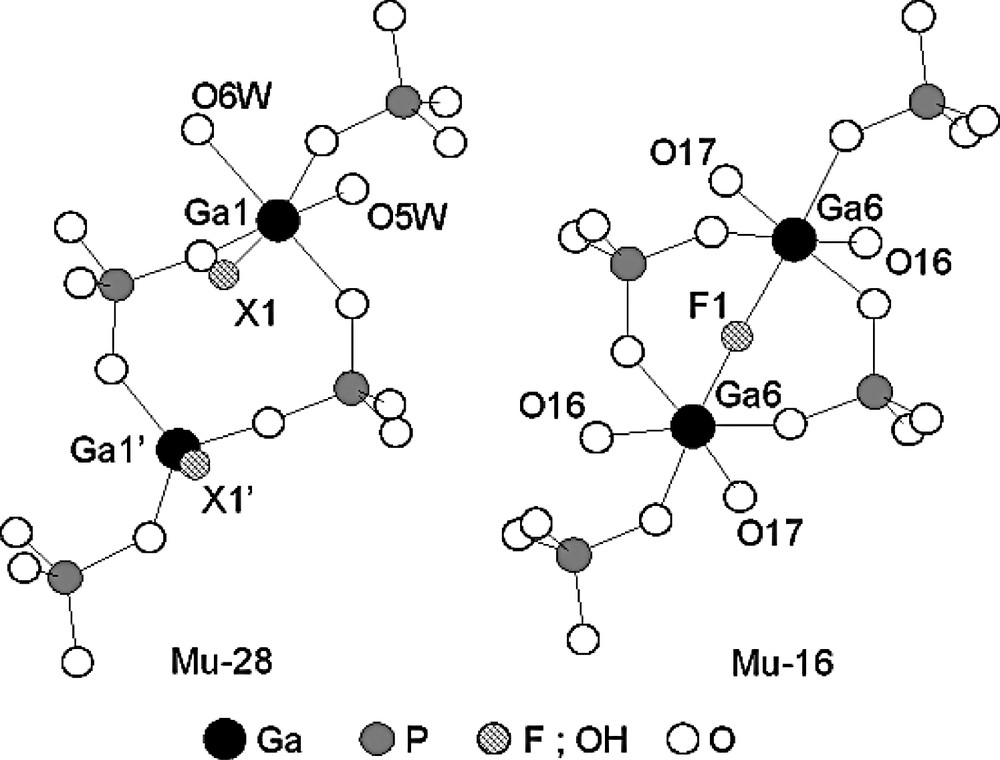
Differences observed in the 4-ring present in the framework of the fluorogallophosphates Mu-28 and Mu-16. In Mu-28, Ga-X and Ga′-X′ bonds correspond to the presence of a 50/50 statistic repartition of hydroxyl groups and fluorine atoms over the two X positions (adapted from [135]).
Three other three-dimensional fluorogallophosphates Mu-12 [136], Mu-17 [137] and Mu-20 [138] for which the synthesis conditions are reported in Table 7 show Ga–F–Ga bonds in their framework. The corresponding bond lengths for Mu-20, Mu-17 and Mu-12 are 2.01, 2.01 and 2.17 Å, respectively.
Synthesis conditions of the fluorogallophosphates Mu-12, Mu-17 and Mu-20 obtained at 170 °C
| Material | Starting molar composition | Crystallization time (days) | Chemical formula | References |
| Mu-12 | Ga2O3:P2O5:0.5 HF:0.5 3DTD a:80 H2O | 4 | Ga20P16O72F4[C2N2H10]4 | [136] |
| Mu-17 b | Ga2O3:P2O5:1 HF:1 APA c:80 H2O | 5 | Ga12P14O50(OH)10F4-[C10N6H34]2 | [137] |
| Mu-20 | Ga2O3:P2O5:2 HF:1 EN d:80 H2O | 2 | Ga12P8O36F12[C2N2H10]4·12H2O | [138] |
a DTD: 4,9-dimethyl-5,8-diaza-dodeca-2,4,8,10-tetraen-2,11-diol.
b Not obtained as pure phase.
c APA: 3-(2-aminoethylamino)propylamine.
d EN: ethylenediamine.
4 Conclusion
The fluoride route, mainly developed in our laboratory, was successfully applied to the synthesis of, first, silica-based microporous materials and, afterwards, of phosphate-based microporous materials, especially gallophosphates.
In a general way, the F– ion is able to play, besides its mineralizing role, a structure-directing or templating role.
As a consequence of lower supersaturation degrees and thus of smaller crystallization rates when F– is used as a mineralizer, the high-silica zeolites prepared via this route display a very low concentration or even no connectivity defects. The latter effect is also probably related to the occlusion of F–, which affords charge balance of the used organic cation without recourse to SiO– defects. The resulting highly hydrophobic character of the obtained pure-silica zeolites led to an original application in the field of energetics, as ‘molecular springs or bumpers’.
Beside several other synthesis parameters, such as, for instance, the nature of the organic agent or the dilution of the reaction mixture, the F– ion can also play, a structure-directing role, as shown by the discovery of several new framework topologies. In some cases it plays a true templating role, as it is found occluded in the small double four-membered ring unit (D4R) of the inorganic framework. Examples were found in the field of silica-based zeolites as well as in the field of the gallophosphate microporous materials. It is worthy to note here that the partial substitution of Si by Ge also stabilizes the D4R cage and thus allowed the formation of other novel topologies displaying this unit. The fluoride route appeared actually very fruitful for the Ga2O3–P2O5 system, due to the fact that Ga can easily adopt four, five or six coordination, which enables a great flexibility in the building of the inorganic framework. In these gallophosphate materials, fluorine is usually part of the framework contributing strongly to the stabilization of the structure, for instance trapped in D4R units as mentioned before, but also bonded to Ga atoms as terminal or bridging species.
Acknowledgments
The authors would like to thank the PhD students involved in this work: Jean-Louis Grieneisen, Bogdan Harbuzaru, Ludovic Josien, Alain Matijasic, Philippe Reinert, Laurence Schreyeck, Loïc Vidal.