

1 Introduction
Although studies related to the coordination of bismuth are not widespread, the synthesis of bismuth(III) complexes with nitrogen-donor macrocycles [1,2] has been investigated and it appears that bismuth(III) ion has a high affinity for these ligands. Additionally, compounds containing radioactive isotopes of bismuth such as 212Bi or 213Bi are now under investigation due to the potential of alpha emitting nuclides in radio-immunotherapy [3,4]. However, a major issue remains the stable in vivo binding of the metal to the ligand without any dissociation. This has induced studies concerning the complexation [5,6] of this ion with polyaminoacid ligands derived from diethylenetriaminepentaacetic acid (DTPA). In this type of complex, the metal exhibits a coordination sphere composed of carboxylate groups and nitrogen atoms. During the last decade, a better understanding of the coordination of bismuth in porphyrins has also arisen whether in organic or aqueous medium [7–11]. Indeed, due to the different positions of functionalisation and also to the possibility of locking a spacer specifically on one side of a porphyrin – ca. selecting the topology – this ligand appears as a plausible candidate for medical applications in radio-immunotherapy (Fig. 1). In this methodology, a chelate (e.g. a porphyrin) is linked to a targeting species (for instance, a monoclonal antibody directed against an antigen motif expressed specifically at the surface of a cancer cell). The resulting molecule is called an immunoconjugate. If the chelate possesses a high affinity for a radionuclide, for instance 213Bi, and if the reaction of coordination proceeds rapidly, the radioactive immunoconjugate, being able to link the cancer cell, should destroy the latter. This concept prompted our team to design porphyrins which are able to complex bismuth in a short period of time without subsequent decomplexation.
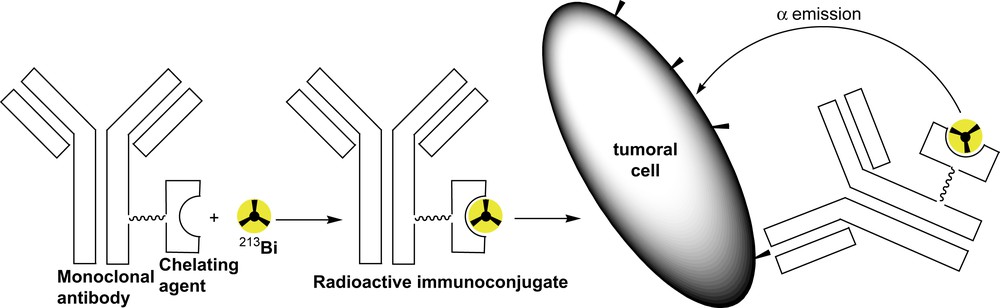
Schematic representation of alpha-radio-immunotherapy with 213Bi.
2 Results and discussion
As our research aims a biomedical application, these ligands were synthesized to address two questions. First, how the kinetics of bismuth insertion can be increased to be fast enough as both isotopes 212 and 213 of bismuth exhibit very short half-lives of 60 and 46 min, respectively? Second, what are the structural features which could lead to stable bismuth porphyrins? Indeed, with unfunctionalised porphyrins such as octa-ethyl porphyrin (OEP) or tetra-tolyl porphyrin (TTP), the kinetics of metalation are very slow and require at least 24 h of heating the ligand with the bismuth salt. Moreover, the stability in organic solvents of the resulting complexes is poor and significant decomplexation is usually observed during the purification process. To solve this problem, a simple concept consists of tethering on the macrocycle flexible arms which could a) deliver around the coordination sphere of bismuth oxygen atoms; b) induce a steric hindrance around the porphyrin centre thereby decreasing the direct access to the coordinated bismuth.
Typically, the preparation of the various targeted armed-porphyrins proceeds according to the synthetic route described in Fig. 2, which allows the synthesis of porphyrins bearing either one or four picket(s) terminated by an ester or an acid function. Thus, as previously reported [12], the four ester picket porphyrin 1 was synthesized by reaction of the desired acyl chloride on tetra-2-aminophenyl porphyrin (TAPP), and was further transformed in the four carboxylic acid picket porphyrin 2 via a conventional saponification reaction. The same type of reactions applied on N-{2-[15,20-bis-(2-acetylamino-phenyl)-10-(2-amino-phenyl)-porphyrin-5-yl]-phenyl}-acetamide (TriAcTAPP) led to the single-picket porphyrins 3 and 4. Actually, porphyrin 4 was also prepared as its picket possesses one more carbon atom than in the case of porphyrin 3 and allows direct comparisons with the latter. Bismuth insertion was performed by adding bismuth nitrate to a solution of free-base porphyrin. In the case of porphyrin 1 bearing four ethyl succinate pickets, heating the solution at 50 °C with 10 equiv of bismuth nitrate for 2 h was necessary to obtain 1Bi in 85% yield. Conversely, with porphyrins bearing one or four carboxylic acid picket(s) 2, 3 and 4, the metalation reaction was completed after 5 min, at room temperature using only 1 equiv of bismuth nitrate. Fortunately, we were able to obtain the X-ray structure of both bismuth complexes 1Bi and 3Bi [13,14] (Fig. 3a and b, respectively).
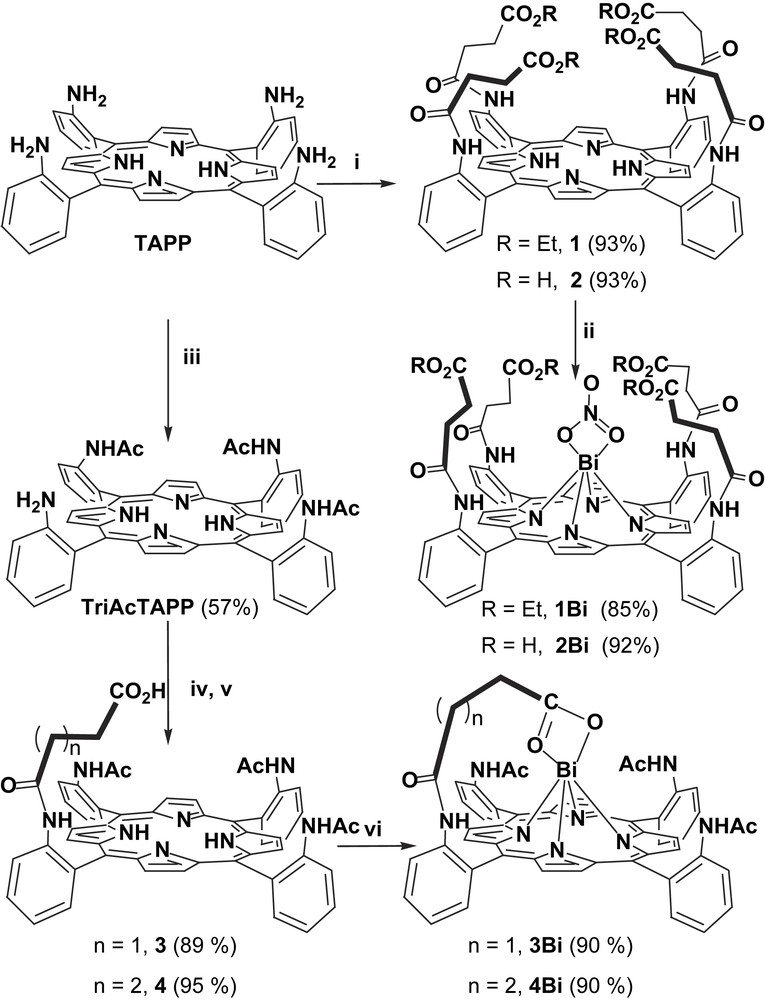
Synthesis of bismuth porphyrins bearing either one or four ester/acid pendant arm(s). Reagents and conditions: i, ClCO(CH2)2CO2Et (5 equiv), NEt3, THF for 1; then KOH, EtOH, 55 °C for 2; ii, Bi(NO3)3·5H2O: 10 equiv, 50 °C, pyridine, 2 h for 1Bi, 1 equiv, RT, pyridine, 5 min for 2Bi; iii, CH3COCl (3 equiv), NEt3, THF; iv, ClCO(CH2)2CO2Et (1.1 equiv), NEt3, THF for 3 or ClCO(CH2)3CO2Me (1.1 equiv), NEt3, THF for 4; v, KOH, EtOH, 55 °C; vi, Bi(NO3)3·5H2O, 1 equiv, RT, pyridine, 5 min.

ORTEP diagrams (30% thermal ellipsoids) of bismuth complexes 1Bi (a) and 3Bi (b). For the sake of clarity, the pyridine solvated molecule has been omitted in 3Bi.
In 1Bi, the Bi(III) is eight-coordinate with an approximate square antiprismatic geometry. The four-nitrogen atoms of the macrocycle form a square, the distorted other square being formed by four oxygen atoms described as follows: two oxygen atoms (O13 and O15) of the nitrate anion, the oxygen atom of a water molecule (Ow1) and a carbonyl oxygen atom (O8′) of the terminal ester group belonging to an arm attached to a symmetrically related macrocycle, forming in this way, a centrosymmetric dimer in the solid state (Fig. 3a). The Bi atom lies 1.125 Å above the four-nitrogen plane and the mean Bi–N bond length is 2.34(2) Å, these values are similar to those observed in (OEP)Bi(SO3CF3) [9] which also adopts a centrosymmetric dimeric form (Δ4N = 1.07 Å and 〈Bi–N〉 = 2.31(1) Å). The Bi–O bond lengths are larger than the sum of their ionic radii (1.13 + 1.40 = 2.53 Å) [15] 2.706(5) and 2.789(4) Å for the bonds with the nitrate anion (respectively with O13 and O15), 2.816(5) Å with the oxygen of the water molecule (Ow1) and 3.018(5) Å with the oxygen atom of the carbonyl group (O8′). This latter value is large but, to the best of our knowledge, this type of bond between a Bi atom and an ethoxycarbonyl group is unprecedented, however, such a large distance is observed in the (OEP)Bi(SO3CF3) dimer [9] where a Bi–O distance of 2.98(2) Å was found between the Bi atom and a triflate oxygen atom. A second water molecule (Ow2) is present in the cage and as shown, both water molecules are engaged in an intra-and inter-molecular hydrogen bond net, contributing to the stability of the dimer. One arm of the molecule does not participate in any interaction and thus exhibits a regular conformation. As reported above, one arm participates to the coordination sphere of the Bi atom – belonging to the second molecule of the dimmer – and the two remaining arms involved in the hydrogen bond net with Ow1 and Ow2, adopt folded conformations.
At the opposite, in 3Bi, the main difference with all bismuth porphyrins reported to date, for which X-ray data were obtained, is the mononuclear structure with the counter-anion delivered by the unique arm of the ligand (Fig. 3b). Indeed, the two oxygen atoms of the carboxylate group (O4 and O5) bound to the metal centre are stabilised by two hydrogen bonds with the neighbouring nitrogens from the amide linkage, N7 and N8, respectively. The two other coordination sites are occupied by two water molecules (Ow1 and Ow2), the bismuth being eight-coordinate and lying 1.145 Å above the four-nitrogen plane. The mean Bi–N bond length is 2.343(2) Å, a value that compares well with 2.340(2) Å in 1Bi. The two coordinated water molecules are also included in a hydrogen bonding net. The first one, Ow1 is hydrogen bonded to N10 of a solvated pyridine molecule (omitted for the sake of clarity) and to another water molecule (Ow3) itself hydrogen bonded to N6, also from an acetamide residue. The second one, Ow2 is hydrogen bonded to O5 from the carboxylate group and to N5 from an acetamide residue.
The bismuth atom is coordinated in a distorted antiprismatic geometry. This distortion is particularly appreciable with the Ow2–Bi–Ow1 and O4–Bi–Ow1 angles which are 76.87° and 76.31°, respectively, where the O4–Bi–O5 and O5–Bi–Ow2 are 46.97° and 58.30°. It is reasonable to correlate this distortion to the deformation of the porphyrin plane itself which adopt ‘saddle-shaped’ and ruffled conformations [16]. To the best of our knowledge, this type of deformation of the macrocycle has never been reported for bismuth porphyrins. Indeed, the carbon atoms in the opposite meso positions (5,15 and 10,20) are not located in the 24-atom least-squares plane but above or below. More precisely, the deviations of the four meso carbons from the mean porphyrin plane (Cm) are 0.297 Å, −0.150 Å, 0.243 Å and −0.164 Å, leading to an average value of 0.213 Å, smaller than that observed for highly distorted nickel porphyrins [17].
This significant distortion in 3Bi led us to prepare the analogous bismuth complex 4Bi in which the acidic arm bears one more carbon atom, as it was prepared from the methyl glutarate acyl chloride instead of the ethyl succinate acyl chloride for 3Bi. Indeed, with a longer arm, the distortion of the macrocycle should be released and the bismuth complex was expected to be more stable. As these bismuth complexes are not water-soluble, we needed a simple way to probe their stability in organic solution. Hence, we chose to expose a solution of each bismuth complex to 2500 equiv of trifluoroacetic acid during 3 h. Indeed, although this medium is really different from the physiological medium in which acid-catalyzed dissociation of radionuclide from the metal chelate should be minimal [18], it allows to compare the stability of different bismuth complexes over a reasonable period of time. Thus, the decomplexation of bismuth was easily monitored by the decrease of the typical Soret absorption at 470 nm [7]. The results are depicted in Fig. 4 and different conclusions can be drawn. First, in the succinyl series, the acid picket porphyrins 2Bi and 3Bi are more stable than the ester picket complex 1Bi. Second, for the succinic acid picket porphyrins, the stability is proportional to the number of pickets. These two observations are consistent with an interaction between the intramolecular carboxylate group(s) and the bismuth cation observed in the X-ray structure. Third, and surprisingly, the glutaric acid picket porphyrin 4Bi is the least stable bismuth complex, even in comparison with the ester picket porphyrin 1Bi. In the absence of a solid-state structure of 4Bi, one plausible explanation is the following. Although longer than the succinyl motif, and presumably due to the arrangement of dihedral angles along the propyl skeleton, the glutaryl arm cannot bend over the centre of the porphyrin as easily as in 3Bi, leading to a weaker interaction between the embedded counter-anion and the bismuth inside the porphyrin.
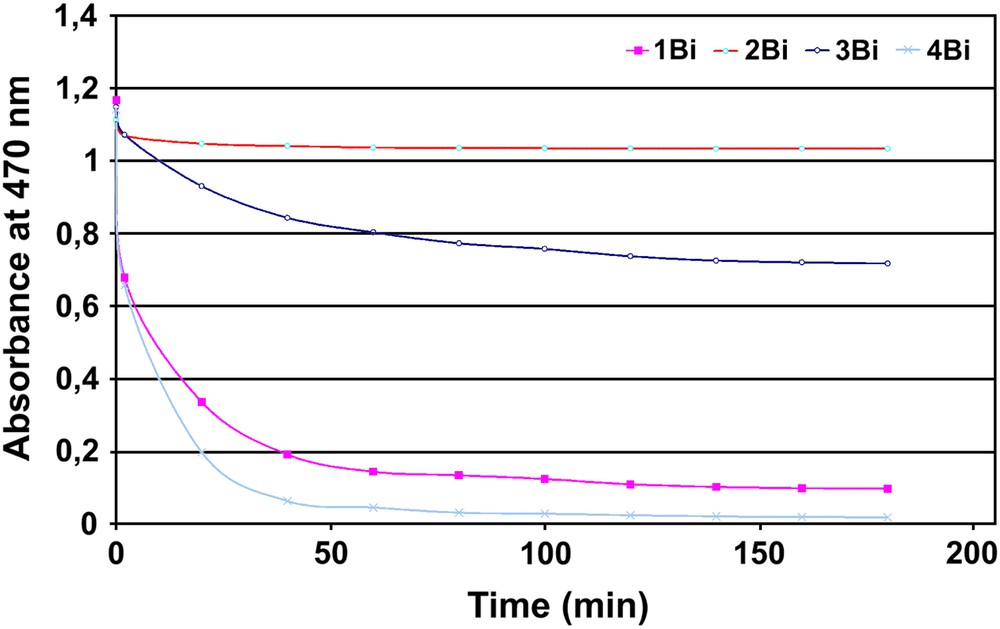
Bismuth decomplexation of four bismuth complexes in acidic medium monitored by UV–vis spectroscopy (CH2Cl2, 2500 equiv TFA).
In conclusion, the work described herein demonstrates that simple and easy synthetic functionalisations of the main porphyrin core change significantly the coordination properties of the basic macrocyle. Indeed, two properties, crucial for further developments in biomedical applications have been investigated. The first one consists of the kinetics of metalation which are clearly improved by tethering to the porphyrin a flexible arm delivering an intramolecular counter-anion to the bismuth cation. The second parameter influenced by the functionalisation of the macrocyle is the stability of the resulting complex. We have shown that this stability is proportional to the number of acidic pickets around the porphyrin. Therefore, for further applications, a balance has to be maintained between the kinetics of metalation and the stability of the complex by tuning the number of pickets attached to the porphyrin.
3 Experimental procedures
The full experimental procedures for syntheses of 1 and TriAcTAPP were reported in Ref. [12].
3.1 Typical saponification procedure
For instance, for the transformation of 1 to 2, compound 1 (0.1 g, 0.08 mmol) was dissolved in THF (20 mL). KOH (0.5 g, 8.9 mmol) in ethyl alcohol (5 mL) was added and the mixture was heated to 50 °C. After 4 h, the reaction mixture was cooled to room temperature, and ether was added until precipitation. The obtained precipitate was filtered and washed with dichloromethane. The resulting solid was dissolved in water. Then, HCl 6 M was added dropwise until porphyrin precipitation. The resulting precipitate was filtered and washed with water. The product was obtained in quantitative yield.
3.1.1 N-(2-{α-10,15,20-Tris-[2-(3-carboxy-propionylamino)-phenyl]-porphyrin-α-5-yl}-phenyl)-succinamic acid, 2
FAB-MS: m/z = 1074.8 [M]+ for C60H50N8O12. FTIR (KBr, cm−1): 1715 (CO)acid, 1670 (CO)amide. 1H NMR (DMSO-d6, 323 K, 500 MHz): δH 10.71 (4H, s, NHCO), 8.67 (8H, s, βpyr), 8.40 (4H, dd, J = 8.6 Hz, J = 1.1 Hz, aro), 7.98 (4H, dd, J = 7.6 Hz, J = 1.7 Hz, aro), 7.77 (4H, td, J = 7.6 Hz, J = 1.6 Hz, aro), 7.48 (4H, td, J = 7.5 Hz, J = 1.1 Hz, aro), 1.67 (8H, t, CH2CH2), 1.33 (8H, t, CH2CH2), −2.91 (2H, s, NHpyr). 13C NMR (DMSO-d6, 323 K, 126 MHz): δC 175.9 (Camide), 173.8 (Cacid), 140.9 (CH aro), 136.0 (CH aro), 132.8 (CH aro), 131.9 (CH βpyr), 129.5 (CH aro), 123.9 (CH aro), 122.9 (CH aro), 116.9 (CH aro), 34.8 (CH2), 33.8 (CH2). Microanalysis: C60H50N8O12·H2O, found (calc): C, 64.93 (64.83); H, 4.79 (4.89); N, 10.25 (9.81)%.
3.1.2 N-{2-[α-10,15,20-Tris-(2-acetylamino-phenyl)-porphyrin-α-5-yl]-phenyl}-succinamic acid, 3
ESI-HRMS: calcd m/z = 939.3020 [M + K]+ for C54H44KN8O6Na, found 939.3017. FTIR (KBr, cm−1): 1710 (CO)acid, 1670 (CO)amide. UV–vis (MeOH/pyridine: 6%): λmax/nm (log ɛ, dm3 mol−1 cm−1): 418 (362.3), 514 (16.7), 547 (4.7), 588 (4.4), 643 (1.5). 1H NMR (CDCl3, 298 K, 300 MHz): δH 11.85 (1H, s, COOH), 8.81 (2H, br s, NHCO), 8.78 (2H, br s, NHCO), 8.70 (8H, s, βpyr), 8.14 (4H, d, J = 7.5 Hz, aro), 8.10 (2H, d, J = 7.5 Hz, aro), 7.94 (4H, d, J = 7.0 Hz, aro), 7.83 (4H, t, J = 8.0 Hz, aro), 7.57 (4H, t, J = 6.5 Hz, aro), 1.99 (2H, t, J = 5.0 Hz, CH2CH2), 1.64 (2H, br s, CH2CH2), 1.24 (3H, s, CH3), 1.22 (6H, s, CH3), −2.72 (2H, s, NHpyr). 13C NMR (CDCl3, 298 K, 75 MHz): δC 136.4 (CH aro), 136.1 (CH aro), 129.4 (CH βpyr), 125.5 (CH aro), 124.7 (CH aro), 124.2 (CH aro), 40.0 (CH3), 31.3 (CH3), 29.7 (CH2), 23.2 (CH2). Microanalysis: C54H44N8O6·CH3OH·H2O, found (calc): C, 69.58 (69.46); H, 4.87 (5.30); N, 11.42 (11.78)%.
3.1.3 4-{2-[α-10,15,20-Tris-(2-acetylamino-phenyl)-porphyrin-α-5-yl]-phenylcarbamoyl}-butyric acid, 4
FAB-HRMS: calcd m/z = 937.4438 [M + Na]+ for C55H46N8O6Na, found 937.3437. FTIR (KBr, cm−1): 1709 (CO)acid, 1669 (CO)amide. UV–vis (CH2Cl2): λmax/nm (log ɛ, dm3 mol−1 cm−1): 418 (134.9), 512 (7.1), 546 (1.6), 586 (2.2), 643 (0.5). 1H NMR (DMSO-d6, 298 K, 500 MHz): δH 11.43 (1H, br s, CO2H), 8.83 (1H, br s, NHCO), 8.78 (1H, br s, NHCO), 8.74 (2H, br s, NHCO), 8.69 (8H, br s, βpyr), 8.12 (4H, d, J = 7.2 Hz, aro), 7.94 (4H, d, J = 6.9 Hz, aro), 7.82 (4H, t, J = 7.2 Hz, aro), 7.57 (4H, br s, aro), 1.42 (4H, br s, CH2), 1.22 (9H, br s, CH3), 1.09 (2H, br s, CH2), −2.72 (2H, s, NHpyr). 13C NMR (DMSO-d6, 298 K, 125 MHz): δC 168.7(Cacid), 138.6 (CH aro), 136.6 (CH aro), 129.4 (CH βpyr), 125.3 (CH aro), 124.2 (CH aro), 34.9 (CH2), 32.4 (CH2), 20.3 (CH2).
3.1.4 N-(2-Bismuth(III) nitrate-{α-10,15,20-tris-[2-(3-carboxy-propionylamino)-phenyl]-porphyrin-α-5-yl}-phenyl)-succinamic acid, 2Bi
In a 50 mL flask, 20 mg (0.01 mmol) of 2 was dissolved in 5 mL pyridine. Then 5.3 mg (0.01 mmol) Bi(NO3)3·5H2O was added. The reaction was stirred at room temperature. After 10 min the solvent was evaporated and dissolved in CH2Cl2 and filtered. The solution was concentrated to dryness and dissolved in a minimum CH2Cl2 to which pentane was added. This allowed precipitation of the product that was filtrated, washed with more pentane and finally dried under vacuum overnight. The product was obtained in 92% yield. FAB-MS: m/z = 1281.7 [M − NO3]+. FTIR (KBr, cm−1): 1384 (NO3), 990 (Bi–Np). UV–vis (CH2Cl2/pyridine: 1/1): λmax/nm (%): 351 (31), 471 (100), 596 (8), 644 (7). 1H NMR (pyridine-d5, 300 K, 500 MHz): δH 8.98 (8H, s, βpyr), 8.81 (20H, d, J = 4.7 Hz, βpyr), 8.67 (4H, d, J = 8.2 Hz, aro), 8.62 (4H, s, NHCO), 8.31 (10H, t, J = 7,8 Hz, Pyr), 7.96 (4H, d, J = 6.5 Hz, aro), 7.83 (24H, t, J = 6.1 Hz, aro, Pyr), 7.50 (4H, t, J = 7.0 Hz, aro), 2.05 (8H, br s, CH2), 1.77 (8H, br s, CH2). 13C NMR (pyridine-d5, 300 K, 125 MHz): δC 175.4 (Camide), 171.5 (Cacid), 149.7 (CH aro), 146.1 (CH aro), 142.9 (CH aro), 141.2 (CH aro), 134.9 (CH βpyr), 133.0 (CH aro), 131.5 (CH aro), 130.2 (CH aro), 126.7 (CH aro), 123.1 (CH aro), 121.8 (CH aro), 118.9 (CH aro), 31.5 (CH2).
3.1.5 N-{2-Bismuth(III) nitrate-[α-10,15,20-tris-(2-acetylamino-phenyl)-porphyrin-α-5-yl]-phenyl}-succinamic acid, 3Bi
The same experimental procedure to that of compound 2Bi was applied on porphyrin 3. Product 3Bi was obtained in 90% yield. ESI-HRMS: calcd m/z = 1129.2863 [M − NO3 − H + Na]+ for C54H41BiN8O6Na, found 1129.2850. FTIR (KBr, cm−1): 1670 (CO), 990 (Bi–Np). UV–vis (CH2Cl2): λmax/nm (log ɛ, dm3 mol−1 cm−1): 350 (4.8), 472 (13.5), 598 (1.7), 646 (1.5). 1H NMR (DMSO-d6, 300 K, 500 MHz): δH 9.03 (2H, d, J = 4.5 Hz, βpyr), 9.03 (2H, d, J = 4.5 Hz, βpyr), 8.99 (6H, d, J = 8 Hz, Pyr), 8.97 (2H, d, J = 5 Hz, βpyr), 8.89 (2H, d, J = 5 Hz, βpyr), 8.72 (4H, br s, aro), 8.55 (3H, t, J = 7 Hz, Pyr), 8.33 (1H, br s, aro), 8.33–8.26 (2H, m, aro), 8.07 (1H, t, J = 6.6, aro), 7.99 (6H, t, J = 7.8 Hz, Pyr), 7.90 (3H, m, aro), 7.73 (1H, br s, aro), 7.45 (4H, br s, NHCO), 7.36 (2H, t, J = 7 Hz, aro), 7.16 (2H, m, aro), 1.87 (2H, br s, CH2CH2), 1.63 (2H, br s, CH2CH2), 1.49 (9H, s, CH3). 13C NMR (DMSO-d6, 300 K, 125 MHz): δC 172.5 (Cacid), 149.9 (CH aro), 135.9 (CH aro), 134.3 (CH aro), 132.7 (CH aro), 132.1 (CH βpyr), 131.9 (CH aro), 129.3 (CH aro), 123.9 (CH aro), 122.2 (CH aro), 121.8 (CH aro), 31.3 (CH2), 29.7 (CH2), 23.2 (CH3).
3.1.6 4-{2-Bismuth(III) nitrate-[α-10,15,20-tris-(2-acetylamino-phenyl)-porphyrin-α-5-yl]-phenylcarbamoyl}-butyric acid, 4Bi
The same experimental procedure to that of compound 2Bi was applied on porphyrin 4. Product 4Bi was obtained in 90% yield. FAB-HRMS: calcd m/z = 1143.3007 [M − H − NO3 + Na]+ for C55H43BiN8O6Na, found 1143.3005. FTIR (KBr, cm−1): 1384 (NO3), 895 ρ(Bi–Np). UV–vis (CH2Cl2): λmax/nm (log ɛ, dm3 mol−1 cm−1): 351 (253.3), 472 (72.3), 600 (5.0), 646 (4.2). 1H NMR (DMSO-d6, 298 K, 500 MHz): δH 8.95 (8H, br s, βpyr), 8.68 (4H, br s, aro), 8.43 (1H, bs s, NHCO), 8.40 (3H, bs s, NHCO), 7.92 (4H, d, J = 7.5 Hz, aro), 7.86 (4H, t, J = 7.5 Hz, aro), 7.52 (4H, t, J = 8.0 Hz, aro), 1.87 (2H, t, J = 7.0 Hz, CH2), 1.70 (2H, br s, CH2), 1.44 (11H, br s, 1CH2 + 3CH3). 13C NMR (DMSO-d6, 298 K, 125 MHz): δC 175.4 (Camide), 174.6 (Cacid), 165.3 (CH aro), 149.2 (CH aro), 146.2 (CH aro), 135.3 (CH βpyr), 130.1 (CH aro), 122.9 (CH aro), 121.8 (CH aro), 34.9 (CH2), 33.0 (CH2), 24.1 (CH3), 20.3 (CH2).
Acknowledgements
We gratefully acknowledge ‘La Ligue contre le cancer’, the ‘Région Bretagne’, and the CNRS (France) for financial support. We are particularly indebted to the ‘Comité des Côtes-d'Armor de La Ligue contre le cancer’ for the generous help to ZH.