

1 Introduction
After seminal report by Shaw in the 1970s, the chemistry of pincer ligands has undergone a rapid development, especially over the last decade [1]. Systems featuring a central carbon ligand and ancillary heteroatomic ligands (O, N, P) have found many applications in coordination chemistry and catalysis, and their chemistry has been extensively reviewed. Comparatively, only a moderate attention has been paid so far to structures featuring sulfur-based ligands as pendent arms and most studies exclusively focused on the chemistry of S–C–S pincer backbones in which the central carbon ligand belongs to a benzene ring, the sulfur ligands being a thioether, a thioketone or a phosphinosulfide [2]. As part of a new program aimed at exploring the use of new polydentate structures featuring ancillary phosphinosulfide ligands in coordination chemistry and catalysis, we recently developed an access to a new class of monoanionic S–P–S 1 and dianionic S–C–S 2 systems (Scheme 1). In this account we summarize our contribution in this area.
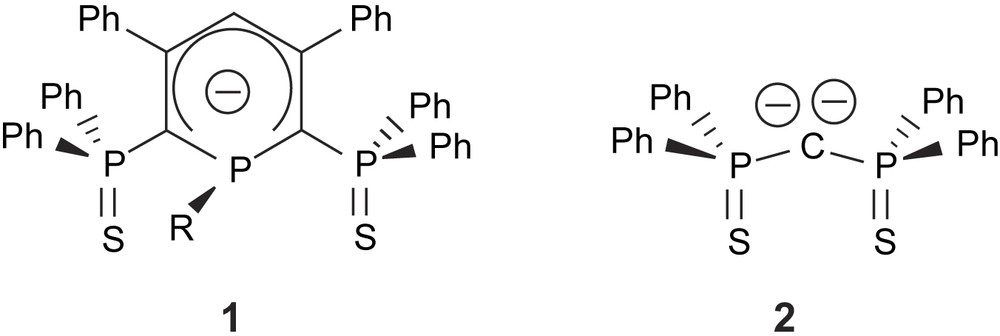
Monoanionic bis(phosphinosulfide) phosphacyclohexadienyl 1 and dianionic bis(phosphinosulfide) methanediide ligands.
2 Results and discussion
2.1 S–P–S pincer ligands
The discovery of ligands 1 arises from our studies on the chemistry of phosphinines. Over the last few years we showed that these aromatic phosphorus derivatives which behave as powerful electron acceptor ligands could be successfully employed to stabilize electron-reduced transition metal complexes and metal surfaces [3–6]. Thus for example, we showed that their very specific electronic properties could be employed to significantly modify optical properties of gold nanoparticles (Scheme 2) [7–9].
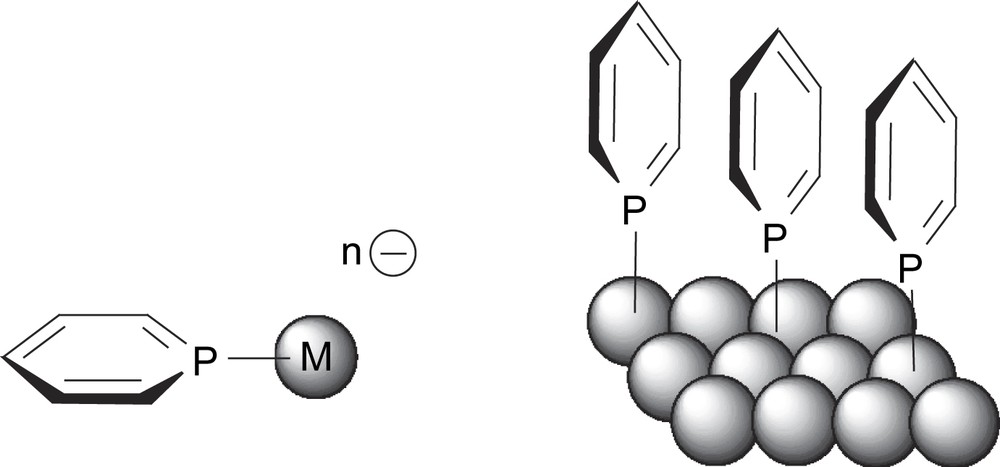
Stabilization of electron reduced transition metal complexes and metallic nanoparticles with phosphinine ligands.
However, like other low coordinate phosphorus derivatives, these ligands are quite sensitive towards nucleophiles and their use in coordination chemistry and catalysis is essentially limited to processes relying on the use of electron-rich metal fragments [10–14].
This limitation mainly results from the presence of a substantial positive charge at phosphorus which renders the ring sensitive towards nucleophiles. However, this property can be exploited to assemble a new type of anionic ligand featuring a delocalized negative charge (Scheme 3). Some of these phosphacyclohexadienyl ligands have found interesting applications in coordination chemistry in the synthesis of π-complexes. Recently we also showed that Rh(I) complexes of these phosphacyclohexadienyl ligands behave as efficient hydroformylation catalysts [15].
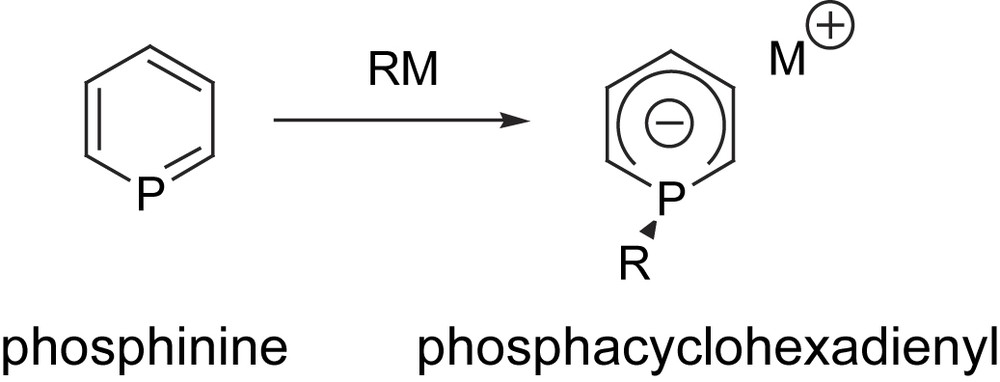
Reactivity of phosphinines towards nucleophilic reagents.
When the ring is substituted by two ancillary phosphinosulfide ligands, the phosphinine ring appeared to be sufficiently reactive to undergo a nucleophilic attack at phosphorus, leading to ligands of type 1 (Scheme 4). Though two mesomeric forms (A and B) can be proposed to rationalize the electronic structure of these ligands, DFT studies unambiguously showed that the negative charge is delocalized over the phosphorus ring (form A).
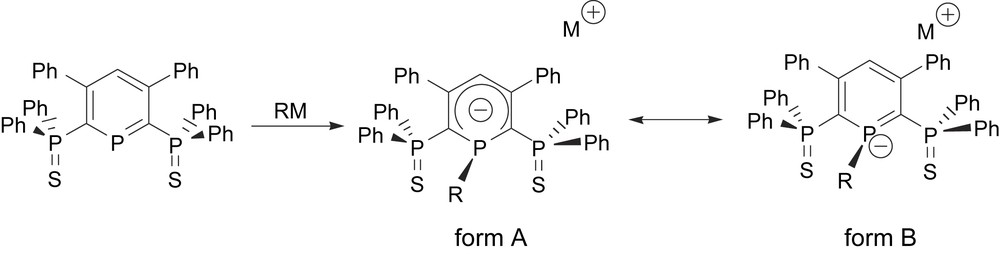
Synthesis of dianions 1 and possible mesomeric forms.
Contrary to classical phosphacyclohexadienyl anions, anions 1 favor σ coordination through the lone pair at phosphorus over π-coordination through the carbocyclic system. This preference can be rationalized taking into account the electronic perturbation induced by the presence of two sulfur atoms. Indeed, as can be seen in Scheme 5, the HOMO of phosphacyclohexadienyl anions is usually localized over the carbocylic system, justifying why coordination through the π-system is favored. On the other hand, in S–P–S anionic ligands, the presence of two lone pairs at sulfur induces a four-electron through-space destabilizing interaction with the lone pair at phosphorus. As a consequence, in these pincer ligands, the HOMO is the lone pair at phosphorus.
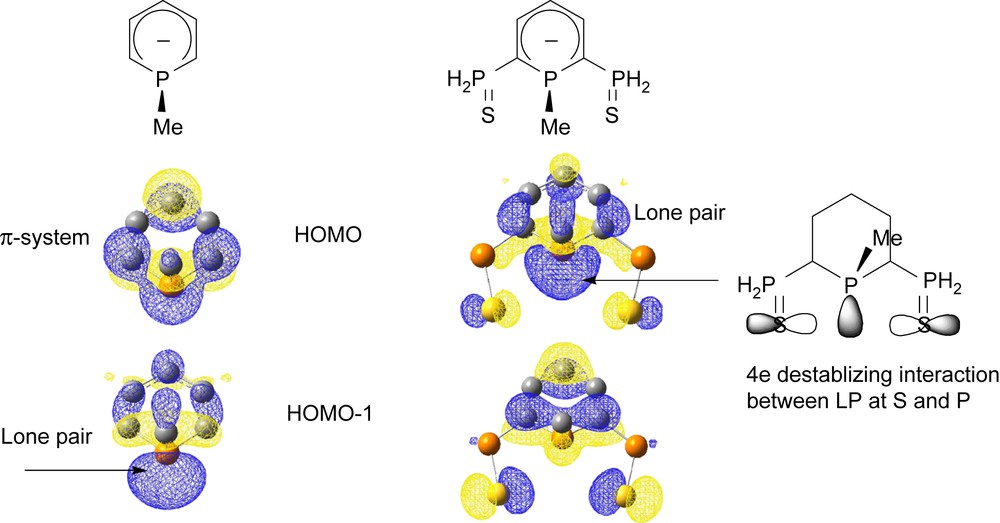
Two highest molecular orbitals of phosphacyclohexadienyl and anionic ligands 1 as given by DFT calculations.
These new systems, which behave as a 6-electron donor, display a very rich coordination chemistry, whose most-significant aspects will be summarized here. Palladium(II) complexes of ligands 1 are easily available through the reaction of the anion with precursors such as L2PdCl2 (L being a 2-electron donor ligand) (Scheme 6) [16]. Palladium chloride complexes such as 3 proved to be particularly robust and could be employed as very efficient catalysts in C–C (Suzuki) and C–B (Suzuki–Miyaura) coupling reactions. Coupling reactions of pinacolborane with iodoarenes yielded the corresponding boronic esters with excellent yields with low catalyst loading (TON between 5 × 103 and 10 × 103) [17].

Synthesis of palladium(II) complexes through a displacement process.
In the course of these studies, we also discovered another possible way to exploit the phosphinine ring in the elaboration of neutral tridentate S–P–S pincer ligands. 2,6-Bis(phosphinosulfide)phosphinines proved to be sufficiently reactive towards electron deficient alkynes to afford phosphabarrelenes such as 4 through a [4 + 2] cycloaddition process. These new S–P–S ligands easily coordinated Pd–Cl fragments and afford very efficient catalyst. Thus complex 4 catalyzes the coupling between Ph-B(OH)2 and bromoarenes with a TON of 9.5 × 104 (TOF [h−1] = 3958). Additionally the palladium complexes featuring P–S ligands (combination of a phosphinosulfide with a phosphinine anion or a phosphabarrelene) could also be successfully employed in the allylation of primary amines with allylic alcohols [18] (Scheme 7).
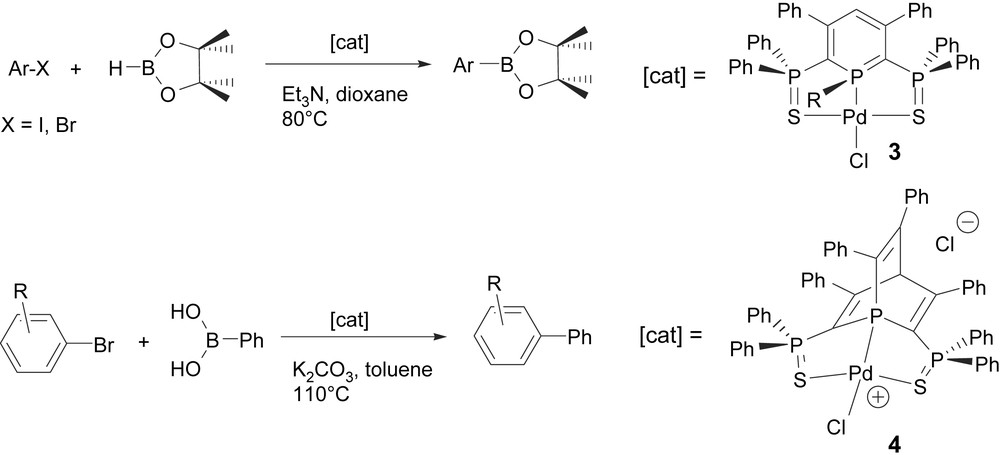
S–P–S palladium(II) complexes and their phosphabarrelene derivatives in catalyzed C–B and C–C bond coupling reactions.
Another striking feature of anionic ligands 1 was discovered in exploring the chemistry of Rh(I) complexes. The 16 VE species 5 was synthesized via the reaction of 1 with the classical Rh(I) precursor [Rh(COD)Cl]2 followed by addition of PPh3. This complex exhibits a high reactivity towards small molecules such as O2, CS2, SO2, MeI, and C2Cl6 [19]. A summary of the reactivity of these complexes is presented in Scheme 8.

Reactivity of complex 5.
As can be seen in Scheme 8, in all cases attack of the substrate occurred with complete regioselectivity on the upper side of the complex. This result was rationalized through DFT calculations on a model reaction with H2 [20]. It was demonstrated that the overall energy associated with this oxidative addition process results from different factors such as the distortion of the metal fragment, a singlet to triplet conversion, the energy of the dissociation of H2 and the energy associated with the formation of two new Rh–H bonds. Using a thermodynamic cycle, taking into account these different data, it was shown that preference for the syn attack essentially results from the ability of the [Rh(SPS)(PPh3)] fragment to form a triplet state in the appropriate geometry to react with the incoming substrate (Scheme 9). A view of the calculated spin density in the model complex [Rh(SPS)(PH3)] is also presented in Scheme 9 (in the model for SPS, all phenyl groups were replaced by H atoms).

Singlet to triplet conversion explaining the regioselectivity of attacks in Rh(I) complex 5.
Contrary to the Rh(I) complex for which the reaction is reversible, the Ir(I) counterpart 11 reacts with dihydrogen to form the corresponding dihydride Ir(III) derivative 13 which was structurally characterized (Scheme 10). A view of one molecule of 13 is presented in Fig. 1. A DFT study has shown that the oxidative addition of H2 onto the Ir derivative 11 is more exothermic (ΔG = −36.1 kcal/mol) than the addition onto the Rh complex (ΔG = −14.9 kcal/mol) which is in equilibrium with the dihydride complex 12. Though the singlet to triplet conversion requires a weaker activation barrier in the case of the rhodium complex, the gain in stabilization energy provided by the formation of two strong Ir–H bond (−171.2 kcal/mol) is the determinant factor (−145 kcal/mol for the Rh complex).
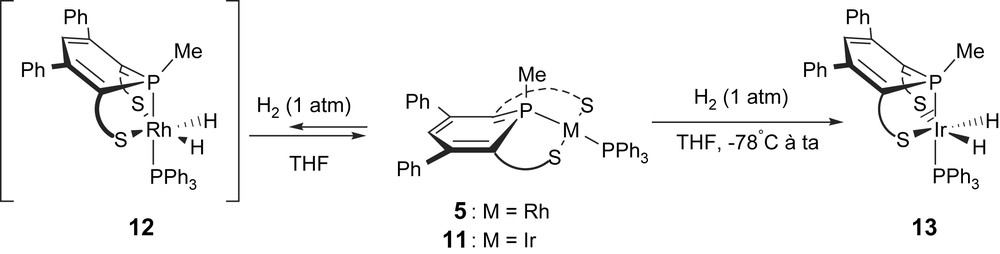
Reactivity of complexes 5 and 11 towards H2.
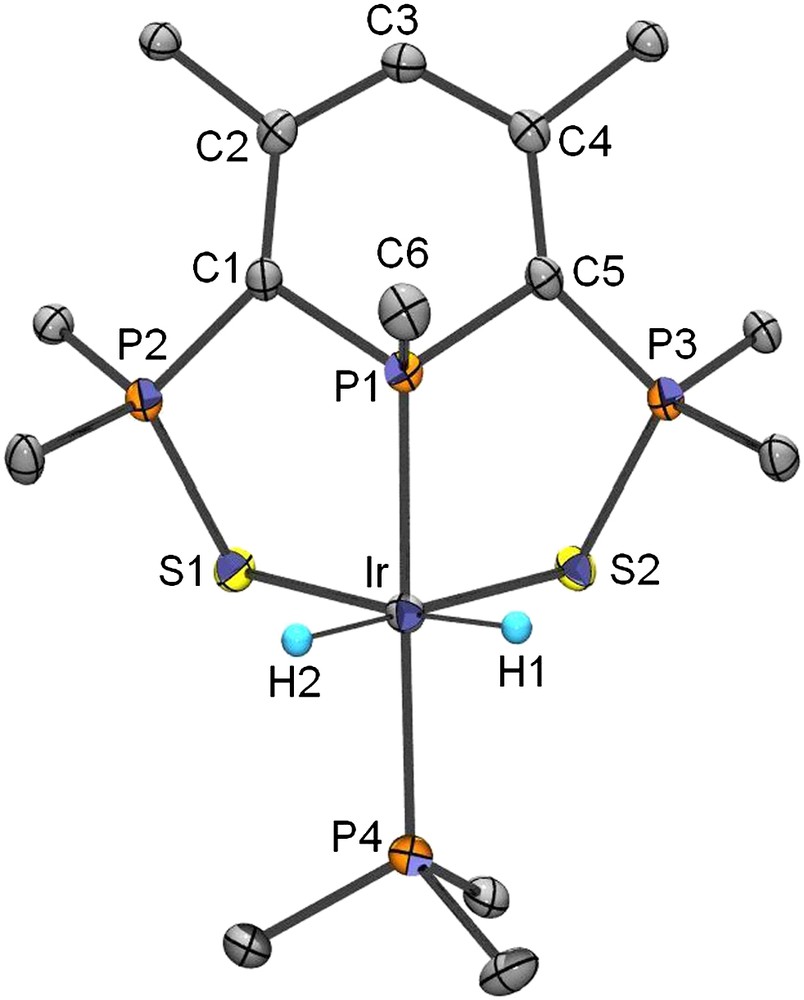
View of one molecule of complex 13 in the solid state. Copyright American Chemical Society 2006.
Anions of type 1 can also allow the stabilization of paramagnetic Rh(II) complexes [21]. In a first step, the homoleptic Rh(III) complex featuring two SPS ligands was obtained by reacting two equivalents of the anion with the [RhCl3(THT)3] precursor. Interestingly, complex 14 showed a reversible reduction wave by electrochemistry to form the 19 VE Rh(II) complex 15. The reduction of 14 was then achieved chemically using Zn as a mild reducing agent. Though complex 15 could not be characterized by X-ray techniques, complete EPR and DFT studies were performed. Combination of the calculations and the experimental data allowed us to conclude that the odd electron is localized in an MO which results from the antibonding combination of the metal AO with two lone pairs at sulfur atoms of different ligands. A view of the SOMO of 15 is presented in Scheme 11.
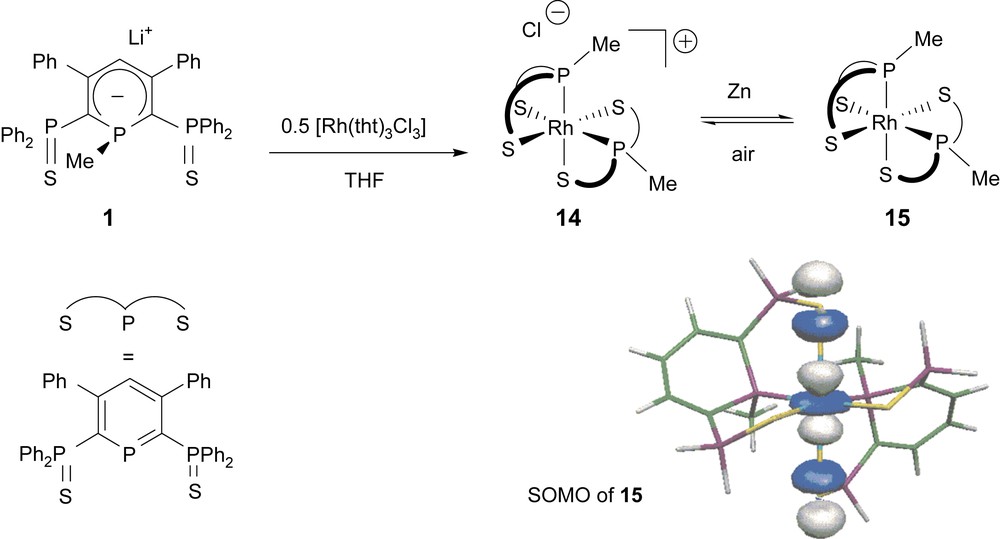
Synthesis and reduction of the homoleptic Rh(III) complex 14 into the paramagnetic 19 VE complex 15. A view of the SOMO of 15 is also presented.
The chemistry of these SPS anionic derivatives is not limited to group 9 and group 10 metal complexes and various other complexes were synthesized with different transition metals: Au(I), Cu(I), Ni(II), Pt(II), Fe(II), Ru(II), Mn(I) and Re(I) and even actinides such as U(IV) [22,23].
2.2 Dianionic S–C–S pincer ligands
So far, as stated in Section 1, only monoanionic SCS pincer ligands were known. Following a similar strategy that was already employed by Cavell et al. and Ong and Stephan for the synthesis of bis-(iminophosphorane)methanediide ligands [24,25], sulfide derivatives of the dppm ligand (dppm = bis(diphenylphosphino)methane) could be easily deprotonated twice to afford the corresponding dianionic ligand 2. This metallation process easily takes places in toluene at room temperature and yields dianion 2 in quantitative yield as a highly moisture sensitive yellow powder. In the solid state, dianion 2 adopts a dimeric structure which bears some resemblance with that of the bis-(iminophosphorane) analogue.
The most significant feature of these dianions is their reactivity towards transition metals since they can potentially act as precursor of transition metal carbene complexes through ligand displacement reactions. In a first series of experiments we explored the reactivity of 2 towards Pd(II) precursors such as [Pd(PPh3)2Cl2] (Scheme 12) [26]. Upon reaction, the two chloride ligands are displaced and the new complex 16 is formed. The X-ray crystal structure of complex 16 is presented in Fig. 2.
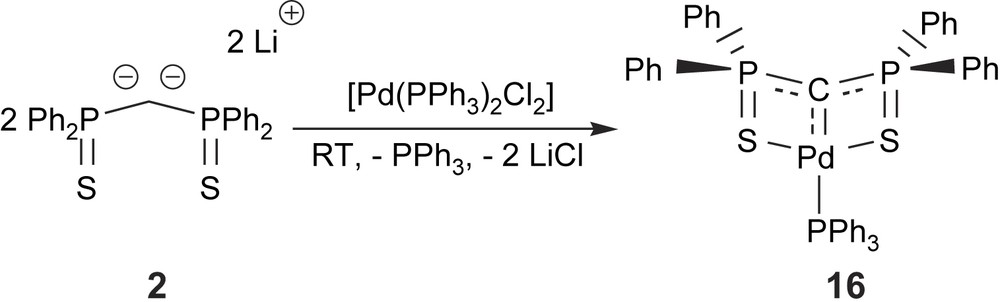
Synthesis of the palladium complex 16 through reaction of dianion 2 with [Pd(PPh3)2Cl2].
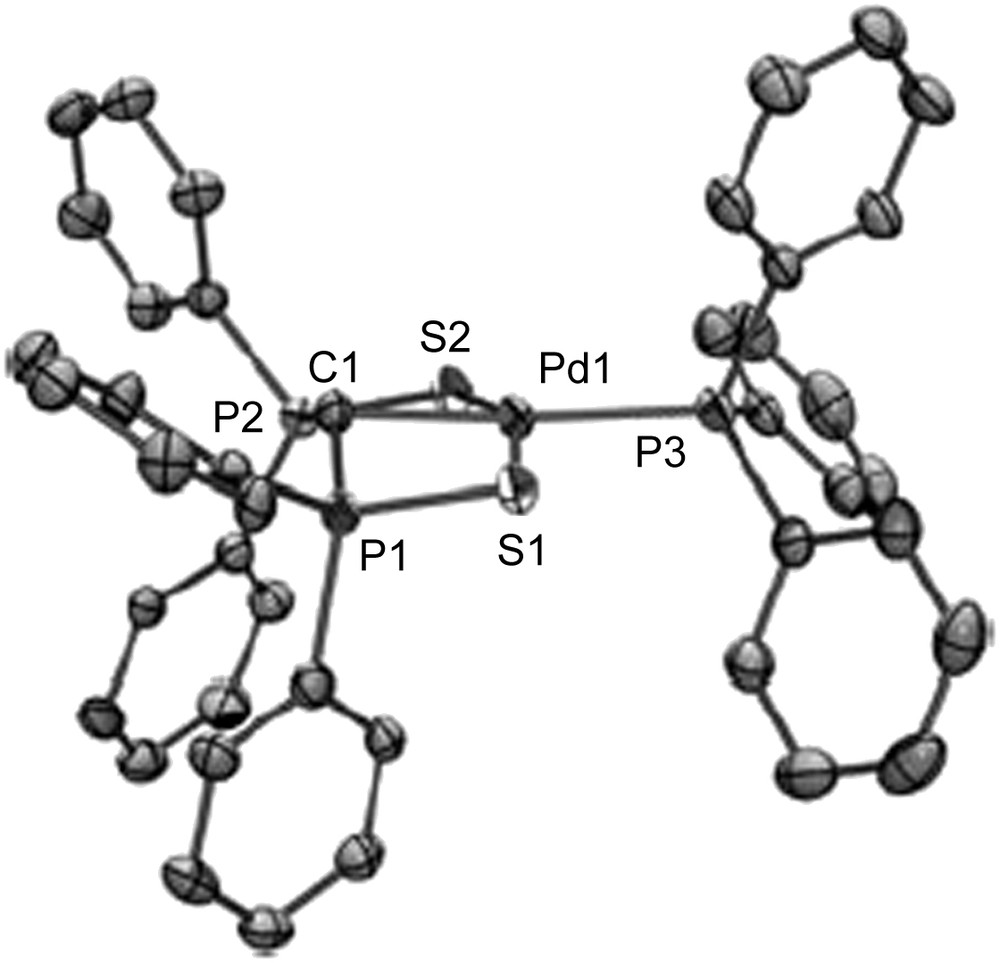
View of one molecule of complex 16 in the solid state. Copyright Wiley 2004.
As shown, the X-ray structure of complex 16 presents a central tricoordinate carbon atom as expected. Unusual features were found. First, a long Pd–C1 bond (2.113(2)) Å ruled out a ‘true’ double bond. Second, the metal center seems to be located in a quasi-perpendicular plane to the carbene fragment (angle of 102.0°). The particular electronic structure of this complex was rationalized through DFT calculations. The HOMO of the complex, which is shown in Scheme 13, consists of a π type antibonding interaction between the nσ orbital of the carbene fragment and the dxz orbital of the metal center. Overall, as both the π bonding and antibonding orbitals are filled, there is no π bonding character between the Pd and C centers, which corroborates the long Pd–C bond.
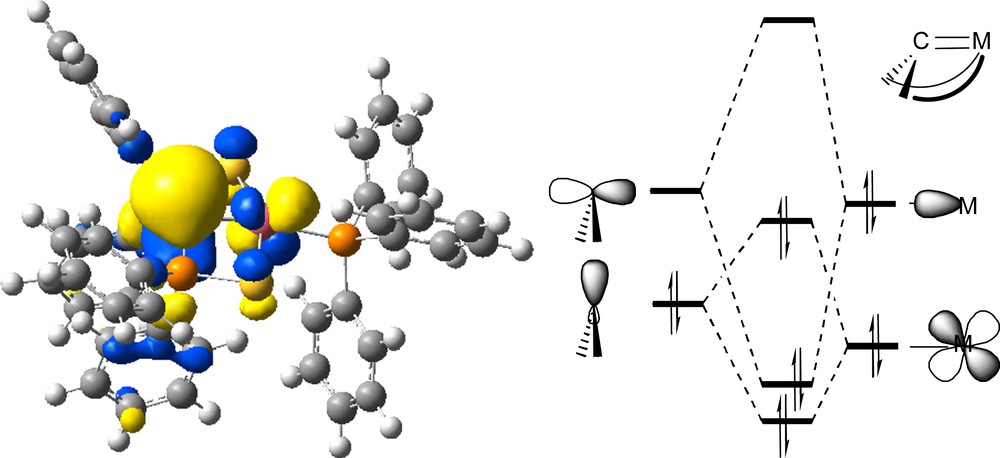
HOMO of complex 16 and simplified MO diagram. Copyright Wiley 2004.
As can be expected from this electronic situation, complex 16 behaves as a nucleophile and it indeed reacts with strong electrophiles such as MeI to afford the cationic complex 17 which features a single bond between the carbon and palladium centers (Scheme 14).

Reaction of complex 16 with methyliodide.
Following a similar strategy, Ru(II) complexes such as 18 were also synthesized [27] (Fig. 3). The X-ray structure of 18 showed a more typical coordination of the carbene fragment with the metal center in the plane of the carbene (Scheme 15). However, here again the C–metal bond length appears to be quite long (Ru–C of 2.053(2) Å), suggesting a very weak double-bond character. This hypothesis was confirmed by DFT calculations which indicate a Wiberg bond index of 0.67 for the Ru–C bond. Moreover, the LUMO of this complex is an antibonding π∗ orbital between the nσ orbital of the carbene fragment with the dxz orbital of the metal center. Contrary to its Pd(II) counterpart, complex 18 proved to be very robust, neither reacting with nucleophiles nor with electrophiles, even under drastic conditions.
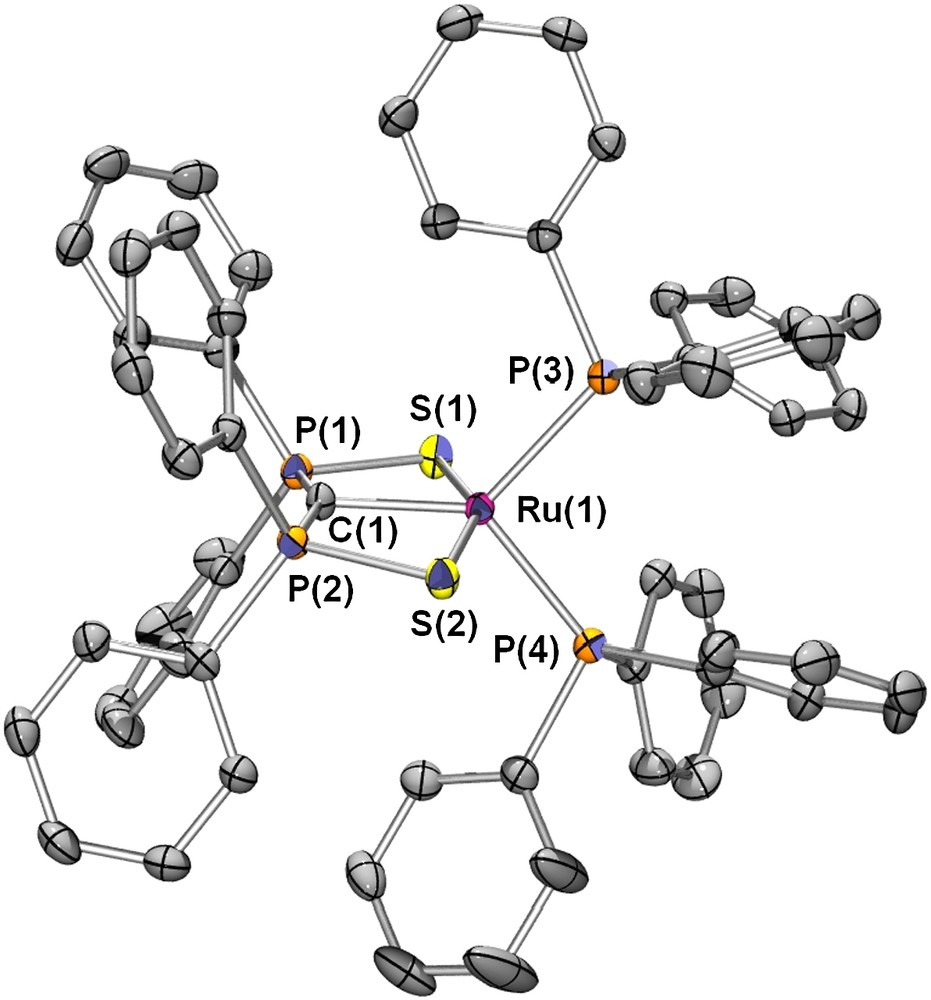
A view of one molecule of complex 18 in the solid state. Copyright American Chemical Society 2005.

Synthesis of the Ru(II) complex 18.
Dianions 2 also proved to be remarkable ligands for lanthanides and, very recently, the strategy employed for the synthesis of Pd(II) and Ru(II) derivatives was successfully extended to the synthesis of rare lanthanide alkylidene complexes (Ln = Sm, Tm) (Scheme 16) [28,29].

Syntheses of dimeric 19, 20 and monomeric homoleptic complexes of Sm and Tm 21 and 22.
The X-ray crystal structures of the iodide bridged dimeric Sm 19 and Tm 20 complexes were recorded. In both structures the geometry at carbon is planar (sum of the angles of 357.8–359.4°), showing the donation of both lone pairs of the dianionic fragment to the Ln center. Unexpectedly, the homoleptic complex 22 adopts two different solid-state structures depending on the temperature, and a low-temperature (177 K) phase transition in the crystal was observed. Two significantly different structures were thus recorded (at 150 K and 230 K). In the low-temperature form (22a), the two carbenic moieties are geometrically different. In one fragment, the carbon is tetrahedral (Σangles = 332°) and the bond distance of 2.423(9) Å falls in the range of single Tm–C bonds. In the other fragment, it is planar and the bond length is shorter (2.378(9) Å). Therefore one can conclude that the overall charge is located on the first carbon atom, which bears a significant sp3 character, whereas the second carbon center behaves as a four-electron donor. In the higher temperature form (22b), the two fragments become identical, the bond distance averages between single and double bond length, and the geometry at the carbon atoms is nearly planar. These data point to a delocalization of the anionic charge over the two carbons and the thulium centers, thereby indicating that a π interaction can develop between these atoms (Scheme 17).

Phase transition in the solid state for complex 22.
As can be expected, both complexes are highly reactive towards electrophiles. Reaction with ketones and aldehydes is a convenient route to form 1,1-bis(diphenylphosphinosulfide) olefin derivatives through a ‘Wittig-like’ process. Monitoring of the process by 31P NMR spectroscopy attested the presence of several intermediates. Fortunately, one of these intermediates, the open metalla-oxetane complex 23 was isolated and structurally characterized, and one molecule is presented in Scheme 18. Metalla-oxetanes have been proposed as intermediates in the reaction of early transition metal carbene complexes with carbonyl derivatives, but few related species have been isolated so far. In the structure, new C–C and O–Sm bonds are formed, while the Sm–alkylidene bond is cleaved. When redissolved in toluene, this complex evolved to the alkene 24 and samarium oxide derivatives, proving this open metalla-oxetane to be an intermediate in the transformation.
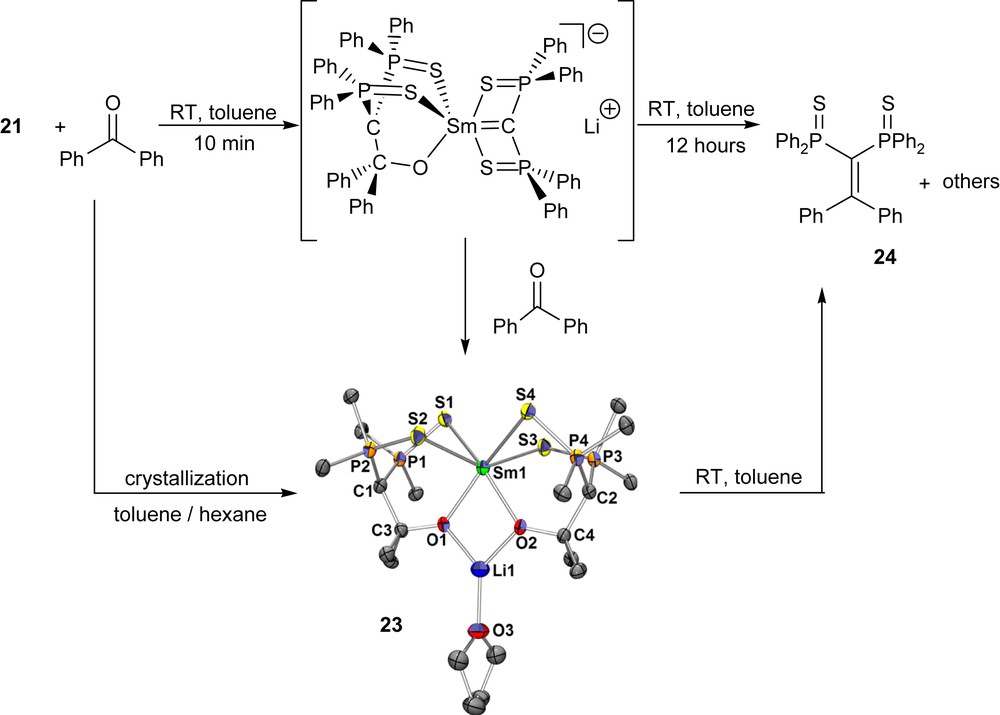
Reaction of the Sm complex 21 with benzophenone. X-ray crystal structure of the samara-oxetane 23 in the solid state. Copyright American Chemical Society 2006.
3 Conclusion
Over the last few years, we have been involved in the development of new S–X–S pincer ligands featuring phosphinosulfide pendent arms, and either phosphorus or carbon central donating moiety. The monoanionic ligand 1 was used to stabilize both electron-rich and electron-poor metal centers: from Cu(I), Au(I) to U(IV). In terms of applications in catalysis, some Pd complexes bearing this ligand not only have shown promising activities, but also proved very robust towards moisture and air. Small molecule activation (CS2, SO2, C2Cl6, MeI…) was achieved with Rh complex in a regioselective manner, which was corroborated by DFT calculations. Most interestingly, this ligand proved quite flexible, being able to accommodate various geometries at the metal center. Finally, electrochemical studies showed ligand 1 to be comparable with the highly electron donating Cp∗ ligand.
Dianionic ligand 2 was obtained quantitatively from the bis-sulfide of the commercially available dppm ligand, which opened the way to the synthesis of new carbene complexes of Pd and Ru centers. DFT calculations have allowed describing in depth the peculiar electronic situation found in these complexes. Alkylidene complexes of the Ln centers Sm and Tm have also been synthesized. Reactivity of these complexes towards ketones has led to the isolation of a rare open metalla-oxetane species, often proposed as intermediate in the reaction of alkylidene complexes of early transition metals with ketones. X-ray studies of the Tm complexes have proved a double bond character for the CTm bond. Thus, in these complexes, the carbon acts as both a σ and a π donor to the lanthanide center.
Acknowledgements
The authors thank the CNRS and the ‘École polytechnique’, for supporting these studies. MD thanks the DGA for a fellowship. IDRISS is gratefully acknowledged for the allowance of computer time (project No. 061616).