

1 Introduction
Dioxygen activation by copper proteins is a very important biological process [1,2]. Oxyhemocyanin, the oxygen transport protein in arthropods and mollusks, contains a dinuclear copper(II) active site that is bridged symmetrically by peroxide [3,4]. Monooxygenase tyrosinase, which activates dioxygen for the oxygenation of phenols to o-diphenols, has been shown to have an active site similar to that of oxyhemocyanin [5]. A large number of dinuclear copper complexes have been reported as model compounds for the active sites in these copper proteins, and they have exhibited effective catalytic activities for O2- and H2O2-based oxidation reactions in homogeneous systems [6–17]. Along with the efforts toward the synthesis of biomimetic copper complexes, various types of active copper–oxygen intermediates have been studied in order to investigate the oxygen-transfer mechanisms of copper proteins [13,14,8,15,17].
Although the implementation of biomimetic dinuclear copper sites in heterogeneous oxidation systems is an intriguing objective, the studies on the heterogeneous catalytic activities of biomimetic single copper sites and the nature of oxidizing species continue to be particularly challenging, given the inherent difficulties in characterizing active sites under the reaction conditions. Despite the existence of considerable efforts with regard to developing effective copper-containing heterogeneous oxidation catalysts, e.g., Cu2+–phthalocyanine incorporated inside Y faujasite and MCM-41 [18], Cu–HMS [19], Cu2+-substituted MCM-41 [20], zeolite-encapsulated Cu2+–salens [21], Cu2+/X and Y zeolites [22], Cu(OH)2/SiO2 [23], and cis-bisglycianato copper(II)/1,6-naphthalenediol oligomer [24], no reports exist on the activities of single copper sites in heterogeneous oxidation systems and the nature of oxidizing intermediates.
Microporous organic–inorganic hybrid coordination polymers have attracted considerable attention because they exhibit the molecular adsorption of not only gas molecules but also organic molecules [25–40]. Among these efficient porous materials, we have particularly studied on the synthesis of transition-metal carboxylate coordination polymers, e.g., dicarboxylates of copper [25,26,34–37], molybdenum [25], and ruthenium [38,39], which are capable of occluding large amounts of gases such as N2, Ar, O2, CH4, and Xe. On these polymer complexes, uniform linear micropores are constructed by the stacking or bonding of two-dimensional lattices of dinuclear transition-metal carboxylates; this feature along with the presence of single-site mono and dinuclear transition-metal centers in uniform linear pores are important characteristics that enable these metal carboxylates to be used as heterogeneous catalysts, as shown in Fig. 1.
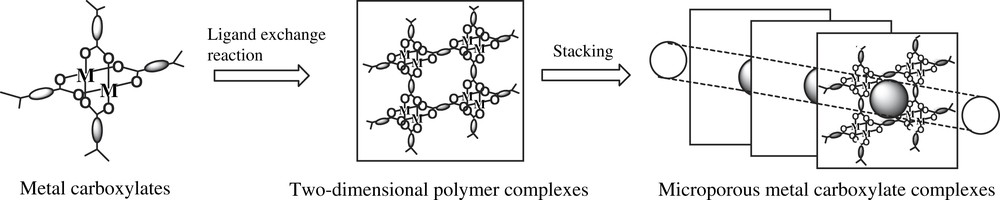
Schematic of the synthesis of microporous metal carboxylate complexes.
In recent years, we have studied the heterogeneous hydrogenation reactions catalyzed by rhodium and ruthenium carboxylate complexes and the heterogeneous oxidation reactions catalyzed by copper and ruthenium carboxylate complexes, and observed the unique catalytic performances [41–48]. Here, we introduce two examples of the catalytic performances for heterogeneous oxidation reactions. One is the H2O2-based oxidation of alcohols catalyzed by a microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate [Cu2II,II(OOCC6H10COO)2]·H2O (1) [47]. During this study, a microporous peroxo copper(II) intermediate, H2[Cu2II,II(OOCC6H10COO)2(O2)]·H2O (2), was also isolated and characterized by elemental analysis, FT-IR, TG/DTA, magnetic susceptibility, electron paramagnetic resonance (EPR), X-ray powder diffraction (XRPD), resonance Raman, diffuse reflectance (DR) UV–vis, BET surface area, pore size, and gas occlusion measurements. Another is the O2-based oxidation of alcohols catalyzed by a microporous ruthenium(II,III) complex containing porphyrin, [RuII,III2(H2TCPP)]BF4 (3) (H2TCPP = 4,4′,4″,4‴-(21H,23H-porphine-5,10,15,20-tetrayl)tetrakis benzoic acid) [48]. Complex 3 exhibited effective catalytic activities for the oxidation of primary aliphatic alcohols at room temperature.
2 H2O2-based oxidation catalyzed by copper(II) trans-1,4-cyclohexanedicarboxylate [Cu2II,II(OOCC6H10COO)2]·H2O (1) [47]
2.1 Oxidation of alcohols with H2O2 catalyzed by complex 1
Complex 1 was used as a heterogeneous catalyst for the oxidation of 2-propanol, cyclohexanol, benzyl alcohol, 1-octanol, and 2-octanol with H2O2 at 293 K, as shown in Table 1. The catalytic activities (turnover frequencies, conversions, and selectivities of the oxidative products) and the catalytic behaviors were influenced by the concentrations of H2O2, i.e., for all oxidation reactions with varying concentrations (less than 46-fold and more than 69-fold excess) of H2O2, the color of complex 1 changed from blue to green and brown, respectively. The green and brown colors persisted for 2 days and 3 weeks, respectively, and then disappeared.
Catalytic activities for oxidation of alcohols catalyzed by complexes 1 and 3 with H2O2 and O2
| Catalysts | Oxidants (mmol) | Alcohol (mmol) | TONb (reaction time/h) | TOF (s−1)c | Product (selectivity/%) |
| 1a | H2O2 (9.7) | 2-Propanol (13.1) | 11 (1) | 1.6 × 10−3 | Acetone (>99) |
| H2O2 (48.5) | 2-Propanol (13.1) | 35 (3) | 5.8 × 10−3 | Acetone (>99) | |
| H2O2 (19.4) | Cyclohexanol (9.5) | 6.5 (168) | 1.1 × 10−4 | Cyclohexanone (>99) | |
| H2O2 (48.5) | Cyclohexanol (9.5) | 9.1 (168) | 1.6 × 10−4 | Cyclohexanone (>99) | |
| H2O2 (9.7) | Benzyl alcohol (9.7) | 7.8 (144) | 7.0 × 10−4 | Benzaldehyde (>99) | |
| H2O2 (48.5) | Benzyl alcohol (9.7) | 12 (144) | 1.5 × 10−3 | Benzaldehyde (>99) | |
| H2O2 (9.7) | 1-Octanol (6.4) | 3.4 (168) | 1.1 × 10−4 | Octyl aldehyde (>99) | |
| H2O2 (48.5) | 1-Octanol (6.4) | 4.1 (168) | 1.6 × 10−4 | Octyl acldehyde (>99) | |
| O2 | Cyclohexanol (9.5) | n.r.d (168) | – | – | |
| 3e | O2 | 1-Octanol (63) | 12 (24) | 1.4 × 10−4f | Octyl aldehyde (>99) |
| Air | 1-Octanol (63) | 1.1 (24) | 1.3 × 10−5f | Octyl aldehyde (>99) | |
| O2 | 2-Octanol (63) | n.r.d (24) | – | – | |
| O2 | 1-Propanol (131)g | 1.4 (5) | 7.9 × 10−5h | Propylaldehyde (>99) | |
| O2 | 2-Propanol (134)g | 0.2 (5) | 1.1 × 10−5h | Acetone (>99) | |
| O2 | Benzyl alcohol (97) | 21 (24) | 2.4 × 10−4f | Benzaldehyde (>95) | |
| Air | Benzyl alcohol (97) | 11 (24) | 1.3 × 10−4f | Benzaldehyde (>95) | |
| O2 | Cyclohexanol (47.3) | 1.9 (24) | 2.2 × 10−5f | Cyclohexanone (>99) |
a Reaction conditions: catalyst 206 μmol, substrate 6.4–13.1 mmol, 30% H2O2 9.7–48.5 mol (23- to 113-fold excess), CH3CN 10 mL, reaction temperature 293 K.
b Turnover number (TON) = [mol of products]/[mol of catalyst].
c TOF = turnover number (TON) s−1 after 1 h.
d n.r. = no reaction.
e Reaction conditions: catalyst 18.6 μmol, substrate 47.3–134 mmol, P(O2) = 1 atm, reaction temperature 298 K, reaction time 24 h.
f After 24 h.
g Reaction time 5 h.
h After 5 h.
In the case of the oxidation of 2-propanol with two different H2O2 concentrations (23- and 113-fold excess of copper atom), the conversions leading to the formation of acetone were observed to be highly selective (100%) for 17% (after 1 h) and 56% (after 3 h) conversion of 2-propanol, respectively, and they occurred with turnover frequencies (TOF = (mol of products)/(mol of complex) s−1) of 1.6 × 10−3 s−1 and 5.8 × 10−3 s−1 (calculated after a reaction time of 1 h), respectively. No induction period was observed for reactions with either H2O2 concentrations (≥10 min). When the green and brown colors disappeared, the reaction stopped, thereby suggesting that the green- and brown-colored were active intermediates for oxidation catalysis. Complex 1 was also observed to catalyze the oxidation of cyclohexanol, benzyl alcohol, 1-octanol, and 2-octanol to the corresponding carbonyl compounds with >99% selectivities. Although complex 1 can adsorb relatively large organic molecules, e.g., o-xylene (0.3 mol/mol of copper), m-xylene (0.4 mol/mol of copper), and p-xylene (0.3 mol/mol of copper), the observed TOFs were lower than those of 2-propanol oxidation with 23- and 113-fold excess H2O2 because it might be easier for 2-propanol, in comparison to cyclohexanol, to move into the micropore and/or for acetone, in comparison to cyclohexanone, to move out from the micropore.
In order to determine whether the active species leached into the solution during a typical catalytic reaction, the mixture of complex 1 and 23-fold excess of H2O2 in acetonitrile was stirred for 3 days at 293 K. The mixture was then filtered through a Büchner funnel (Whatman No. 2) and the filtrate was treated with 2-propanol at 293 K. The samples of this reaction mixture taken after 1, 2, and 3 h contained no oxidation products, thus indicating that the observed catalysis was heterogeneous. Using the solid after filtration as a catalyst, the oxidation of 2-propanol with 23-fold excess of H2O2 was also carried out at 293 K. The TOF of 1.2 × 10−3 s−1 was similar to that for first-time use (1.6 × 10−3 s−1). In addition, no copper species was detected in the filtrate (<0.02 ppm), thus indicating that the leaching of a catalytically active copper species in the solution was negligible. Furthermore, after oxidation catalysis, complex 1 was characterized by FT-IR, elemental analysis, DR UV–vis, XRPD, BET surface area, and pore size distribution measurements. The results are as follows: FT-IR spectrum (cm−1): 1594s, 1511w, 1423s, 1373w, 1332w, 1297m, 1222w, 1045w, 929w, 784m, 767m, 727w, 526m. XRPD pattern (2θ/°): 8.56, 9.20, 10.88, 14.13, 17.24, 18.55, 19.25, 20.10, 21.90, 22.66, 23.92, 25.22, 26.08, 28.63 with relative intensities of 100, 75, 9, 4, 23, 10, 13, 10, 2, 6, 7, 6, 7, 5, respectively, in 2°–30° angles. BET surface area: 301.2 m2/g (pore size diameter was 5.1 Å). DR UV–vis spectrum: 261, 375, and 660 nm. Elemental analysis: found: C, 38.33; H, 4.97%. Calcd for C16H24O10Cu2 = [Cu2(C8H10O4)2]·2H2O: C, 38.17; H, 4.80%. These results were consistent with those for as-prepared complex 1. Thus, complex 1 was stable and reusable under the present oxidation conditions. On the contrary, the organic carboxylate ligands of other microporous dinuclear copper(II) carboxylates, e.g., copper(II) terephthalate and copper(II) fumarate, were decomposed to H2O2.
2.2 Synthesis, structure, and compositional characterization of microporous peroxo copper(II) complex, H2[Cu2II, II(OOCC6H10COO)2(O2)]·H2O (2)
As mentioned above, green- and brown-colored intermediates were observed by adding different concentrations (less than 46-fold and more than 69-fold excess, respectively) of H2O2 in the studies of catalytic oxidations. Here, the green-colored intermediate was synthesized and isolated according to Eq. (1).
| [Cu2II,II(OOCC6H10COO)2] (1) + H2O2 → [Cu2II,II(OOCC6H10COO)2(O2)]2− (2) + 2H+ | (1) |
The solid green-colored oxidizing copper(II) intermediate, H2[Cu2II,II(OOCC6H10COO)2(O2)]·H2O (2), was obtained in 87.8% (94.5 mg scale) yield. This intermediate was prepared by the reaction of complex 1 in acetonitrile suspension with 20-fold excess of H2O2. After the reaction, the product was washed with excess amounts of acetonitrile and methanol and then dried under vacuum for 2 h at 298 K. It was noticed that the pH of complex 1 changed from neutral to acidic by H2O2 addition, thus suggesting that the Cu–OO–Cu species was formed by a heterolytic H–OOH bond cleavage in complex 1. According to the elemental analyses results, pure complex 2 could not be obtained with less than 20-fold excess of H2O2, which might be due to the hydrophobicity in the micropore of complex 1.
The XRPD patterns of complexes 1 and 2 were recorded, and the crystal structures were determined and refined by the Rietveld method. Fig. 2 shows the perspective views of stacking complexes 1 and 2 along the a and c axes, respectively. The molecular structure of 1 was isomorphous to several [CuII2(μ-carboxylate)] complexes, the crystal structures of which have been previously reported [49]. The intermolecular coordination of an oxygen atom of carboxylate ligand to the vacant site of copper atom caused the stacking of the two-dimensional [Cu2(O2CC6H10CO2)] layers to form three-dimensional lattice structures. In contrast, the X-ray structure analysis of complex 2 revealed that the microporous structure of complex 2 was constructed by the bridging of μ-1,2-trans Cu–OO–Cu species between two-dimensional [Cu2(O2CC6H10CO2)] layers. The Cu(1)–O(9) and Cu(2)′–O(10) distances were 1.877(2) Å and 1.879(1) Å, respectively, which were quite similar to those of the μ-1,2-trans-copper(II) peroxo complex determined by X-ray crystal analysis (1.852 Å) [50] and slightly smaller than those of the reported side-on copper(II) peroxo complexes (1.892–1.941 Å) [15]; however, the distances were considerably greater than those of copper(III) complexes (ca. 1.80 Å) [16,51]. The intermolecular Cu(1)⋯Cu(2)′ separation stretched to 4.572(4) Å, which was much greater than that of the Cu(1)⋯Cu(2)′ separation (2.992(6) Å) for complex 1 because of the insertion of an interperoxide ligand between the two-dimensional layers. The size of the micropore structure for complex 1 was slightly altered by the bridging of μ-1,2-trans Cu–OO–Cu bonding between the two-dimensional [Cu2(O2CC6H10CO2)] layers, and complexes 1 and 2 exhibited a high BET surface area of 393.9 m2/g and 328.4 m2/g, respectively, and a high micropore porosity of 4.9 Å, as shown in Fig. 3. In addition, the absorption of nitrogen occurred at temperatures below 200 K for both complexes, and the maximum amounts of occluded N2 gas for 1 and 2 were 1.27 and 1.09 mol/mol of Cu at 77.5 K, respectively (Fig. 4). These results suggested that the micropore for 1 was retained after the formation of Cu–OO–Cu species between the layers; this was consistent with the results analyzed by the Rietveld method. To our knowledge, complex 2 is the first example of a microporous copper(II) peroxo complex.

Unit-cell packing of complex 1 viewed along the (a) a and (b) c axes and that of complex 2 viewed along the (a)′ a and (b)′ c axes.
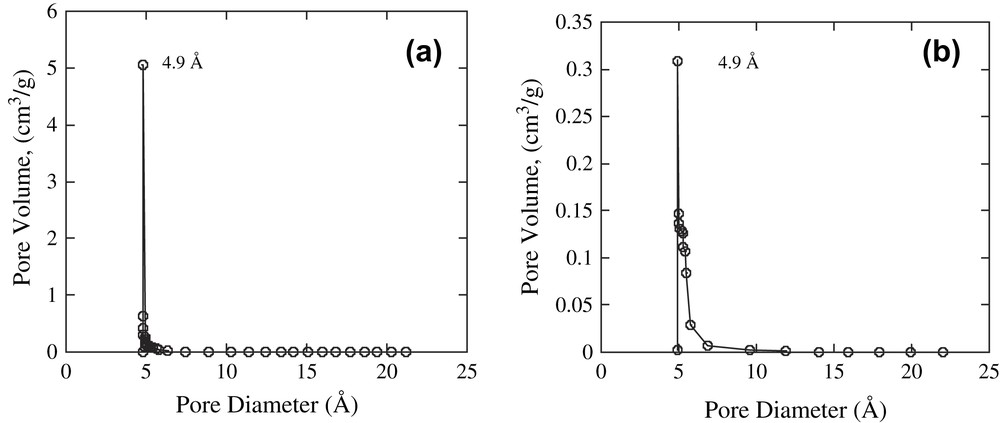
Pore size distribution of (a) complex 1 and (b) complex 2.
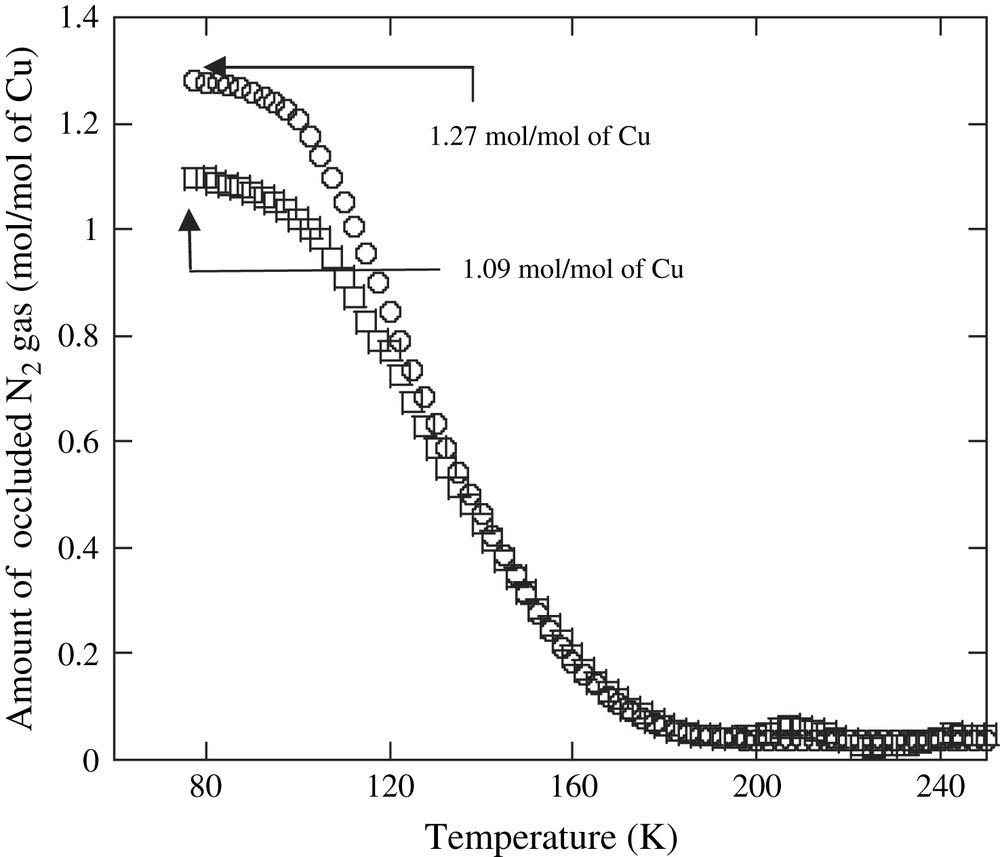
Temperature dependence of the amount of occluded N2 gas for complex 1 (○) and complex 2 (□).
Other characterization results obtained by elemental analysis, TG/DTA, DR UV–vis, ESR, resonance Raman spectra, and magnetic susceptibility measurements of complex 2 were consistent with that of the Rietveld analysis. The C and H elemental analysis data was as follows: found: C, 37.00%; H, 4.66%. Calcd. for H2[Cu2(C8H10O4)2(O2)]·H2O = C16H24O11Cu2: C, 37.07%; H, 4.47%. TG/DTA measurement for complex 2 performed under atmospheric conditions showed a weight loss of 5.35% with an exothermic peak at 405.8 K, which corresponded to the decomposition of the peroxo ligand (7.0%). The thermal stability of the peroxo copper intermediate, 2, was considerably higher than those of the reported copper peroxo complexes [6,11–14,8,15]. Above 473 K, the decomposition of the organic ligands in complexes 1 and 2 began at around 513 K with an exothermic peak at 511.0 and 515.7 K, respectively.
Resonance Raman spectra of complexes 1 and 2 in the range of 700–850 cm−1 are shown in Fig. 5. A new peak at 805 cm−1 was observed for complex 2. This peak was attributed to the O–O stretch of the bridging peroxide on the basis of its frequency. The 805 cm−1 O–O stretch was in the range of 800–840 cm−1, which was also observed in the case of μ-1,2-trans CuII–OO–CuII complexes [6,11,52,53]. Further, it was in the upper range of the O–O stretching frequencies observed in side-on μ-η2:η2 CuII–OO–CuII (741–760 cm−1) [6,11] and CuIII(μ2-O)2CuIII (590–616 cm−1) complexes [51,16] and in the lower range of the O–O stretching frequencies observed in copper(II) hydroperoxide species (892 cm−1) [12].

Resonance Raman spectra, using an excitation wavelength of 532 nm, of complex 1 (– – –) and complex 2 (—) in the range of 700–850 cm−1.
The DR UV–vis spectrum of 1 showed two absorption bands at 384 and 655 nm due to ligand-to-metal charge transfer (CT) and d–d transition bands, respectively, as shown in Fig. 6 [23,54]. The DR UV–vis spectrum of 2 also exhibited a CT band at 385 nm and a d–d band at 665 nm. The UV–vis spectra of μ-1,2-trans CuII–OO–CuII complexes, [CuII2(bpman)(O2)]2+ (bpman = 2,7-bis[bis(2-pyridylmethyl)amiomethyl]-1,8-naphthyridine) [52] and [{LCuII}2(O2)]2+ (L = tris[(2-pyridyl)methyl]amine) [50] showed two new bands at 505 and 620 (br) nm and 525 and 590 (br) nm, respectively, due to the coordination of the peroxo ligand to the dicopper sites. In contrast, complex 2 showed only one broad band at around 665 nm, owing to the presence of an intense band of copper-carboxylate fragments. However, the position at 665 nm was shifted to longer wavelength, thereby suggesting the coordination of H2O2 to the Cu atom.
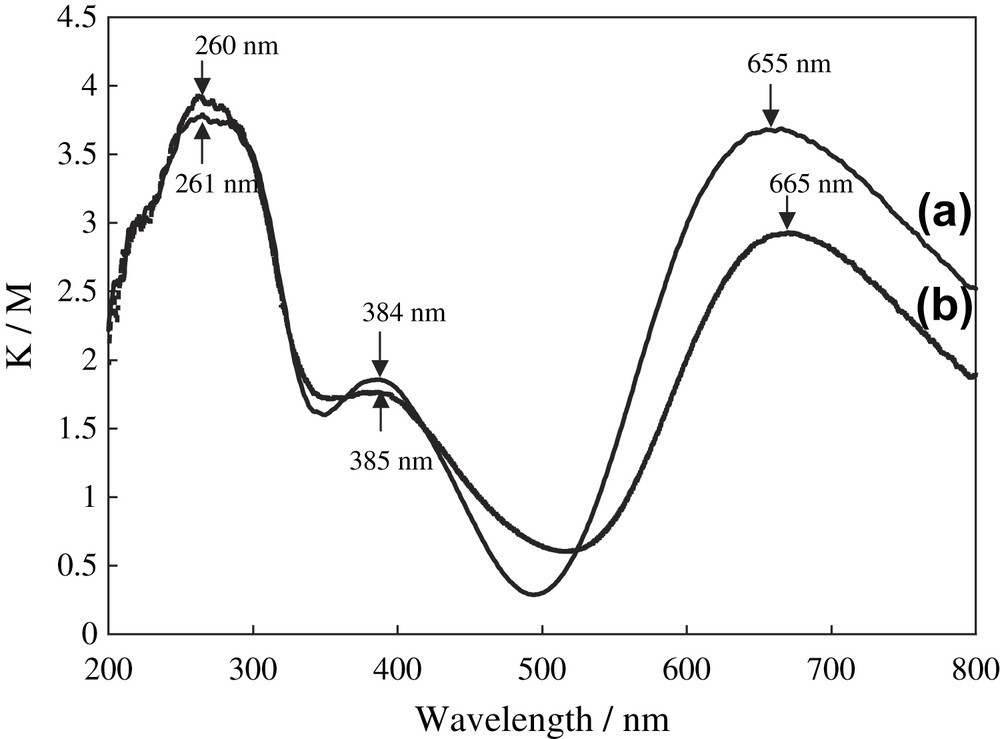
Diffuse reflectance UV–vis spectra of (a) complex 1 and (b) complex 2.
Temperature dependence of magnetic susceptibility of complex 1 is shown in Fig. 7a. It is indicated that the effective magnetic moment μeff was 1.29 B.M. at 300 K, thereby suggesting the presence of antiferromagnetic coupling between two copper(II) atoms in a copper center. Based on the best fit of the χA values to the Bleaney–Bowers equation [55], shown below (Eq. (2)), the magnetic parameters can be estimated as g = 1.96, −2J = 320.4 cm−1 emu/mol, and P = 1.4% (impurity).
| χA = Ng2β2/kT[(1 − P)/(3 + (−2J/kT) + 3/4P)] | (2) |
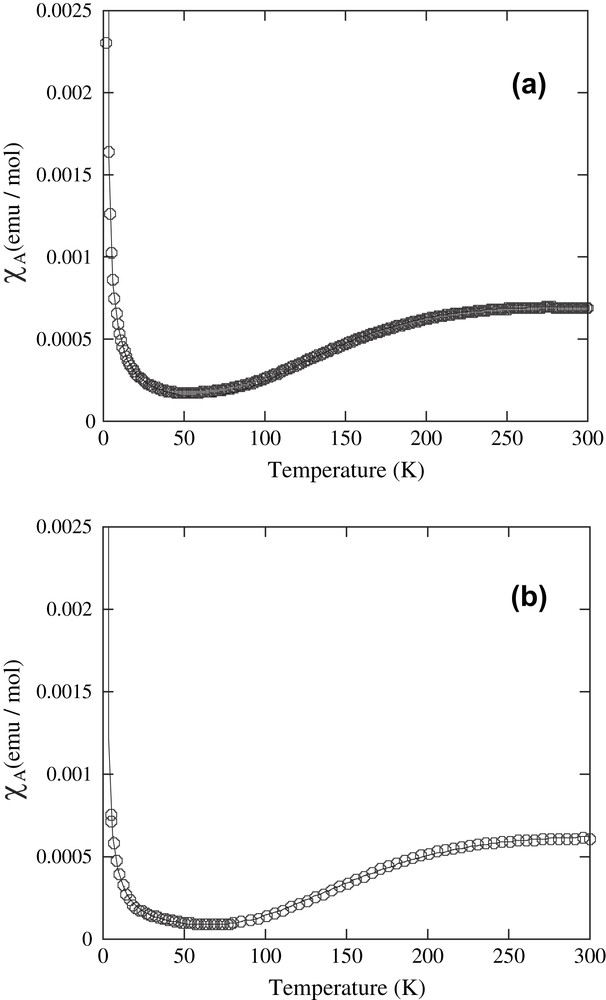
Temperature dependence of the magnetic susceptibilities of (a) complex 1 [the solid lines are the best-fit curves of the Bleaney–Bowers equation with 2J = −320.4 cm−1, g = 1.96, and P = 0.014] and (b) complex 2 [2J = −358.9 cm−1, g = 2.01, and P = 0.009].
The EPR spectra of both complexes 1 and 2, which were measured at room temperature, showed EPR silent states due to strong antiferromagnetic coupling between the two copper(II) atoms.
Finally, in order to determine whether the oxidizing complex 2 is actually an active intermediate for heterogeneous oxidation catalysis, its characteristic reaction with 2-propanol was studied in a suspension of 2 in acetonitrile-d3 in an NMR tube at 293 K to which 3 equivalents of 2-propanol were added. After being allowed to stand for 4 h, the solution was analyzed using an 1H NMR spectrometer. Acetone was detected for complex 2 in the absence of H2O2, thus indicating that complex 2 was actually an oxidative intermediate in a heterogeneous system.
3 O2-based oxidation catalyzed by ruthenium(II,III) complex containing porphyrin [RuII,III2(H2TCPP)]BF4 (3) [48]
3.1 Oxidation of alcohols with O2 and air catalyzed by complex 3
[Ru2II,III(H2TCPP)]BF4 (3) (Fig. 8) was also used as a heterogeneous oxidation catalyst for the oxidation of various alcohols with 1 atm dioxygen and/or air without any additives such as reductants or radical initiators at 298 K, as shown in Table 1. It has been shown that only a few solid catalysts such as Fe(NO3)2–FeBr3 [56] and MnFe1.5Cu0.15O4 [57] are active for the heterogeneous oxidation of alcohols at room temperature under atmospheric pressure of dioxygen or air without any additives. Under these conditions, these catalysts exhibited fast initial rates for alkenic and benzylic alcohols, while the rates for primary and secondary aliphatic alcohols were significantly low.
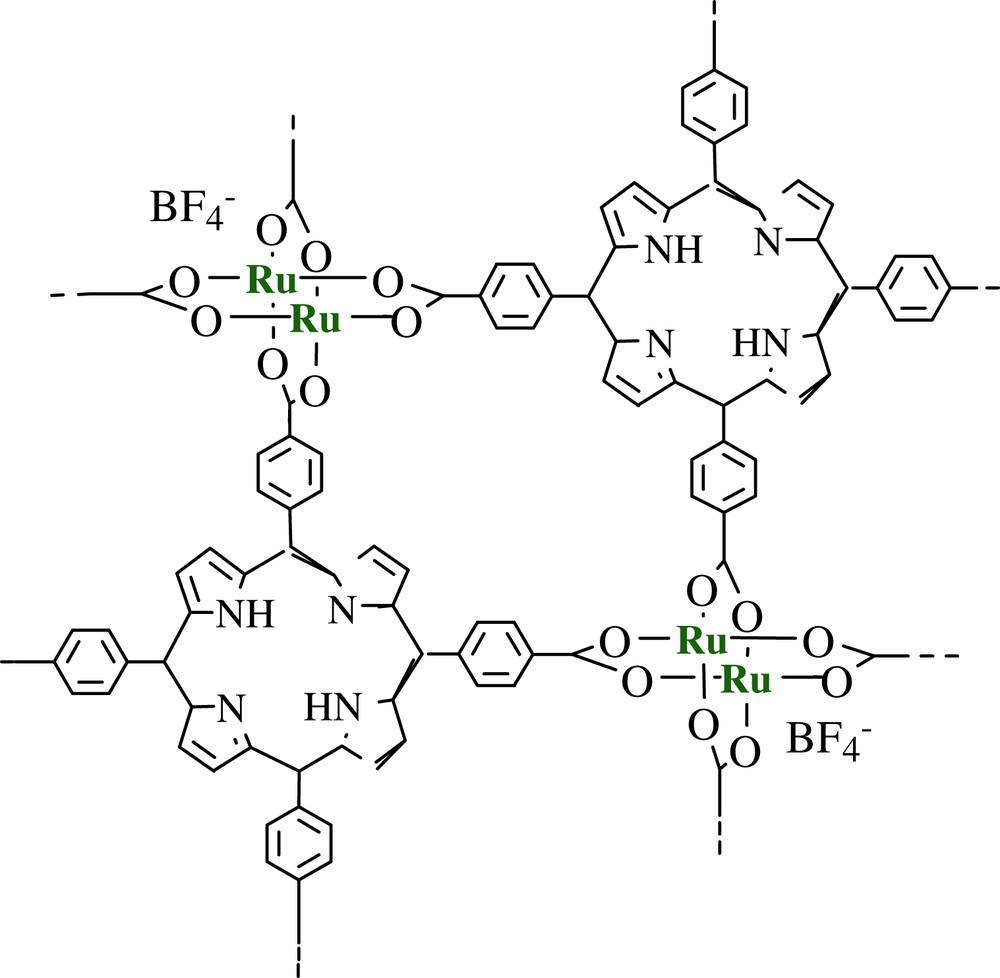
Proposed 3-D body-centered, interpenetrating network structure for complex 3.
In the oxidation of primary aliphatic alcohols 1-octanol and 1-propanol, catalyzed by complex 3 with 1 atm dioxygen, the conversions to octyl aldehyde and propylaldehyde were observed to be highly selective (>99%) with TOFs of 1.4 × 10−4 s−1 and 7.9 × 10−5 s−1 after 24 and 5 h, respectively. The TOFs were considerably higher than those for the oxidation of 2-octanol (no reaction) and 2-propanol (TOF of 1.1 × 10−5 s−1 after 5 h). Furthermore, the TOF of 1-octanol oxidation, which was catalyzed by complex 3 in air, was 1.3 × 10−5 s−1; this was >10 times higher than those of the reported Fe(NO3)2–FeBr3/air system (no reaction) and MnFe1.5Ru0.35Cu0.15O4/air system (9.0 × 10−7 s−1). In contrast, the TOFs for the oxidation of benzyl alcohol catalyzed by complex 3 with 1 atm dioxygen and air were 2.4 × 10−4 s−1 and 1.3 × 10−4 s−1, respectively, which were higher than that of the Fe(NO3)2–FeBr3/air system (9.3 × 10−5 s−1) but lower than that of the MnFe1.5Ru0.35Cu0.15O4/air system (1.1 × 10−3 s−1). In addition, the TOF for the oxidation of cyclohexanol catalyzed by complex 3 with 1 atm dioxygen was 2.2 × 10−5 s−1, which was lower than those of the Fe(NO3)2–FeBr3/air system (9.3 × 10−5 s−1) and the MnFe1.5Ru0.35Cu0.15O4/air system (5.2 × 10−4 s−1). Therefore, effectively, complex 3 was the most active complex for the oxidation of primary aliphatic alcohols.
The ability of complex 3 to adsorb various types of alcohols was examined by TG/DTA measurements [58]. Complex 3 adsorbed 1-propanol (3.6 molecules/Ru2 unit), 2-propanol (3.1), benzyl alcohol (1.3), 1-octanol (1.3), 2-octanol (1.3), and cyclohexanol (1.3), as shown in Fig. 9, which indicates that the adsorption capability of complex 3 decreased with an increase in the steric hindrance and/or the length of the alcohol molecules; this behavior was similar to that observed in complex 1. Although the shape and size of the guest molecules significantly influenced the adsorption capability of complex 3, it is evident that the initial rates for the oxidation of primary aliphatic alcohols were considerably higher than those for secondary aliphatic alcohols. In addition, the result that nonporous ruthenium(II,III) fumarate and complex 1 exhibited no catalytic activities showed that both micropore and dinuclear ruthenium(II,III) sites of complex 3 were indispensable for the oxidation reaction with dioxygen.
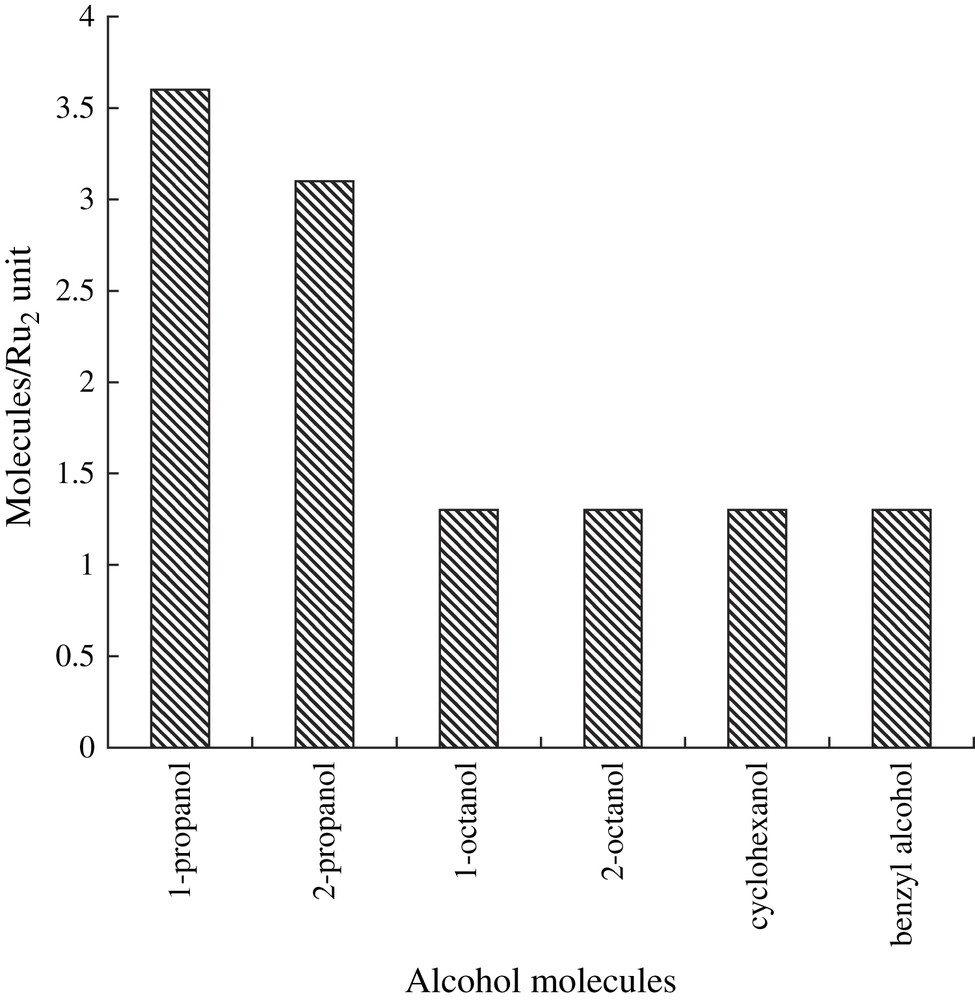
Capability of complex 3 to adsorb various types of alcohols.
In order to determine whether complex 3 is reusable, the oxidation of 2-propanol catalyzed by complex 3 at 323 K for 24 h was repeated twice with the same catalyst. Acetone was produced at the same rate (2.7 × 10−4 s−1) as that during the first run (2.7 × 10−4 s−1). In addition, the UV–vis spectrum of the reaction solution showed no band due to the Ru species, thereby indicating that the leaching of a catalytically active ruthenium species in the solution was negligible. Moreover, after the oxidation reaction, complex 3 exhibited a high BET surface area of 292 m2/g and a high micropore porosity of 5.0 Å, as shown in Fig. 10, similar to that of as-prepared complex 3 (BET surface area: 427 m2/g, pore size: 4.9 Å). These results suggested that the micropore of 3 was retained after the oxidation reaction and complex 3 was reusable under the present reaction conditions.

Pore size distribution of (a) as-prepared complex 3 and (b) 3 after oxidation reaction. Reaction conditions: catalyst 18.6 μmol, 2-propanol 134 mmol, P(O2) = 1 atm, reaction temperature 50 °C, reaction time 24 h.
3.2 ESR studies for ruthenium–oxygen radical species generated by complex 3 under O2 atmosphere
In the oxidation of benzyl alcohol catalyzed by complex 3, the rates decreased with a decrease in the dioxygen partial pressure. In addition, the rates decreased to a half in the presence of a radical scavenger, 2,6-di-t-butyl-4-methylphenol, for the oxidation of benzyl alcohol with 1 atm dioxygen. These results suggested that an oxidizing radical species was formed as an intermediate during the oxidation reactions. In order to observe the oxidizing species, the ESR spectra of complex 3 at 4 K under 20-torr helium, 100-torr oxygen, and 1-atm dioxygen were measured, as shown in Fig. 11. Under 20-torr helium atmosphere, two signals were observed at g = 4.3 and 1.95, which were consistent with an S = 1/2 ground state due to the strong antiferromagnetic coupling between the diruthenium(II,III) cores [59,60]. Under the dioxygen atmosphere, a new signal was observed at g = 2.03, and the signal intensities increased with an increase in the dioxygen pressure. The position of the new signal was very close to that of [RuII,III(μ-O2CEt)4(DM-DCNQI)]n (DM-DCNQI = bis(2,5-dimethyl-N,N′-dicyanoquinonediimine)) (g = 2.02), which was due to the bridging of a radical DM-DCNQI anion between the diruthenium(II,III) cores [61], while it was slightly different from the signal at g = 2.33 due to the strong antiferromagnetic coupling between the diruthenium(II,III) cores and the bridging nitroxide radical species of [Ru2II,III(μ-O2CBut)4(tempo)2][Ru2II,III(O2CCBut)4(H2O)2](BF4)2 (tempo = 2,2,6,6-tetramethylpiperidine-1-oxyl) [59,60]. Thus, the observed signal at g = 2.03 might be due to the axial bridging of the oxygen radical species between the diruthenium(II,III) cores of complex 3.
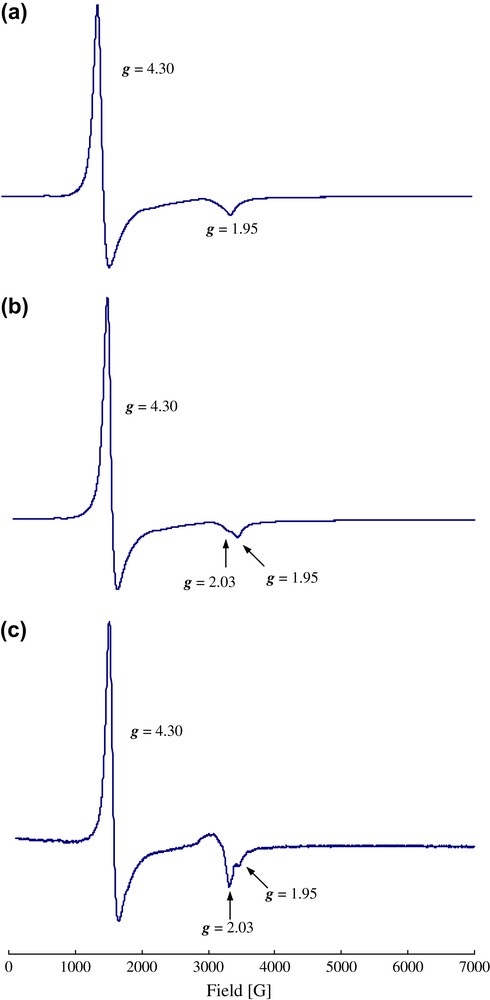
ESR spectra of (a) as-prepared complex 3, (b) complex 3 under 100 torr dioxygen, and (c) complex 3 under 1 atm dioxygen at 4 K.
Finally, in order to observe the affinity of the diruthenium(II,III) cores toward dioxygen, the dioxygen absorption–desorption isotherm of complex 3 was observed at 77.4 K, as shown in Fig. 12. The dioxygen molecules were physically adsorbed into the micropore of complex 3. The isotherm exhibited hysteresis in the desorption curve of dioxygen, while no hysteresis was observed in the Ar adsorption–desorption isotherm at 87.3 K, as shown in Fig. 12a and b. In addition, complex 1 also showed no hysteresis in the dioxygen adsorption–desorption isotherm, as shown in Fig. 12c. Thus, the high affinity of dinuclear ruthenium(II,III) sites toward dioxygen molecules might accelerate the formation of the active ruthenium–oxygen radical species.
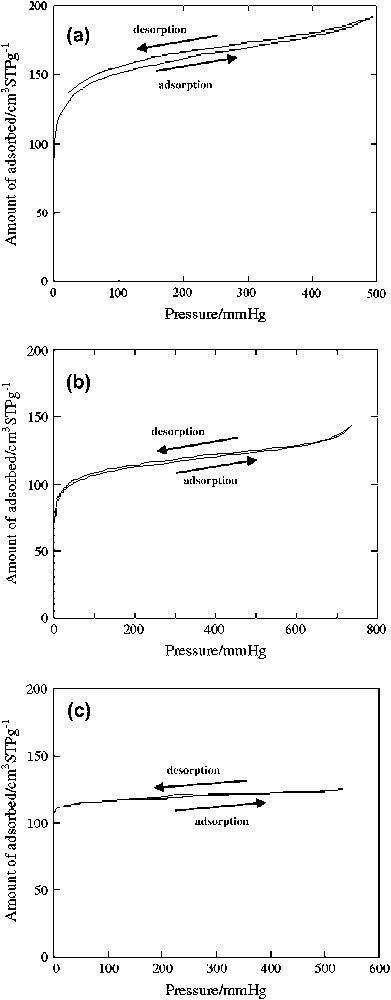
(a) Dioxygen adsorption–desorption and (b) argon adsorption–desorption isotherms of complex 3 and (c) dioxygen adsorption–desorption isotherm of complex 1. Dioxygen and argon adsorption–desorption measurements were carried out at 77.4 and at 87.3 K, respectively.
4 Conclusion
Here, we successfully demonstrated a new approach for using microporous organic–inorganic hybrid materials as heterogeneous oxidation catalysts. The results for the heterogeneous oxidation of various alcohols with H2O2 catalyzed by biomimetic microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate complex (1) showed that (1) the catalytic reactions were quite selective with high conversions and turnover frequencies for heterogeneous oxidation of not only benzylic and secondary alcohols but also primary alcohols with H2O2 via the formation of the green- and brown-colored active oxidizing intermediates, (2) the micropore structure of 1 was stable toward H2O2, and (3) complex 1 was reusable for heterogeneous oxidation catalysis. We also obtained a green-colored microporous complex, H2[Cu2II,II(OOCC6H10COO)(O2)]·H2O (2), which was synthesized by the reaction of complex 1 with 20-fold excess of H2O2 in acetonitrile and characterized by elemental analysis, magnetic susceptibility, FT-IR, TG/DTA, DR UV–vis, EPR, XRPD, resonance Raman, BET surface area, pore size distribution, and gas occlusion measurements. The results revealed that (4) μ-1,2-trans Cu–OO–Cu bridging between two-dimensional layers formed a microporous structure, (5) the peroxo ligand was stable under 405.8 K in a solid state, (6) complex 2 had a high micropore porosity of 4.9 Å, high surface area, and high gas occlusion property comparable to that of as-prepared complex 1, and (7) complex 2 was the first example of an active copper peroxo intermediate for heterogeneous oxidation catalysis. Unfortunately, the brown-colored oxidizing species could not be obtained as a pure solid.
[Ru2II,III(H2TCPP)]BF4 (3) can act as an efficient heterogeneous catalyst for the oxidation of various alcohols, specifically primary aliphatic alcohols, with 1 atm dioxygen and/or air at room temperature without any additives. Such a unique reactivity has never been reported in heterogeneous oxidation reactions with dioxygen or air. The reaction pathway included the formation of an Ru–oxygen radical intermediate, which might be due to the high affinity of diruthenium(II,III) sites toward dioxygen.
Microporous organic–inorganic hybrid materials described here would be crucial for the development of novel heterogeneous oxidation catalysts.
Acknowledgements
This work was supported by Grant-in-Aid for Specially Promoted Research No. 15350088, and by a High-Tech Research Center Project, both from the Ministry of Education, Culture, Sports, Science and Technology, Japan. One of the authors (CNK) is grateful for the support received in the form of a Grant-in-Aid for Young Scientists (B) No. 16750126 from the Ministry of Education, Culture, Sports, Science and Technology, Japan.