1 Introduction
The copper-catalyzed polymerization of 2,6-dimethylphenol (DMP) was first reported by Hay et al. in 1959 [1]. Thus, the reaction of DMP (8.0 mmol) with copper(i) chloride (0.4 mmol; DMP/Cu = 20) in nitrobenzene (30 mL), in the presence of a large excess of pyridine (111.3 mmol; pyridine/Cu = 278), leads to the formation of poly(1,4-phenylene ether) (PPE; also known as PPO) along with small amounts of 4-(3,5-dimethyl-4-oxo-2,5-cyclohexadienylidene)-2,6-dimethyl-2,5-cyclohexadienone (DPQ), which result from the C–C coupling of two DMP molecules (Fig. 1): PPO resin was born and was commercialized by General Electric Co. in the early sixties [2].
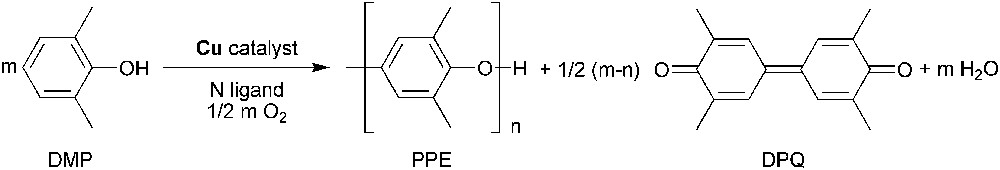
Reaction scheme for the copper-catalyzed oxidative coupling of DMP.
Industrial investigations were then carried out to improve the properties of this polymer, and Gowan showed that PPE could be blended with polystyrene (PS) [3], since both polymers are completely miscible [4]. Hence, the Noryl® resin (PPE/PS blend) was introduced to the marketplace in the late 1960s, and it is still one of the most important commercial thermoplastics produced nowadays, with a yearly increasing turnover (around one billion dollars at the end of last century [5]). Indeed, Noryl® exhibits a broad range of outstanding properties, like low moisture absorption, excellent dimensional stability, good impact resistance, high dielectric strength, excellent flammability rating, and good strength and stiffness over a wide range of service temperatures. As a result, Noryl® has numerous applications in computers and business equipment (housings), automotive (spoiler, cowl vent grilles), electrical insulation (wire coatings), telecommunications (mobile phones), appliances (motor components), electronics (MP3 and DVD players), and many other industries.
Several studies on the mechanism of this polymerization reaction have been described in the literature throughout the years after its discovery [6–12]. Surprisingly, after almost fifty years of research efforts, this mechanism is not entirely elucidated. The present paper is thus meant to give an account on the status of the mechanistic knowledge regarding this reaction, at the beginning of the 21st century.
2 Discussion
Due to the value of PPE as an engineering plastic, its mechanism of formation has been extensively investigated [12–18], and is nevertheless still the subject of debate. Actually, most mechanistic features are accepted by the scientific community, and only the nature of the initial active species is the matter of discussion (Fig. 2).
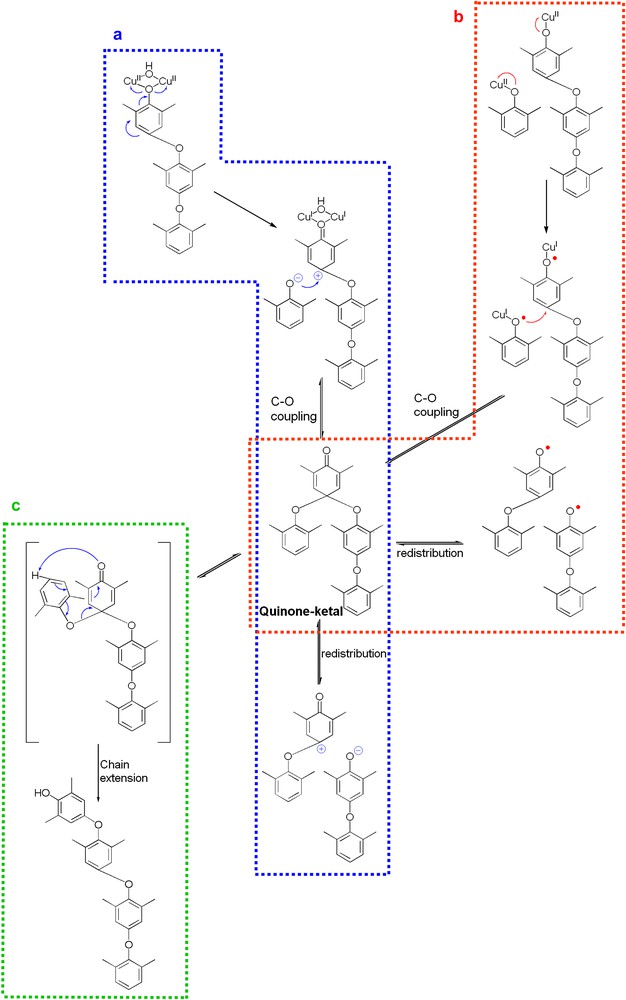
Schematic representation of the proposed mechanisms for the oxidative coupling of DMP: (a) ionic pathway [57]; (b) radicalar pathway [36]; (c) Claisen-type rearrangement of the quinone–ketal intermediate [28,59].
Three alternative pathways for the C–O coupling can be envisaged [19,20]; (i) coupling of free DMP phenoxyl radicals produced via a copper-mediated one-electron oxidation of the phenolic substrate, (ii) coupling of a phenolate anion with a phenoxonium cation stemming from a two-electron transfer from one bridging phenolate to two copper ions (Fig. 2a) [21], (iii) coupling of DMP phenoxyl radicals coordinated to mononuclear copper(i) species, resulting from a monoelectronic transfer from a phenolate to a copper(ii) ion (Fig. 2b) [22].
The involvement of free DMP radicals is unlikely since the use of a one-electron oxidant such as silver oxide, lead oxide or ferricyanide with this substrate leads to the sole formation of DPQ, presumably through the C–C coupling of two 2,6-dimethyl-2,5-cyclohexadienone radicals (Fig. 3) [23]. Furthermore, in the copper-catalyzed polymerization of DMP, radical species have never been detected in significant quantities by EPR; only small amounts of such species were found with rapid flow techniques [24], or when 2,4,6-tri-tert-butylphenol, a phenolic derivative known to give stable radicals, was used [25]. The participation of the copper ions during the coupling reaction is therefore anticipated. Thus, mechanisms (ii) and (iii) are a priori equally possible, and they both yield a quinone–ketal intermediate (Fig. 2).
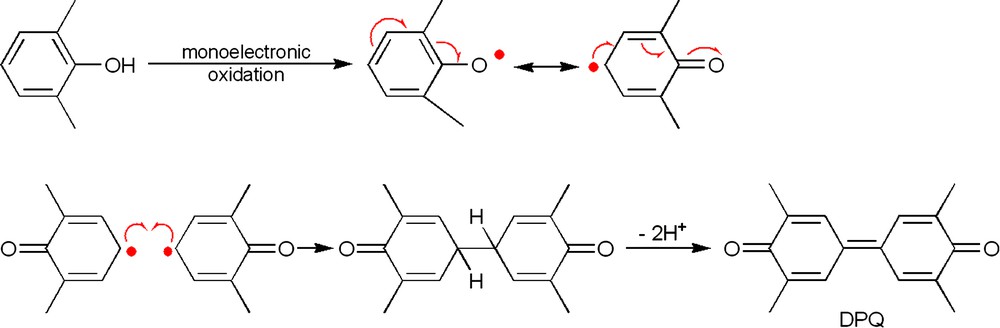
Radicalar pathway for the formation of DPQ [60].
The formation of this quinone–ketal key intermediate (Fig. 2c) is universally accepted [26,27]. The quinone–ketal undergoes a Claisen-type rearrangement, which is a concerted sigmatropic transposition [28]. Accordingly, the critical issue is the way in which the quinone–ketal species are generated. Two schools are thus opposed on mechanistic aspects, namely the homolytic (or radicalar) pathway and the heterolytic (or ionic) pathway. Most of the research groups studying this polymerization reaction consider that a phenoxyl radical is produced by a mononuclear copper species, followed by their coupling (Fig. 2b) [29]. In contrast, Kresta et al. [6] and we [16] believe that a biomimetic dinuclear copper species (the active site of laccase, a copper enzyme that catalyzes the oxidation of phenols, contains a dinuclear copper entity [30]) is involved in the oxidative coupling (Fig. 2a), suggesting a two-electron process (each copper ion participating as one-electron oxidant). In that respect, we think that the formation of DPQ is due to a mononuclear pathway, since only one copper ion (one-electron process [31]) is required to produce a 2,6-dimethyl-2,5-cyclohexadienone radical, which will experience a C–C coupling (Fig. 3).
Recently, ab initio calculations have supported our proposed mechanism where the selective C–O coupling is achieved via the nucleophilic attack of a phenolate on the para-carbon of a phenoxonium cation (Fig. 2a) [32]. Indeed, the cation needs to be in the singlet state to authorize a nucleophilic attack, and this is in total conformity with the mechanism proposal where the phenoxonium cations are produced by means of a double one-electron transfer from a phenolate to both copper centres of a antiferromagnetically, phenoxido-bridged dinuclear copper(ii) complex [33].
Dinuclear copper units as active species have also been lately suggested by the ‘radicalar pathway’ school [34]. Actually, kinetic investigations have resulted in a 1.7–2nd-order rate dependence on the catalyst concentration. Consequently, the mechanism depicted in Fig. 4 has been proposed, where each copper ion of a dinuclear complex oxidizes a coordinated phenolate to the corresponding phenoxyl radicals. Next, a coordinated phenoxyl radical couples with an adjacent mesomeric 2,6-dimethyl-2,5-cyclohexadienone radical. However, this mechanism appears to be highly improbable, as it does not lead to the formation of the quinone–ketal derivative, which is widely accepted [5]. Actually, the redistribution mechanism (see Fig. 2), which has been extensively investigated and essentially proven [27,35], can only be explained through a quinone–ketal intermediate [36,37].
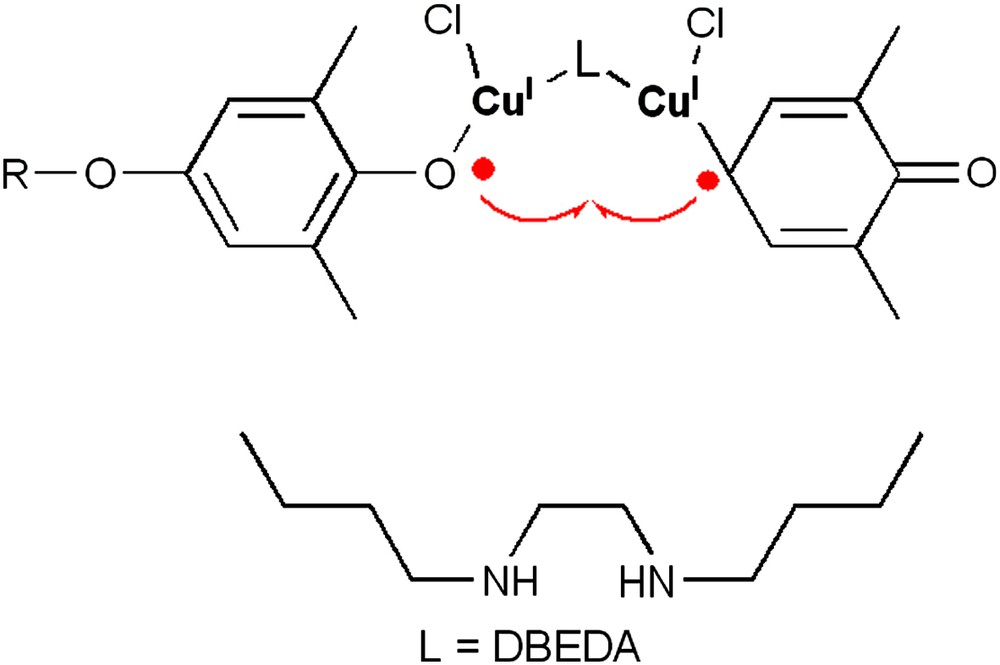
Proposed mechanism for the coupling of 2,6-dimethylphenol catalyzed by a dinuclear copper–DBEDA complex [34].
Our research efforts to further validate our ionic mechanism, namely the attack of a phenolate anion to a phenoxonium cation, has recently given rise to the development of a novel copper-mediated oxidative coupling reaction [38,39]. With the objective to ‘isolate’ important active species present at the early stages of the polymerization reaction, one equivalent of 2,4,6-trimethylphenol, two equivalents of copper(ii) chloride, two equivalents of sodium methoxide, and four equivalents of neocuproine (neo = 2,9-dimethylphenanthroline) were mixed in methanol. The motivation to use 2,4,6-trimethylphenol lies in the fact that the para-position of the phenolic ring is blocked, thus preventing a polymerization reaction. Furthermore, neocuproine is known to form very stable copper(i) species [40], owing to the presence of ortho-methyl groups, which strongly stabilize the tetrahedral geometry of CuI ions, thus precluding their reoxidation to CuII [41]. Finally, two equivalents of base were used, since the goal was to obtain a doubly bridged dinuclear copper(i) species bearing an ‘oxidized’ phenolic moiety, as anticipated (see Figs. 2a and 5b). Surprisingly, a red compound immediately precipitated from this reaction mixture, which was analyzed as [CuI(neo)2]Cl [40]. The formation of copper(i) species clearly suggests the oxidation of at least one of the reactants. 1H NMR analysis of the filtrate reveals the absence of 2,4,6-trimethylphenol. Instead, two new compounds are detected, namely 4-(methoxymethyl)-2,6-dimethylphenol (MDP) and 4-hydroxy-3,5-dimethylbenzaldehyde (HDB) [38].
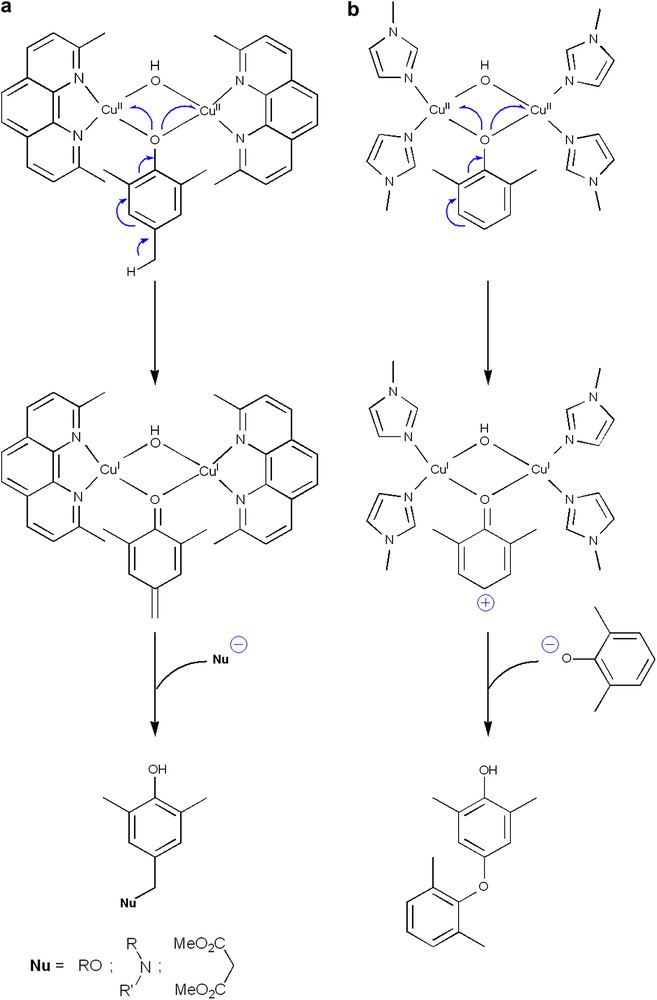
Comparison between (a) the copper-mediated 1,6-nucleophilic addition to 2,4,6-trimethylphenol, and (b) the oxidative coupling of DMP.
A mechanism to explain the formation of these two derivatives is depicted in Fig. 5a. Inspired by the active site found in copper proteins containing the type-3 copper site [42], a dinuclear copper species bridged by a phenolate and a methoxide is proposed as the key intermediate of this unexpected, but remarkable reaction. As a result of the coordination of the bridging phenolate to the metal ions, the para-benzylic proton is activated. The deprotonated 2,4,6-trimethylphenol then reduces each CuII centres to CuI, in a similar way as the one suggested for the polymerization of 2,6-dimethylphenol (Fig. 5b). This two-electron transfer induces the polarization of the benzylic C–H bond, resulting in a rearrangement of the aromatic ring to a quinone methide (Fig. 5a). The nucleophilic addition of methanol (the solvent used during these investigations) to the quinone methide, a highly electrophilic moiety [43,44], then produce 4-(methoxymethyl)-2,6-dimethylphenol (MDP). The formation of the aldehydic derivative HDB can be justified through a second two-electron process on the MDP substrate, yielding a ketal intermediate which is hydrolysed to HDB (Fig. 6).
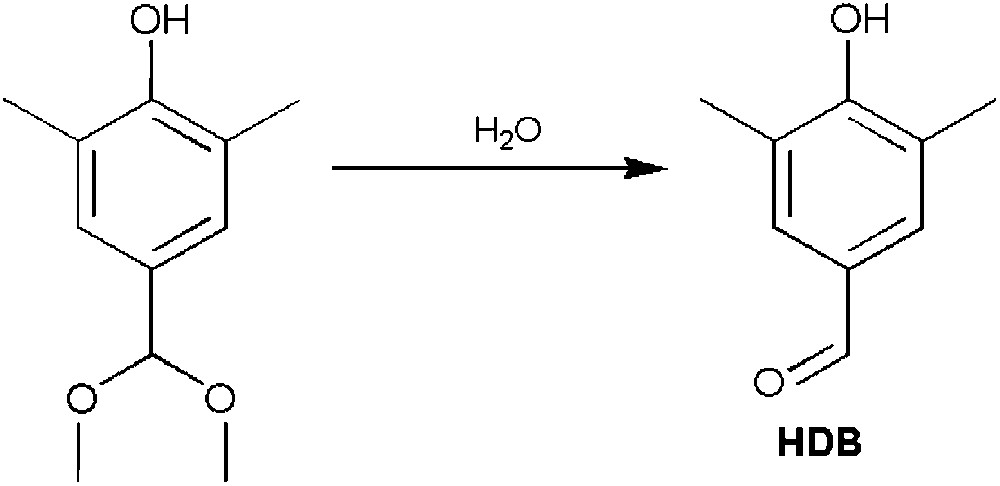
Formation of 4-hydroxy-3,5-dimethylbenzaldehyde (HDB) via hydrolysis of the ketal intermediate.
The preparation of quinone methide by oxidation with large amounts of silver(i) oxide or lead(ii) oxide, and its use with various nucleophiles has been reported [45,46]. Since the last decade, Milstein and co-workers have been extensively studying the formation of intramolecular metal–quinone methide complexes with transition metals [47,48]. For instance, a stable [palladium(quinone methide)] complex has been synthesized and isolated, whose crystal structure is represented in Fig. 7a [49]. The reaction of this complex with dibenzylideneacetone (DBA) in deuterated benzene yields the known [1,2-bis(diphenylphosphino)ethane](η2-dibenzylidenacetone)palladium(0) complex [50], and the free quinone methide BHT-QM, which is characterized by proton NMR (Fig. 7b) [49]. If the substitution of BHT-QM by DBA is performed in methanol, 4-(methoxymethyl)-2,6-di-tert-butylphenol (MDBP) is obtained, which results from the nucleophilic attack of methanol on the unbound quinone methide derivative (Fig. 7b). This result clearly strengthens our mechanism proposal for the formation of MDP and HDB (Figs. 5a and 6).
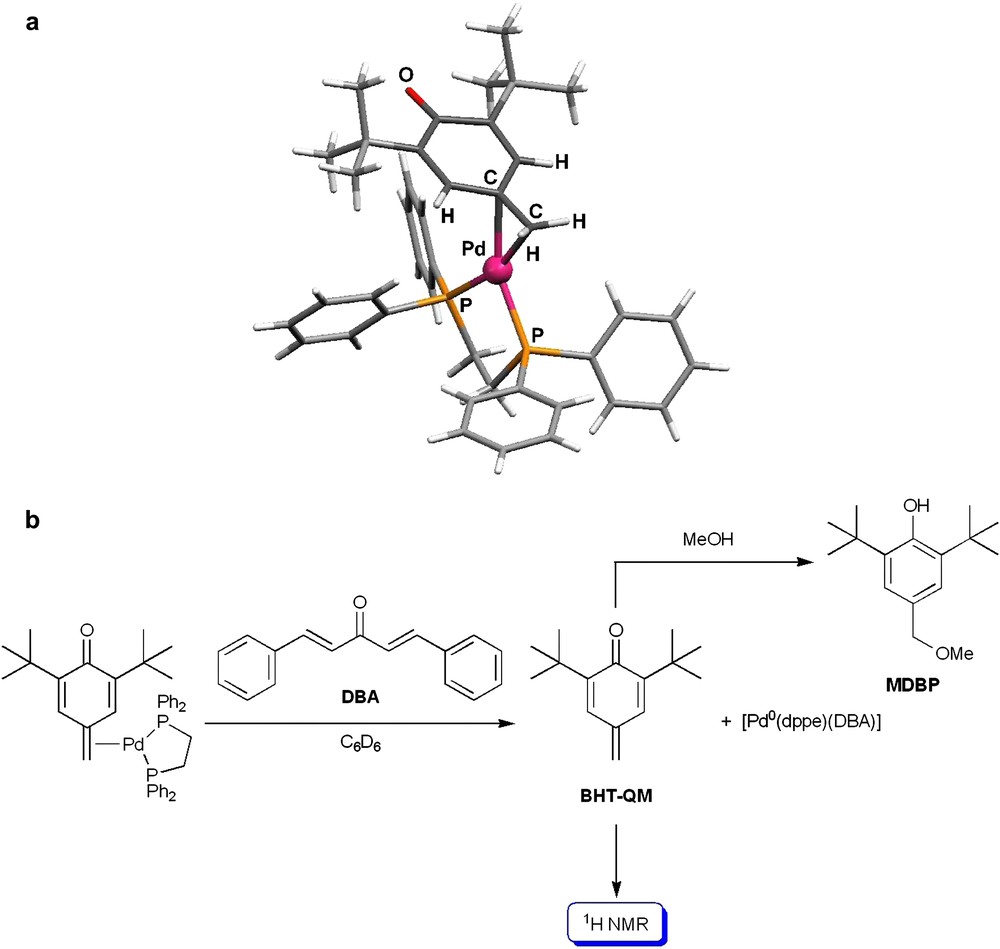
(a) Molecular structure of the quinone methide palladium(0) complex; (b) reactivity of the complex and the quinone methide BHT-QM [49].
The participation of phenoxonium cations in the catalytic cycle of the oxidative coupling of DMP has been disqualified by Kobayashi and Higashimura, on the basis of an experiment whose reaction scheme is depicted in Fig. 8. This experiment takes into account the high electrophilic character of such carbocation [29]. The lifetime of this type of a phenoxonium moiety in the presence of a nucleophile has indeed been theoretically estimated to a few nanoseconds [51]. Thus, the reaction of a mixture of 2,6-dimethylphenol (DMP, 6 mmol) and n-pentylamine (n-PA, 30 mmol) with benzyl chloride (BzCl, 0.6 mmol) in toluene at 40 °C leads, after 9 days under argon, to the sole formation of the benzylated amine (Fig. 8) [52]. On the other hand, if the same DMP/n-PA mixture is used in toluene, at 40 °C under dioxygen, in the presence of the copper complex [Cu(tmed)(OH)]2Cl2 (tmed = N,N′-tetramethylethylenediamine), only PPE is produced with a yield of 75% [29].

Reactivity (nucleophilicity) of 2,6-dimethylphenol (DMP) and n-pentylamine (n-PA) towards benzyl chloride (BzCL) [29].
n-PA is obviously a much better nucleophile than DMP. Unfortunately, the authors have not considered the presence of phenolate anions during the polymerization reaction. Actually, it would have been more appropriate to compare the reactivity of n-PA and sodium 2,6-dimethylphenolate towards benzyl chloride, for instance. n-Pentylamine is clearly not basic enough (pKa = 10.64 [53]) to deprotonate 2,6-dimethylphenol (pKa = 10.63 [54]). The nucleophilicity of sodium 2,6-dimethylphenolate is expected to be superior. For example, the reaction of 2,6-dimethylphenol with an equimolar amount of potassium hydroxide in methanol quantitatively produces potassium 2,6-dimethylphenolate [55]. The subsequent reaction of this potassium salt with benzyl chloride, in DMF at 80 °C, yields 95% of benzyl 2,6-dimethylphenyl ether within 2 h [55].
Furthermore, Milstein and co-workers have recently prepared, isolated, and characterized by single-crystal X-ray diffraction, a metal-stabilized phenoxonium cation (complex 2; Fig. 9a) [56]. This complex 2 has been quantitatively prepared from a solution of complex 1 in CH2Cl2/CH3CN (Fig. 9b), and characterized by 31P, 1H, and 13C NMR, and by ES-MS [56]. Single crystals of complex 2 are obtained from a CH2Cl2 solution layered with hexane. Its crystal structure (Fig. 9a) reveals an unprecedented η2 coordination mode of the iridium(i) ion to the CO double bond of a quinonoid-type moiety.
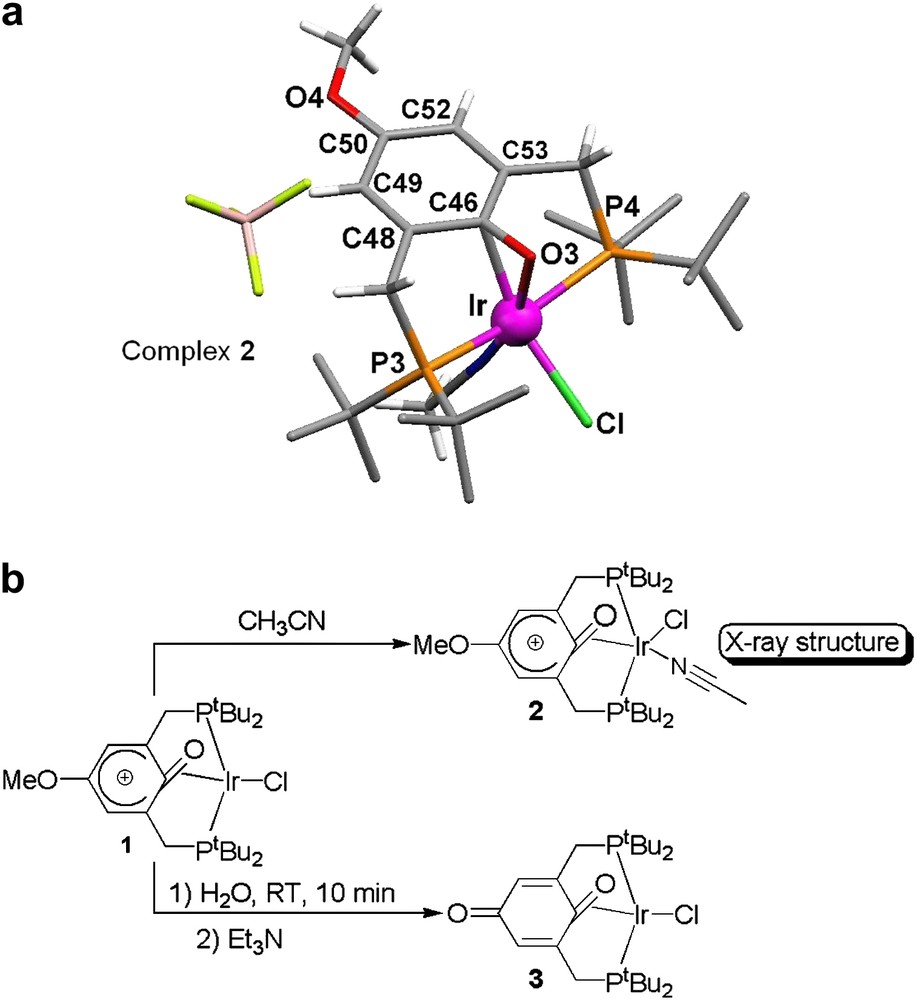
(a) Molecular structure of the phenoxonium iridium(i) complex 2; (b) reactivity of complex 1 [56].
Structure analysis of the six-membered ring (see Fig. 9a and Table 1) shows that the short C48–C49 and C52–C53 bonds have an sp2 character, while the longer C46–C48, C49–C50, C50–C52, and C46–C53 bonds possess an sp3 character. As a result, most of the positive charge is localized on the para-carbon atom, where it is stabilized by the lone pairs of the methoxy substituent.
C–C bond lengths within the coordinated phenoxonium ring [56]
| Bond distances in Å | |
| C48–C49 | 1.362(7) |
| C52–C53 | 1.391(7) |
| C46–C48 | 1.430(7) |
| C49–C50 | 1.395(7) |
| C50–C52 | 1.401(7) |
| C46–C53 | 1.416(7) |
As a matter of fact, reaction of complex 1 with water, followed by the addition of triethylamine leads to the formation of the iridium quinone complex 3 (Fig. 9b), which stems from a nucleophilic attack of the para-carbon atom of the phenoxonium cation. This result is in total agreement with our mechanism proposal for the polymerization of 2,6-dimethylphenol (Fig. 2a).
Finally, the involvement of a (μ-hydroxido)(μ-phenoxido)dicopper(ii) complex in the catalytic cycle of the oxidative coupling of DMP has been questioned by Kobayashi and Higashimura [29]. These authors state that ‘It seems to be obscure for this reaction mechanism that excess N-methylimidazole (NMI) should depress the formation of this dicopper complex, but as NMI increased, the reaction rate and C–O selectivity became higher’. We have indeed reported [57] the beneficial use of an excess of the monodentate N-methylimidazole (30 equivalents per copper ion) which, a priori, should favour the formation of mononuclear species. Therefore, to further prove the formation of such dinuclear species (a crucial intermediate for an ionic pathway where a two-electron process is required; Fig. 5b), the following experiment has been carried out. As the use of 2,4,6-trimethylphenol has led to some oxidative coupling reactions (see above and Fig. 5a), a more inert substrate, namely pentafluorophenol, has been used applying the same experimental conditions. Thus, one equivalent of pentafluorophenol, two equivalents of copper(ii) chloride, two equivalents of sodium methoxide, and two equivalents of neocuproine were simply mixed in dimethylformamide/methanol at room temperature [38]. As expected, in contrast to the same reaction with 2,4,6-trimethylphenol, the formation of the characteristic [CuI(neo)2]Cl complex did not occur. After a few days, single crystals of [Cu2Cl2(neo)2(μ-CH3O)(μ-C6F5O)] were produced with a yield of 47% (Fig. 10).

Molecular structure of [Cu2Cl2(neo)2(μ-CH3O)(μ-C6F5O)] [38].
The exclusive formation of this dinuclear complex (no mononuclear species have been detected) has been also observed in solution by UV–vis–NIR and EPR spectroscopies [39]. The spontaneous self-assembly of a μ-methoxido-μ-phenoxido-bridged dinuclear copper complex from a mixture of four different reactants, one of them being a mononucleating ligand, illustrates the tendency of copper to form dinuclear species in the presence of oxido ligands, and supports our proposed active species for the polymerization of DMP (Fig. 5b). The need of an excess of ligand is most likely due to its role as stabilizer of essential intermediate reactive species. Gao and co-workers [58] have recently suggested that imidazole stabilizes the phenoxyl radicals by forming an intermediate complex and thus facilitates the C–O coupling process (Fig. 11).
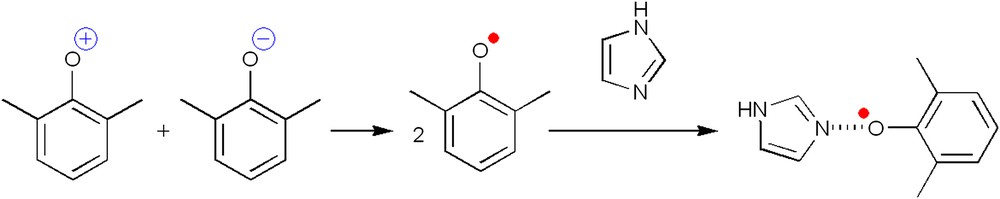
Stabilization of phenoxyl radicals through the formation of a complex intermediate with imidazole [58].
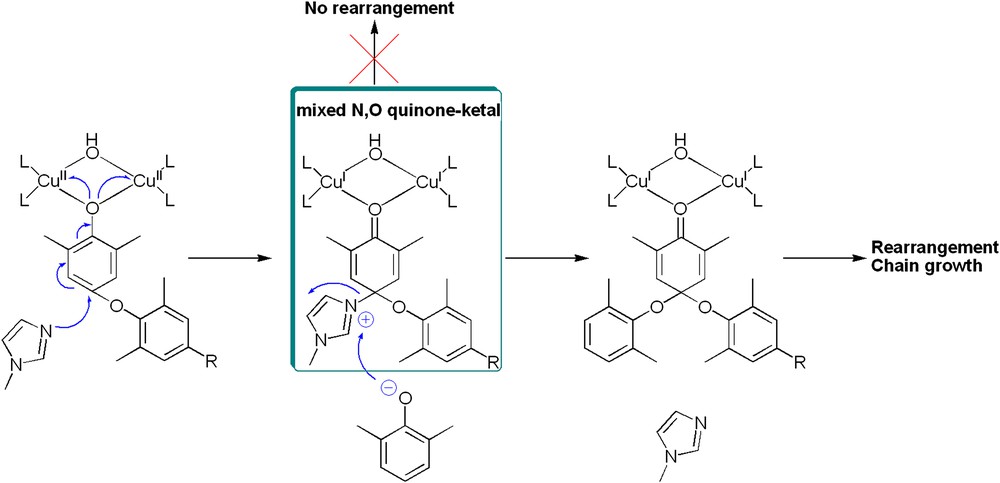
We rather believe that N-methylimidazole stabilizes the key intermediate species of our mechanism proposal, namely the quinone–ketal resulting from the nucleophilic attack of a phenoxonium cation (Fig. 12). The imidazolium quinone–ketal1 cannot undergo the rearrangement step, but its redistribution process with a phenolate anion gives rise to a quinone–ketal which can produce an elongated polymer. N-Methylimidazole thus facilitates the pre-arrangement of the key active species.
3 Concluding remarks
Since the discovery of the oxidative coupling of 2,6-di-substituted-phenols catalyzed by copper/amine catalysts at the end of the 1950s, a number of reports have been published. While most of the mechanism of the reaction is elucidated and accepted by the scientific community, the coupling mechanism, and particularly the selectivity for C–O/C–C coupling, is still unclear. Some groups consider that the polymerization proceeds via the coupling of phenoxyl radicals generated by the copper catalyst, whereas other ones believe that dinuclear copper species produce phenoxonium cations that couple with phenolate anions. In 2003, the ‘radical’ school has ruled out the ‘ionic’ pathway, emphasizing the impossibility to have phenoxonium cations in the reaction mixture. Nevertheless, significant recent experimental data from the literature, compiled in the present paper, tend to support the possible existence of such carbocationic species during the course of the polymerization reaction. Based on the experimental evidence currently available, both polymerization pathways, i.e. the radicalar and the ionic one, cannot be absolutely excluded. One can even consider that the polymerization of 2,6-dimethylphenol proceeds through two (or more) competitive mechanisms, or by means of a combination of both (or more) pathways. To our point of view, further mechanistic investigation is still needed in order to firmly establish the mechanism of this reaction.
Acknowledgements
Support from the NRSC Catalysis (a Research School Combination of HRSMC and NIOK) is kindly acknowledged. The research is financially supported by the Dutch Economy, Ecology, Technology (EET) programme, a joint programme of the Ministry of Economic Affairs, the Ministry of Education, Culture and Science, and the Ministry of Housing, Spatial Planning and the Environment. Former Ph.D. students and postdocs whose names are given in the list of references are thanked for the input. Prof. Dr. Ger Challa, retired from Groningen University, is thanked for many stimulating discussions.
1 Mass spectroscopy evidences of such species have been recently obtained in our group, which will be published elsewhere.