

1 Introduction
L’étude de la nature coordinante des oxoanions tétraédriques, pyramidaux ou substitués a fait l'objet de plusieurs travaux dans notre laboratoire [1–10]. De même, l'étude des dicarboxylates y a été initiée récemment par la synthèse de composés malonato, succinato et maléato [11–12]. Tiekink [13] a fait une synthèse des travaux sur les carboxylates. De façon générale, peu de travaux ont été publiés quant aux complexes organostanniques glutarato, 3,3-diméthylglutarato et hydrogénooxalato. L'objet du présent travail réside dans la synthèse et la caractérisation par spectroscopies infrarouge et Mössbauer de cinq dérivés organostanniques contenant des dicarboxylates.
2 Partie expérimentale
2.1 Synthèse des ligands
Les ligands L1 = CO2(CH2)3CO2TMN2·5H2O, L2 = CO2HCH2C(CH3)2CH2CO2TMN·5H2O et L3 = (HC2O4)2NH3(CH2)2NH3·H2O sont obtenus par neutralisation totale ou partielle des acides CO2H(CH2)3CO2H, CO2HCH2C(CH3)2CH2CO2H et H2C2O4·2H2O par TMNOH (20% en solution aqueuse) ou éthylène diamine (98%). Le mélange obtenu est agité pendant 2 h environ à température ambiante. Pour L1 et L2, les solutions limpides obtenues ont été mises à l'étuve à 60 °C pendant quelques jours, et les cristaux blancs obtenus ont été recueillis et séchés sur P2O5, alors que pour L3 le précipité blanc obtenu a été lavé à l'éthanol et séché sur P2O5.
2.2 Synthèse des complexes
Tous les complexes sont obtenus en mélangeant à température ambiante les sels L1, L2 et L3 avec SnR2Cl2 (R = Me, Bu) dans des stœchiométries indiquées dans le Tableau 1.
Sels utilisés, avec leurs stœchiométries
| Dérivés/stœchiométrie | %C | %H | %N | NPO | Température de fusion |
| CO2(CH2)3CO2TMN2·5H2O (L1) | 42,39 (42,19) | 10,87 (11,02) | 7,60 (7,46) | CB | – |
| CO2HCH2C(CH3)2CH2CO2TMN·5H2O (L2) | 40,86 (40,76) | 10,21 (10,05) | 4,33 (4,82) | CB | – |
| (HC2O4)2NH3(CH2)2NH3·H2O (L3) | 28,36 (28,02) | 4,72 (5,02) | 11,02 (11,92) | PB | – |
| (SnMe2Cl)2CO2(CH2)3CO2·H2O (A1) (1:2) | 22.03 (21,62) | 4,07 (4,39) | – | PB | >260 °C |
| SnBu2ClCO2H(CH2)3CO2·H2O (A2) (1:1) | 37,39 (36,91) | 6,47 (6,42) | – | CB | 106 °C |
| SnMe2ClCO2HCH2C(CH3)2CH2CO2·2H2O (A3) (1:1) | 28,48 (28,36) | 5,53 (5,52) | – | PB | >260 °C |
| SnBu2CO2CH2C(CH3)2CH2CO2·2H2O(A4) (1:1) | 42,18 (42,26) | 7,39 (6,95) | – | CB | 104 °C |
| SnBu2(HC2O4)2 (A5) (1:1) | 35,06 (35,80) | 4,86 (4,71) | – | PB | >260 °C |
Les mélanges donnant des solutions limpides sont agités pendant 2 h environ et laissés à évaporer lentement pendant quelques jours, au terme desquels des cristaux blancs obtenus sont pêchés et séchés sur P2O5, alors que ceux donnant des précipités blancs sont agités pendant 2 h, filtrés, lavés à l'éthanol à chaud, puis séchés sur P2O5.
Les analyses élémentaires, effectuées au laboratoire de microanalyses de l'université de Padoue (Italie) sont reportées dans le Tableau 1.
- A1 = (SnMe2Cl)2CO2(CH2)3CO2·H2O
- A2 = SnBu2ClCO2H (CH2)3CO2·H2O
- A3 = SnMe2ClCO2HCH2C(CH3)2CH2CO2·2H2O
- A4 = SnBu2CO2CH2C(CH3)2CH2CO2·2H2O
- A5 = SnBu2(HC2O4)2.
3 Résultats et discussion
Les données infrarouge sont reportées dans le Tableau 2.
Données infrarouge
| Attributions | A1 | A2 | A3 | A4 | A5 |
| νOH | – | 3425 L | 3396 L | 3425 L | |
| 2729 f | 2725 f | 2728 f | 3394 L | ||
| 2670 f | 2650 f | 2653 f | 3327 ép | ||
| 3395 L | – | – | – | – | |
| 1699 ép | 1740 m | 1672 F | 1734 f | ||
| 1625 tF | 1621 F | 1634 ép | 1636 m | 1687 F | |
| 1566 tF | 1568 tF | 1616 F | 1554 F | 1621 tF | |
| 1559 m | |||||
| 1340 F | 1297 m | 1261 m | |||
| 1673 ép | 874 F | 1257 m | 1239 m | 794 tF | |
| 796 F | 851 ép | 1227 m | 1156 m | ||
| 1157 ép | |||||
| 598 ép | 600 m | ||||
| 578 m | – | 589 ép | 552 m | 483 F | |
| 543 F | 532 m | 474 F | |||
| 495 F | |||||
| 481 f | 453 m | ||||
| 461 f | 468 F | 416 ép | 393 f | 385 F | |
| 445 f | 380 F | ||||
| 351 m | 339 m | ||||
| 304 m | |||||
| 561 ép | 682 tF | 568 F | 639 F | 683 m | |
| 520 m | 605 F | 518 ép | 606 F | – | |
| νSnCl | 275 F | 285 F | 273 tF | – | – |
| νSnO | 227 m | 212 m | 202 F | 291 F | 240 F |
| 213 m | 208 f |
3.1 A1 = (SnMe2Cl)2CO2(CH2)3CO2·H2O
La présence d'une bande à 520 cm−1 attribuée à
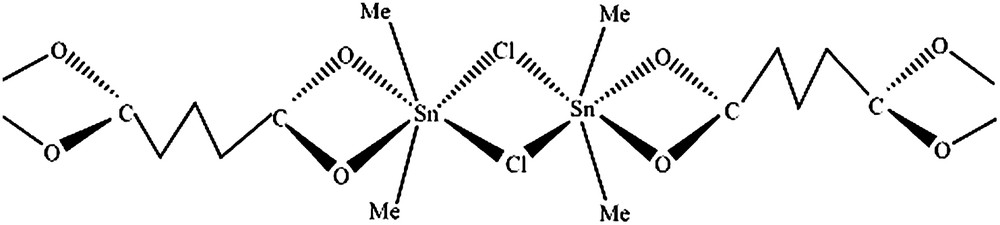
Structure de A1 = (SnMe2Cl)2CO2(CH2)3CO2·H2O.
3.2 SnR2ClL·n H2O [L = CO2H(CH2)3CO2 (A2), L = CO2H(CH2)2C(CH3)2CH2CO2 (A3)], n = 1,2 ; R = Bu, Me
La présence, respectivement, d'une bande à 605 cm−1 sur le spectre infrarouge du complexe A2 et à 518 cm−1 sur celui du complexe A3, attribuée à
La structure découlant de ces données spectroscopiques proposée est dimère, avec des groupements SnR2 dissymétriquement transcoordinés, d'une part, par les oxoanions chélatants et, d'autre part, par les chlorures pontants et contenant des liaisons hydrogène, du fait de la largeur de la bande correspondant à νOH (Fig. 2).
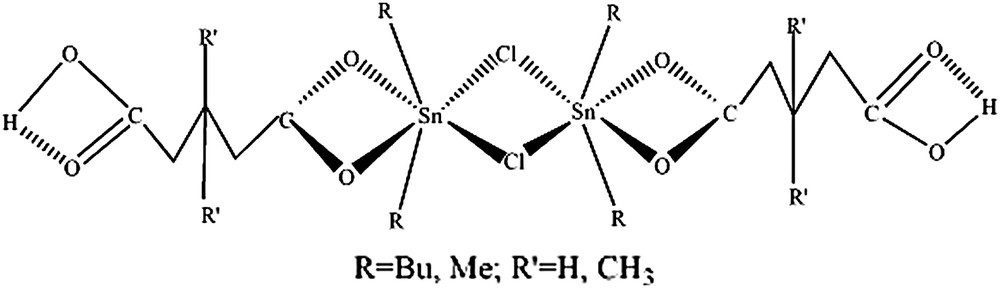
Structure de SnR2ClL·n H2O [L = CO2H(CH2)3CO2 (A2), L = CO2H(CH2)2C(CH3)2CH2CO2 (A3)], n = 1,2 ; R = Bu, Me.
3.3 A4 = SnBu2CO2CH2C(CH3)2CH2CO2·2H2O
La présence d'une bande moyenne à 606 cm−1 attribuée à

Structure de A4 = SnBu2CO2CH2C(CH3)2CH2CO2·2H2O.
3.4 A5 = SnBu2(HC2O4)2
La valeur de l'éclatement quadripolaire (Q.S. = 4,46 mm s−1 supérieur à 4 mm s−1) indique une transcoordination symétrique du résidu SnBu2 et un environnement octaédrique autour de l'étain comme dans SnBu2SO4 (Q.S. = 4,78 mm s−1) [21] et SnBu2SeO3 (Q.S. = 4,18 mm s−1) [16].
La structure découlant de ces données spectroscopiques est discrète, avec une transcoordination symétrique du résidu SnBu2 (Fig. 4) et des liaisons hydrogène intermoléculaires, en raison de la largeur de la bande due à νOH.
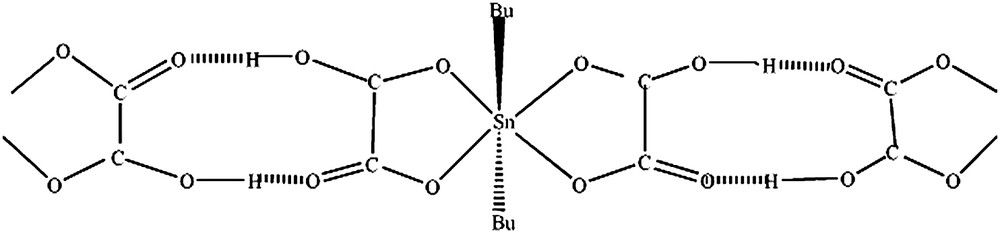
Structure de A5 = SnBu2(HC2O4)2.
4 Conclusion
Dans les composés étudiés, les anions glutarate et 3,3-diméthylglutarate se comportent comme bichélatants, le glutarate acide et le 3,3-diméthylglutarate acide comme monochélatant, les premiers donnant des composés polymères et les seconds des composés à structure discrète ; quant à l'oxalate acide, il se comporte comme monochélatant et donne une structure à chaîne infinie, avec des liaisons hydrogène associant les dérivés SnBu2(HC2O4)2 entre eux.
Remerciements
Nous remercions les professeurs M. Vidali et U. Russo (université de Padoue, Italie) pour les analyses élémentaires et l'enregistrement des spectres infrarouge et Mössbauer, ainsi que le Pr. Libasse Diop (UCAD) pour sa relecture du manuscrit.
Vous devez vous connecter pour continuer.
S'authentifier