

1 Introduction
La céramique traditionnelle est encore abondamment utilisée sous la forme de poteries en terre cuite dans de nombreux pays du tiers monde. Elle est usitée pour la collecte et la conservation de l'eau de boisson, la conservation des céréales et pour la cuisson des aliments. Toutefois, ces matériaux présentent souvent de piètres caractéristiques mécaniques (faible contrainte de rupture, accompagnée d'une grande fragilité), imputables en partie à la nature chimique et minéralogique fondamentalement complexe des matières brutes argileuses, et surtout au manque de maîtrise de ces matières et des différentes étapes du procédé céramique. Ainsi donc, par exemple, la calcite est souvent ajoutée aux mélanges argileux dans le but de mieux maîtriser les variations dimensionnelles qui accompagnent la cuisson. La nature des phases minérales formées consécutivement à la décomposition de la calcite en CaO dépend de la température de cuisson, de la distribution en taille des grains de calcite et de sa concentration [1–6]. Par ailleurs, un excès de CaO libre peut générer une porosité importante [6].
Nous examinons ici l'influence de la calcite sur la microstructure, les propriétés physiques et mécaniques d'une argile kaolinitique du Burkina Faso utilisée depuis de nombreuses années pour la confection de poteries à usage domestique. Les méthodes d'analyse d'image ont été mises en œuvre pour quantifier les modifications microstructurales ; l'analyse du comportement mécanique a été menée en termes de ténacité et de contrainte à la rupture, en procédant par des essais de flexion trois points.
2 Matériaux et méthodes expérimentales
2.1 Analyses physicochimiques
La matière première de base est une argile kaolinitique extraite dans des dépôts allivionnaires de la région de POA au Burkina Faso, entre 2 et 3° O, d'une part, et 12 et 13° N, d'autre part [7]. Cette région est renommée depuis toujours pour son savoir-faire dans le domaine de la poterie. Nous avons effectué un prélèvement d'une dizaine de kilogrammes de terre argileuse en cinq endroits différents du site. Les cinq prélèvements ont ensuite été rassemblés en un seul ; après homogénéisation de l'ensemble, nous obtenons une cinquantaine de kilogrammes pour le site POA [7]. La composition chimique élémentaire, détaillée dans le Tableau 1, indique clairement la nature silico-alumineuse de la poudre POA, avec une prédominance de la silice. L'importance de la perte au feu s'explique par le caractère naturel de cette poudre (elle contient notamment des résidus organiques) et la proportion élevée de kaolinite. Des investigations plus poussées, à l'aide de différentes techniques, ont révélé les aspects essentiels suivants [7] :
- – les analyses X indiquent que POA contient essentiellement la kaolinite et le quartz comme phases cristallines majeures. La composition minéralogique de cette poudre, déterminée à partir de ces données et de l'analyse chimique élémentaire, indique des taux massiques de 71,3% de kaolinite et 21,1% de quartz. Par ailleurs, une comparaison avec Kg2 (une argile pure contenant surtout une kaolinite à faible degré de cristallinité) révèle que la kaolinite dans POA est peu ordonnée, comme on peut le voir sur la Fig. 1 ;
- – la courbe d'analyse thermique différentielle (ATD) de POA met en évidence une décomposition thermique en deux étapes [8] : (i) une déshydroxylation des minéraux argileux dans la plage de température 400–600 °C ; (ii) une réorganisation structurale entre 900 et 1000 °C. Sur la courbe ATD de la Fig. 2, le renflement exothermique aux environs de 320 °C correspondrait à la décomposition des résidus organiques ;
- – l'importante dilatation thermique associée à ces transformations atteste la teneur élevée de POA en minéraux argileux [9] ;
- – l'analyse des spectres Mössbauer de la Fig. 3 indique que POA contient presque exclusivement du fer structural, disposé en site octaédrique, substitué à l'aluminium [10].
Analyse chimique quantitative de la poudre POA et de Kg2 une kaolinite de référence de faible cristallinité provenant de Géorgie (États-Unis)
| Al2O3 | Fe2O3 | Na2O | K2O | SiO2 | CaO | MgO | TiO2 | Total | Perte au feu | |
| POA (% masse) | 28,2 | 4,5 | 0,1 | 0,7 | 54,3 | 0,2 | 0,3 | 1,4 | 89,6 | 10,4 |
| Kg2 (% masse) | 38,5 | 1,1 | – | < 0,1 | 43,9 | – | < 0,1 | 2,4 | 85,9 | 13,8 |
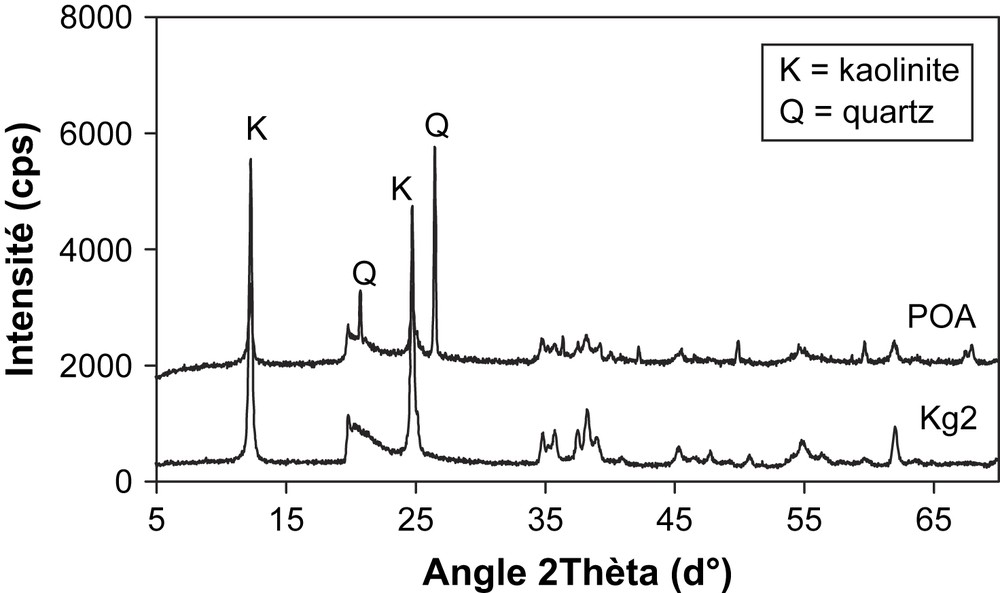
Diffractogrammes X des poudres des argiles POA et Kg2 : le spectre de l'argile POA indique les pics du quartz en plus de ceux de la kaolinite.
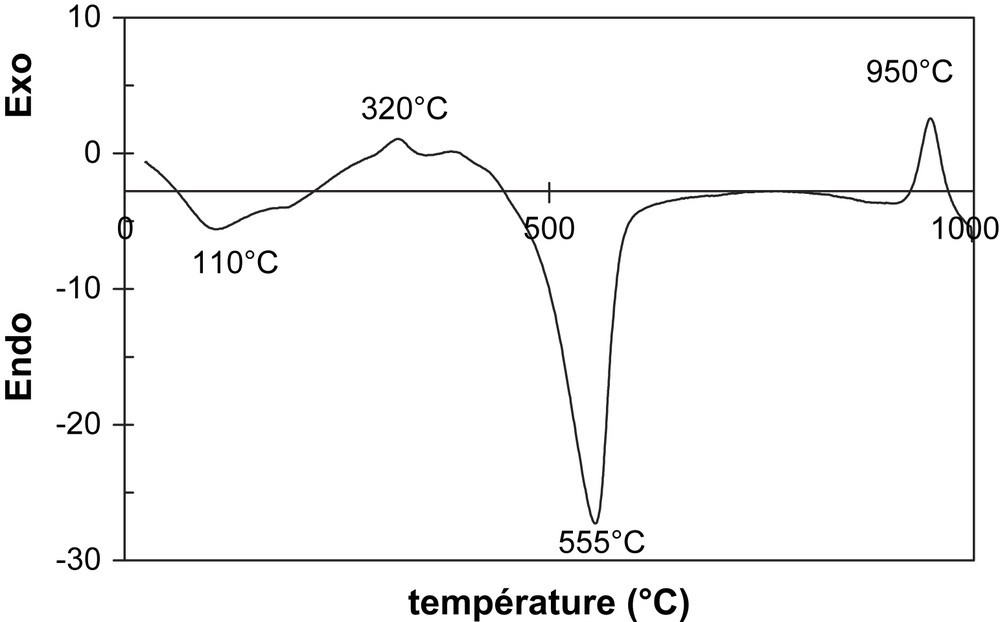
Courbe ATD de l'argile POA.
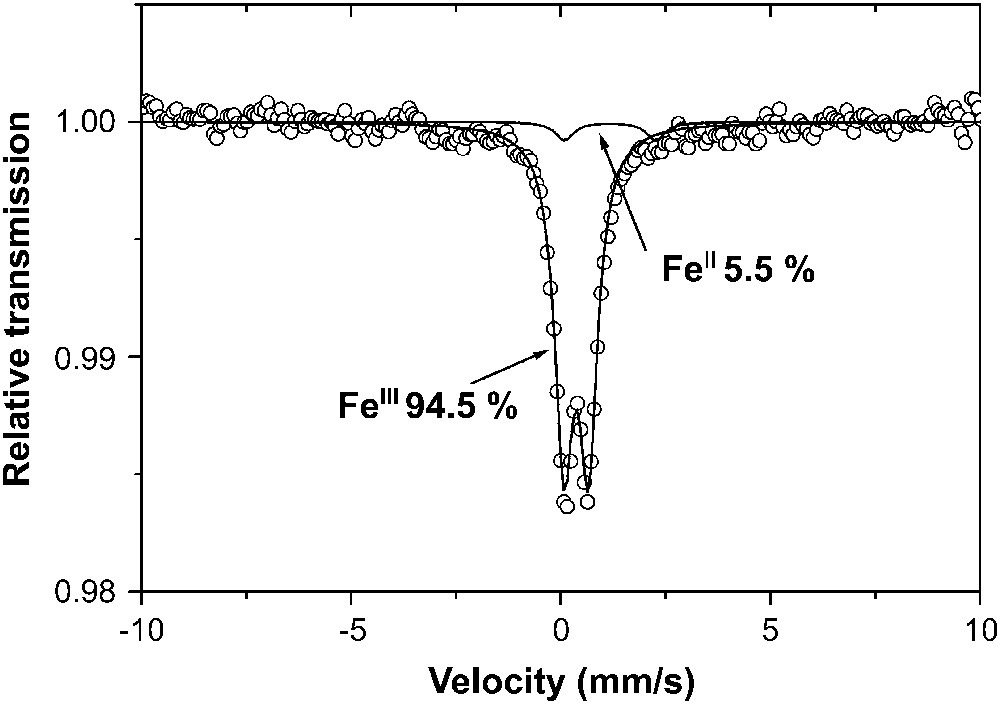
Spectre Mössbauer expérimental (○) et calculé (—) de l'argile POA à la température ambiante de 25 °C.
2.2 Élaboration des matériaux
Différents lots de poudre, obtenus en remplaçant 5, 10, 15, 20 et 25% en poids de POA par de la calcite (Tableau 2), ont été humidifiés à l'eau et broyés finement à l'aide de billes en alumine. Les barbotines ont ensuite été étuvées (100 °C), broyées de nouveau et granulées avant le pressage (15 MPa) pour obtenir des plaques de dimensions 12 × 5 × 1 cm3. Ces plaques furent séchées pendant 24 h à 40 °C, puis portées à 1100 °C pendant 1 h, sous air en adoptant une vitesse de montée de 3 °C min−1.
Composition des différents mélanges de POA avec la calcite
| Mélanges | POA (% masse) | Calcite (% masse) |
| POA | 100 | 0 |
| MEL5 | 95 | 5 |
| MEL10 | 90 | 10 |
| MEL15 | 85 | 15 |
| MEL20 | 80 | 20 |
| MEL25 | 75 | 25 |
2.3 Caractérisation mécanique
Des éprouvettes d'essai mécanique de forme parallélépipédique, de dimensions l = 80 mm, B = 8 mm et W = 5,5 mm, ont été découpées dans ces plaques. Les essais de flexion trois points (avec un rapport 4 entre la distance inter-appuis, L, et la hauteur de l'échantillon, W) ont été conduits sur une machine universelle de type Schenck, équipée d'une cellule de charge de capacité 10 kN, avec une vitesse de traverse de 3 mm min−1. La contrainte à la rupture, σR, est calculée à partir de la charge de rupture, FR, à l'aide de la relation (1). Des éprouvettes munies d'une entaille fine de taille a0 ont été rompues par flexion trois points pour la mesure de la ténacité. Ce paramètre, KIc, est calculée à partir de la contrainte de rupture au droit de l'entaille, σt, dans la configuration de chargement L/W = 4, avec un rapport 0,35 entre la taille de l'entaille et la hauteur de l'échantillon, et un facteur Y, qui dépend à la fois de la configuration de l'essai et de la géométrie de l'échantillon, comme l'indique la relation (2).
| (1) |
| (2) |
2.4 Observations micrographiques
Après un dépôt d'or sur les surfaces de rupture, les observations microstructurales ont été conduites à l'aide d'un microscope électronique à balayage (MEB) Philips FEG.
2.5 Caractérisation morphologique
Afin d'entreprendre les analyses morphologiques, un échantillon de chaque nuance de matériau a été enrobé sous vide dans une résine, puis finement poli dans de l'éthanol absolu. Pour chaque échantillon, six champs de mesure (de taille 764 × 572) ont été capturés à l'aide d'une caméra CCD, avec une résolution de 0,102 μm. Les images en niveaux de gris à faible grossissement sont transformées en images binaires dans lesquelles la phase solide apparaît en noir et les pores en blanc. Les paramètres morphologiques suivants ont été évalués sur ces images binarisées à l'aide d'un logiciel conçu et validé au laboratoire [11] (la phase solide est notée S et la phase poreuse P) :
- – la fraction volumique des pores, VV(P), qui correspond au volume occupé par la phase poreuse par unité de volume de matériau. Comme les deux phases sont complémentaires, on a la relation : VV(P) + VV(S) = 1 ;
- – la surface spécifique des pores, SV(P), qui correspond à l'aire occupée par la phase poreuse par unité de volume du matériau ;
- – le libre parcours moyen dans la phase poreuse, Lm(P), ou dans la phase solide, Lm(S), représente la longueur moyenne des intersections d'une ligne d'analyse avec la phase considérée ;
- – le libre parcours moyen dans la phase solide pondéré en mesure, L∗(S), est obtenu à partir des distributions des tailles pondérées linéairement [12].
3 Résultats et discussion
3.1 Comportement mécanique
Les résultats des essais mécaniques sont rassemblés dans le Tableau 3. Pour chaque nuance de matériau, la contrainte moyenne de rupture a été calculée en testant au moins 71 échantillons. En comparaison avec des matériaux similaires de la bibliographie, la résistance à la rupture de POA apparaît relativement élevée, et elle n'est pas sensiblement diminuée par l'ajout de calcite. Au contraire, un renchérissement accompagne l'ajout de 5% en poids de calcite. La ténacité décroît fortement dans la plage de concentration de calcite 0–15%, puis se stabilise autour d'une valeur palier comparable à celles reportées pour des verres sodiques et des schistes bitumineux [13].
Caractéristiques mécaniques des différentes nuances de matériaux
| POA | MEL5 | MEL10 | MEL15 | MEL20 | MEL25 | |
| σR (MPa) | 26,57 ± 3,18 | 30,04 ± 4,4 | 27,7 ± 3,38 | 23,1 ± 3,32 | 21,6 ± 4,55 | 26,6 ± 4,17 |
| KIc (MPa m1/2) | 0,78 ± 0,04 | 0,67 ± 0,3 | 0,66 ± 0,02 | 0,61 ± 0,04 | 0,62 ± 0,04 | 0,63 ± 0,05 |
3.2 Corrélation comportement mécanique–taux de calcite
Le renchérissement de la résistance à la rupture aux alentours de 5% de calcite nous semble dû à la formation de nouvelles phases cristallines, notamment la géhlénite et l'anorthite, au cours du frittage des différents mélanges argile–calcite [7,14]. En effet, les diffractogrammes de poudre des tessons issus des mélanges MEL15 et Kg2-15%calcite (Figs. 4 et 5) indiquent que la géhlénite est la phase cristalline qui se forme la première. Cependant, l'anorthite devient très vite la phase prépondérante, de telle sorte qu'après un palier d'1 h à 1100 °C, on ne décèle pratiquement plus de géhlénite dans les tessons [7].
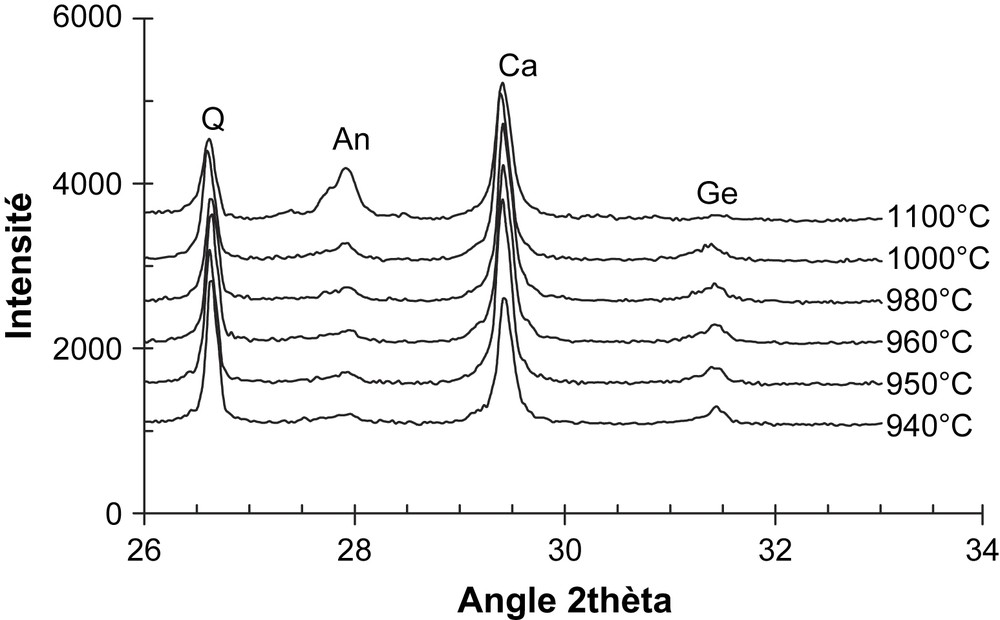
Germination et transformation de phases cristallines dans le mélange MEL15 à différentes températures (sans palier) et à 1100 °C (1 h de palier) : Q = quartz ; An = anorthite ; Ge = géhlénite ; Ca = calcite . La calcite a été introduite dans les tessons comme standard interne [7,14].
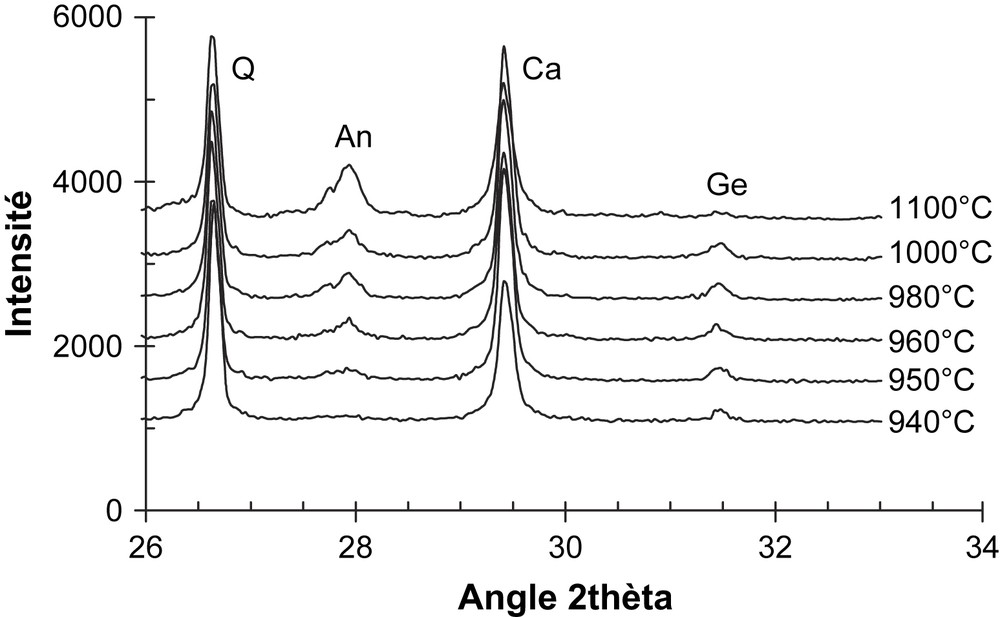
Germination et transformation de phases cristallines dans le mélange Kg2 + 15%calcite, à différentes températures (sans palier) et à 1100 °C (1 h de palier) : Q = quartz ; An = anorthite ; Ge = géhlénite ; Ca = calcite. La calcite a été introduite dans les tessons comme standard interne [7,14].
Par ailleurs, on constate une diminution de la ténacité dans la plage 0–15% de calcite. En considérant le facteur de conversion chaux/calcite (qui est de 0,56), les pourcentages de chaux que l'on a dans les mélanges POA, MEL5, MEL15,et MEL25 sont respectivement 0%, 2,8%, 8,4% et 14%. Le facteur de conversion chaux/anorthite étant de 0,20, on peut déduire que les taux théoriques d'anorthite dans les tessons des mélanges frittés à 1100 °C avec un palier final d'une heure sont respectivement 0%, 14%, 42% et 70%. Des travaux antérieurs ayant montré que les tessons des nuances POA ou Kg2 avec 15% de calcite contenaient en moyenne 30% d'anorthite [7,14], nous pensons qu'au-delà d'un certain taux de calcite, une partie des atomes de calcium ajoutés à l'argile POA pourrait se retrouver en dehors des structures de l'anorthite et de la géhlénite. Par conséquent, nous pensons que la diminution de la ténacité dans la plage 0–15% de calcite serait le reflet des modifications structurales induites par les réactions de la calcite avec les minéraux argileux, d'où la prise en compte des aspects microstructuraux des tessons.
3.3 Aspects microstructuraux
L'influence de la calcite sur la structure de POA frittée à 1100 °C est illustrée sur la micrographie MEB de la Fig. 6a. Le tesson de POA se caractérise par une structure non homogène, avec des tailles de pores de 2 à 3 μm. Les grains de quartz de tailles très variées sont incrustés dans une matrice amorphe. Comparativement, le tesson de la nuance MEL5 de la Fig. 6b comporte une densité plus élevée de grains, alors que la densité de gros pores est notablement réduite. Cette distribution, plus homogène, de grains et de pores est probablement à l'origine de la contrainte à la rupture élevée. La forte porosité qui caractérise le tesson de la nuance MEL10 est associée à une densité élevée de pores, comme le montre la Fig. 6c. Avec un taux massique de calcite de 15%, le caractère individuel des grains n'est plus marqué dans le tesson, et la porosité est accrue, comme le montre la Fig. 6d ; c'est ce qui explique certainement la grande fragilité de cette nuance de matériau. La porosité est très élevée dans les tessons de matériaux à 20 et 25% de taux massique de calcite, comme on peut le constater sur les Fig. 6e et f.
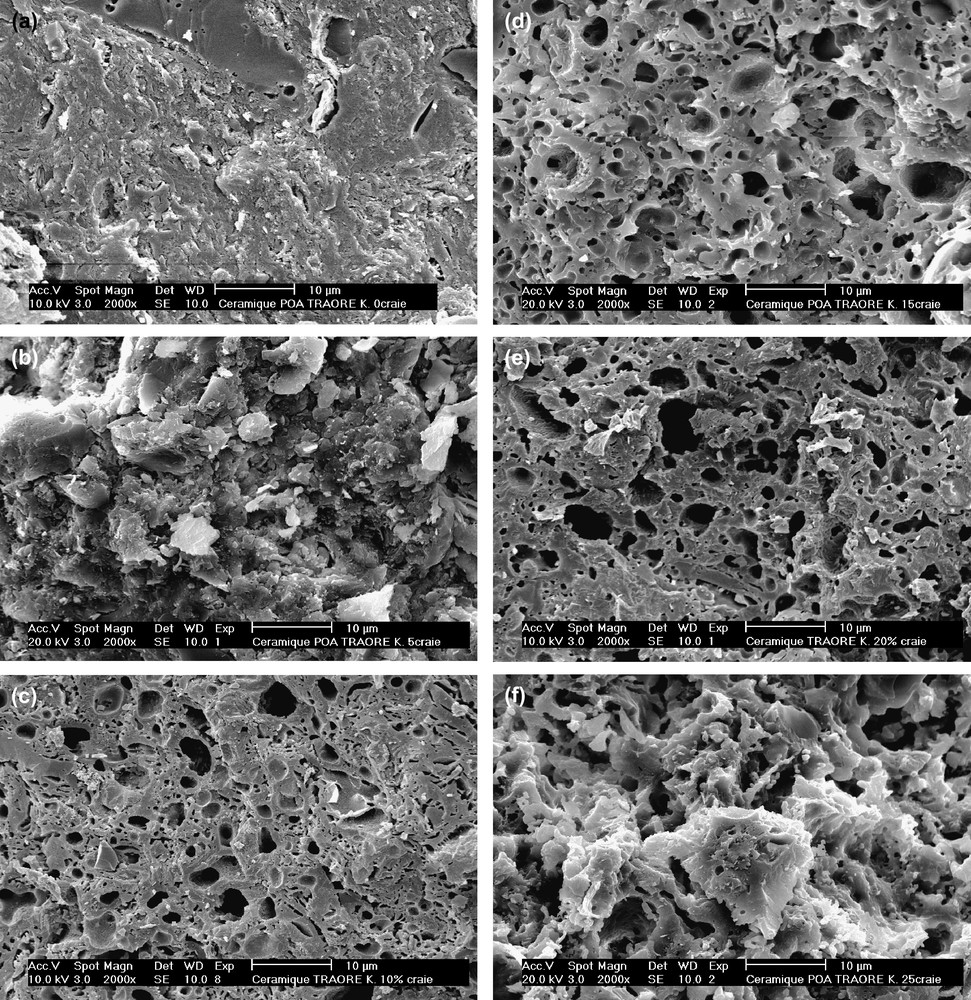
(a) Observation MEB du faciès de rupture du tesson de POA. (b) Observation MEB du faciès de rupture du tesson de MEL5. (c) Observation MEB du faciès de rupture du tesson de MEL10. (d) Observation MEB du faciès de rupture du tesson de MEL15. (e) Observation MEB du faciès de rupture du tesson de MEL20. (f) Observation MEB du faciès de rupture du tesson de MEL25.
3.4 Corrélation propriétés mécaniques–microstructure
Sur les Fig. 7a et b sont respectivement présentées une photographie en niveaux de gris et l'image binarisée associée, utilisées pour des mesures morphologiques du tesson d'un mélange POA-calcite.
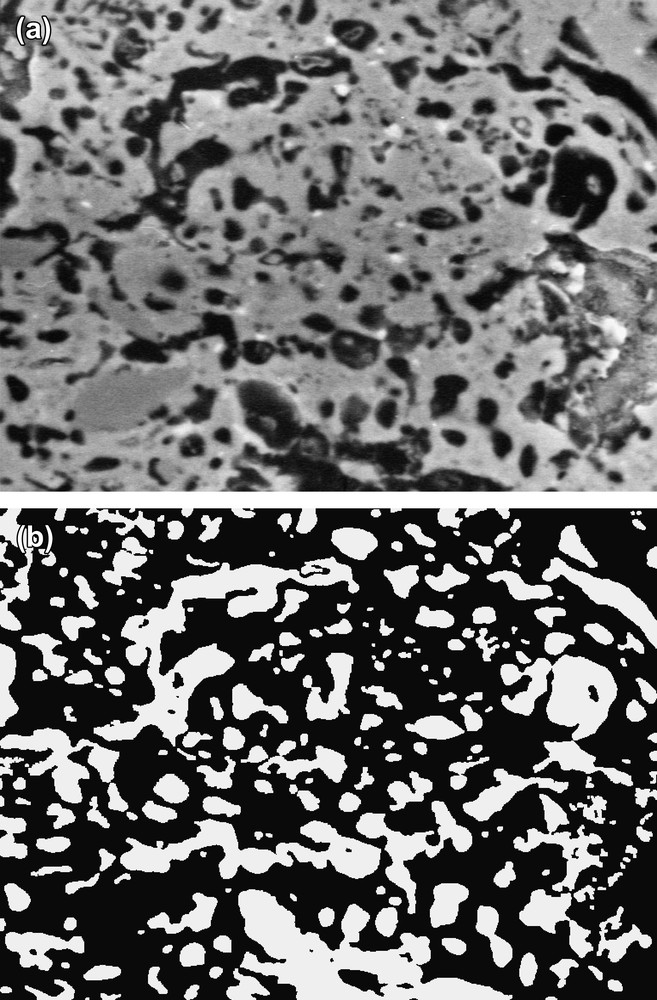
(a) Image en niveau de gris de faible grossissement obtenue sur le tesson d'un mélange POA-calcite. (b) Image binaire, associée à la Fig. 7a : la phase solide apparait en noir et les pores en blanc.
Les résultats des mesures morphologiques sont regroupés dans le Tableau 4. On remarque une faible dépendance du libre parcours moyen dans la phase poreuse vis-à-vis du taux de calcite ; la contribution majeure de la porosité provient des petits pores. Ces résultats indiquent que l'accroissement de la porosité résultant de la formation de nouvelles phases n'est pas corrélé au changement dans la morphologie des pores : leur densité croît avec le taux de calcite, mais pas leur taille moyenne. Ainsi donc, l'accroissement de la surface spécifique de l'interface solide/pore n'est pas dû à la présence de nouvelles structures plus fines, mais simplement à une forte densité de pores. Cette augmentation de la densité de pores se conjugue avec un amincissement des travées entre les pores mitoyens, c'est-à-dire une réduction du libre parcours moyen dans la phase solide.
Principaux paramètres morphologiques mesurés
| VV (P) | SV (S/P) | Lm (P) (μm) | Lm (S) (μm) | L∗(S) (μm) | |
| POA | 0,092 | 204 | 1,81 | 17,79 | 3,69 |
| MEL5 | 0,167 | 344 | 1,94 | 9,68 | 3,66 |
| MEL15 | 0,294 | 598 | 1,96 | 4,72 | 3,87 |
| MEL25 | 0,318 | 582 | 2,18 | 4,68 | 3,77 |
Pour un corps poreux, on peut s'attendre à ce que la ténacité soit dépendante de l'épaisseur des travées entre les pores. Afin de tester cette hypothèse, nous avons reporté sur la Fig. 8 la ténacité en fonction du libre parcours moyen dans la phase solide. La droite de régression tracée sur la figure semble bien décrire une relation linéaire entre la ténacité (paramètre mécanique) et le libre parcours moyen Lm(S) (paramètre morphologique). Le libre parcours moyen dans la phase solide semble donc un paramètre morphologique pertinent pour suivre l'influence du taux de calcite sur la microstructure des céramiques à base POA.
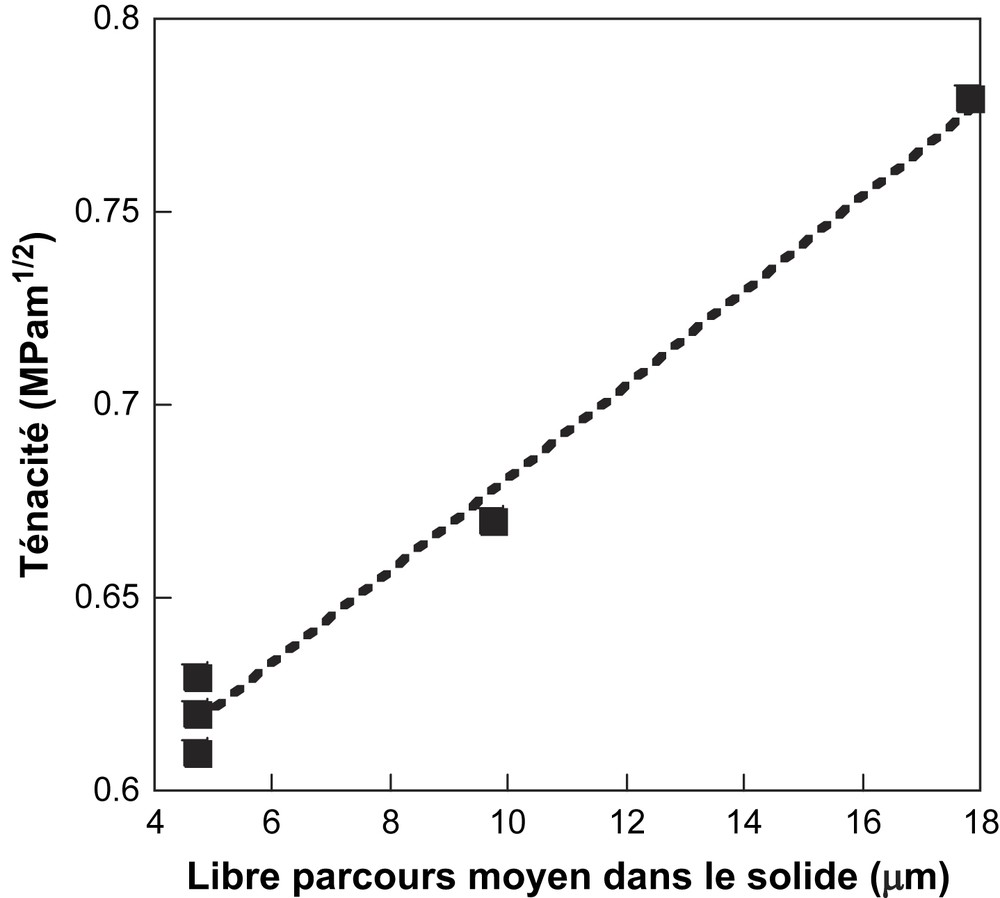
Relation entre la ténacité et le libre parcours moyen dans la phase solide.
4 Conclusion
La microstructure des matériaux céramiques obtenus en mélangeant une argile kaolinitique avec de la calcite a été étudiée par analyse d'image, et corrélée aux propriétés mécaniques. Bien que la présence de calcite réduise le retrait au frittage, la formation de phases cristallines intermédiaires génère une porosité multimode, qui détériore les propriétés mécaniques dès que le taux massique de calcite dépasse 5%. Ces matériaux poreux peuvent être caractérisés par une évolution linéaire de la ténacité en fonction du libre parcours moyen dans la phase solide.
Remerciements
Les auteurs expriment leur gratitude au département DSF de l'IRD (ex-Orstom) et à AIRE Développement pour leur soutien financier au travers de bourses d'étude octroyées à K. Traoré.
Vous devez vous connecter pour continuer.
S'authentifier