

1 Introduction
Mononuclear non-heme iron centers are frequently present in a variety of protein systems which perform important biological functions involving dioxygen [1–4,7,21]. The oxidative cleavage of catechol and other dihydroxy aromatics is a key step in the biodegradation by soil bacteria of naturally occurring aromatic molecules and many aromatic environmental pollutants [3,4]. The mononuclear non-heme iron proteins that catalyze the oxidative cleavage [5] of catechol or its derivatives with the incorporation of molecular oxygen are exemplified by catechol dioxygenases (Scheme 1). If two of the hydroxyl substituents in the catechol substrate are in the ortho positions then the ring cleavage (Scheme 2) can occur either between the two groups (intradiol) or between one hydroxyl group and an adjacent carbon atom (extradiol) [1,5–7,21,36]. The X-ray crystal structure of the intradiol-cleaving protocatechuate 3,4-dioxygenase (PCD) from Pseudomonas putida reveals a trigonal bipyramidal iron(III) site with four endogenous protein ligands (Tyr408, Tyr447, His460 and His462) and a solvent-derived ligand [5,9,10–12]. A very similar active site has been found [8,36] recently for another member of the intradiol dioxygenase family, namely, catechol 1,2-dioxygenase (CTD). The spectroscopic properties of the Fe(III) center are altered by substrate binding to an active site. In contrast, the extradiol dioxygenases have an iron(II) in the active site with two histidine and a glutamate ligand that form a square pyramidal coordination around the iron with two ligated water molecules [5,14].
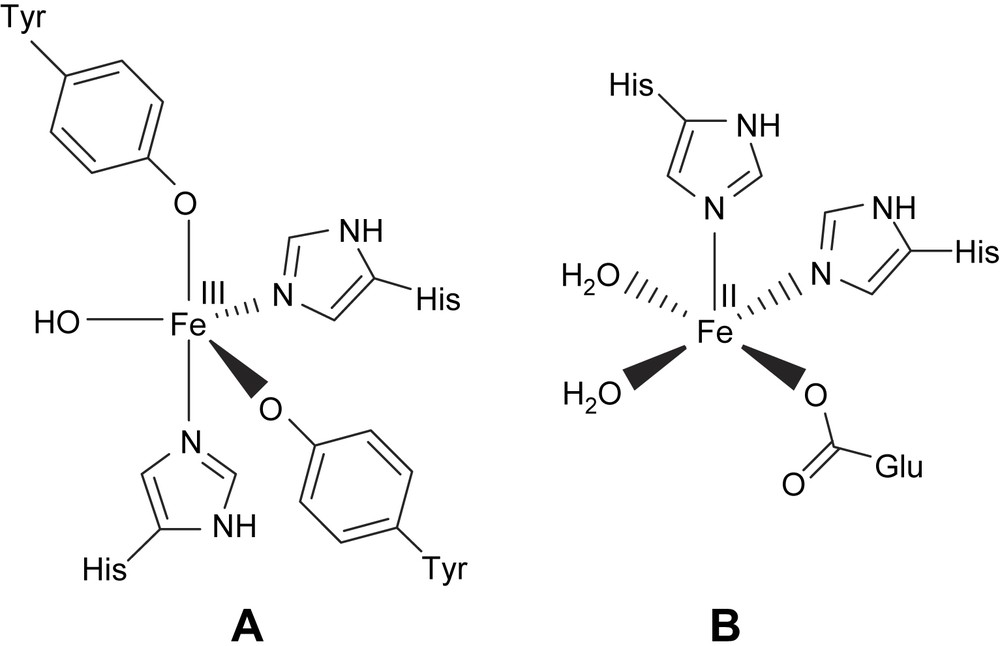
Active site structures of intradiol (A) and extradiol dioxygenase (B) enzymes.

Modes of cleavage of catechol.
As the interaction of iron(III) with phenolate moieties of tyrosine residues plays important roles in the enzyme function and in stabilizing the active site geometries of CTD and PCD enzymes, iron(III) complexes of phenolate ligands have attracted much interest as models to mimic the enzyme active sites and function. Initially Funabiki et al. reported the catalytic intra- and extradiol oxygenation of 3,5-di-tert-butylcatechol (H2DBC) by py/bipy/FeCl3 complexes [13]. In earlier studies Que and co-workers [5,14–17,49] synthesized a series of nitrogen-, carboxylate- and phenolate- containing iron(III) complexes, the catalytic properties of which have been explored. Indeed they found a clear relationship between the reactivity of the adducts and Lewis acidity of the iron(III) centers as modulated by the tripodal ligand, which plays an important role in dictating the catecholate-to-iron(III) charge-transfer absorptions occurring in the visible region. The tripodal ligand complexes reported by Nishida et al. [18] and Que and co-workers [15,16,49] contain a coordinated phenolate group, in addition to pyridyl moieties and they effect oxidative cleavage of catechols. Que and co-workers proposed a novel substrate rather than an oxygen activation mechanism [5,14,17] (Scheme 3) for the dioxygenase reaction, wherein coordination to iron(III) renders the substrate susceptible to dioxygen attack because of delocalisation of unpaired spin density from iron onto catecholate. The substrate catechol loses both of its protons upon coordination to the iron site and becomes susceptible to the reaction with oxygen to yield a peroxide intermediate which then decomposes to the product. The substrate activation mechanism implies that the yield of the desired cleavage product would increase with increase in Lewis acidity of the iron center.
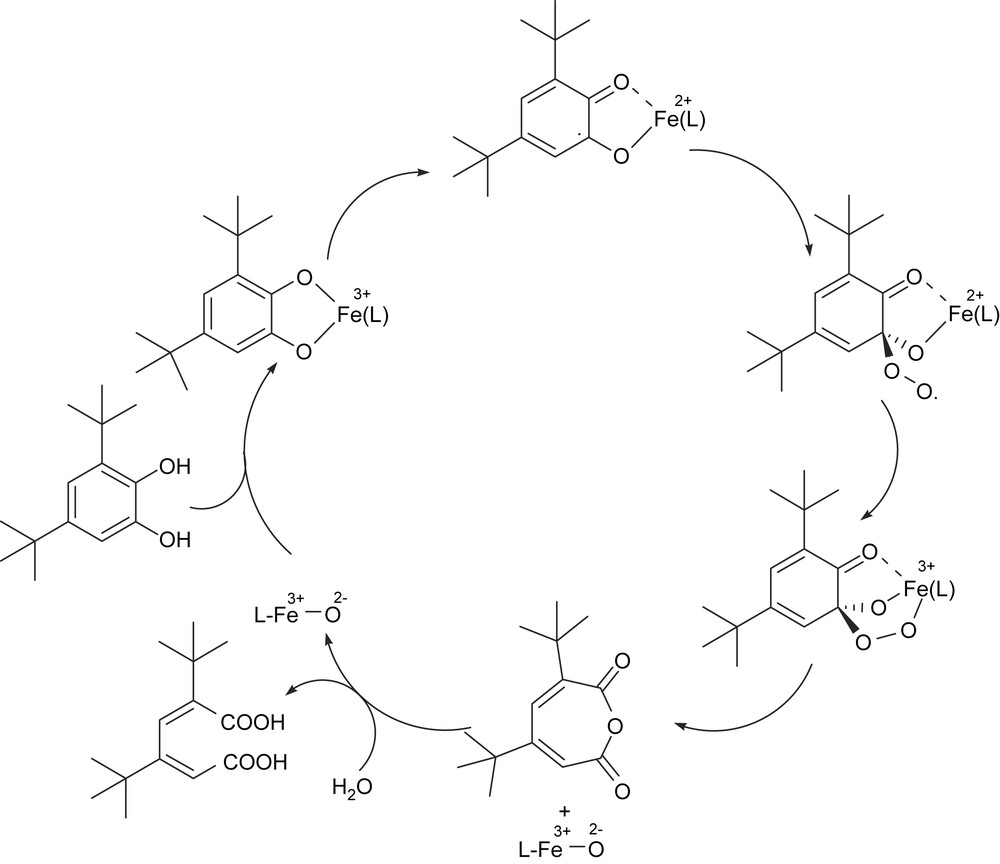
Proposed substrate activation mechanism for intradiol-cleavage of H2DBC.
In the present review, we summarize our work [19,20,22–25,38] on synthetic analogues for intradiol-cleaving catechol dioxygenases. We have isolated mononuclear iron(III) complexes of simple tridentate and linear and tripodal simple/sterically hindered tetradentate mono- and bisphenolate ligands [19,20,22–25], which closely mimic the enzyme active site structure and function. These ligands provide a reasonable analogue to histidine and tyrosinate coordination in CTD enzyme via the bzim and phenolate moieties. The study of iron(III) complexes of certain simple tridentate ligands with both phenolate and imidazole functionalities also provided the information pertinent to understanding of structure/spectra correlations and the function and reactivity of the active site. We have used 3,5-di-tert-butylcatechol (H2DBC) as the model substrate and the advantages are the relatively high stability of the main cleavage product and the fast reaction of the catecholate complexes with dioxygen.
2 Functional models for 1,2-CTD
2.1 Iron(III) complexes of linear tridentate 3N ligands
The mononuclear iron(III) complexes of the linear tridentate 3N ligands (Scheme 4) bis(pyrid-2-ylmethyl)amine (L1), N,N-bis(benzimidazol-2-ylmethyl)amine (L2), N-methyl-N′-(pyrid-2-ylmethyl)ethylenediamine (L3), N,N-dimethyl-N′-(pyrid-2-ylmethyl)ethylenediamine (L4) and N-phenyl-N′-(pyrid-2-ylmethyl)ethylenediamine (L5) have been found to be functional models for catechol dioxygenases [25]. The differently substituted amine functions in the ligands are expected to tune the Lewis acidity of the iron(III) center and hence the electrochemical properties and dioxygenase activity of the complexes. The X-ray crystal structures [22,23,25] of [Fe(L1)Cl3] and [Fe(L4)Cl3] (Fig. 1) reveal that the 3N ligands are cis-facially coordinated and the cis-coordinated chloride ions can be replaced by bidentate catechols. In fact, hyperfine broadening observed from O17-enriched water for the 4-HBA–PCD complex [26] [4-HBA = 4-hydroxybenzoate] has shown that at least two coordination sites of Fe(III) appear adjacent and are accessible to exogenous ligands. Thus on the addition of even neutral H2DBC rather than H2CAT (catechol), two charge-transfer bands are exhibited with relatively low absorptivity, and on the addition of Et3N the absorptivity is enhanced due to the deprotonation of H2DBC. This reveals the spontaneous deprotonation of H2DBC rather than H2CAT, to bind strongly to Lewis acidic iron(III) center. The two new bands arise due to involvement of different ligand orbitals of catecholates in ligand-to-metal charge-transfer (LMCT) transitions. The energies of both the bands are shifted to higher region as the substituents on the catecholate ring are varied from electron donating to electron withdrawing: H2DBC > H2TBC (4-tert-butylcatechol) > H2CAT > H2TCC (3,4,5,6-tetrachlorocatechol). Electron-donating substituents would be expected to raise the energy of the catecholate frontier orbitals and thus minimize the ligand-to-metal energy gap. The position of the low-rather than high-energy LMCT band exhibits remarkable dependence on the nature of the tridentate ligand [15,27–29], and the magnitude of energy of this band reflects the Lewis acidity of the iron(III) center as modified by the 3N ligands. The incorporation of –NMe2 group as in [Fe(L4)Cl3] shifts the bands to longer wavelengths, illustrating that steric hindrance rather than electron-releasing effect of –NMe2 group is important in catecholate adduct formation. The similarity between the electronic spectra of the catecholate adducts of the complexes and the catechol-bound enzymes suggests that the catecholatodianion is chelated to iron(III) center in the enzymes.
Structures of tridentate 3N ligands.
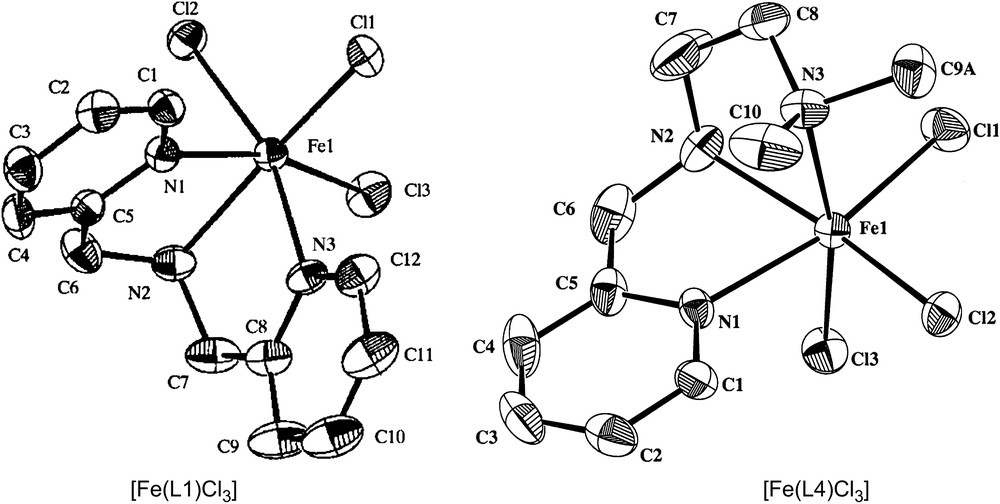
Structures of [Fe(L1)Cl3] and [Fe(L4)Cl3].
The E1/2 values of FeIII/FeII redox couple in methanol solution follow the trends [Fe(L1)Cl3] > [Fe(L2)Cl3]; [Fe(L5)Cl3] > [Fe(L4)Cl3] > [Fe(L3)Cl3] reflecting the decrease in Lewis acidity of the iron(III) center on replacing (i) the pyridyl moieties in [Fe(L1)Cl3] by the more basic benzimidazole moieties as in [Fe(L2)Cl3], (ii) the electron-attracting N-Ph group in [Fe(L5)Cl3] by the electron-releasing amine N-Me group as in [Fe(L3)Cl3], and (iii) the amine N–H atom in [Fe(L3)Cl3] by the electron-releasing amine N-Me group as in [Fe(L4)Cl3]. On coordination of DBC2−, the E1/2 values decrease enormously (800 mV) reflecting considerable decrease in Lewis acidity of iron center [27] on substrate binding and supporting the suggestion that the iron(III) center in the dioxygenases not only takes part in activation of the substrate molecules, but also favors the later stages of the reaction.
The ability of the complexes to catalyze the cleavage was studied by reacting the complexes with equimolar amount of H2DBC in DMF in the presence of molecular oxygen. The oxidative intradiol products 2–9 (Table 1, Scheme 5) were identified and quantified by GC and GC–MS techniques. The very low abundance of the oxidative extradiol-cleavage products observed is very much in contrast to the nearly quantitative yield of the oxidative extradiol-cleavage products observed [30,31] for the reaction of [FeL(DBC)Cl] (L is the cis-facially coordinated macrocyclic ligand 1,4,7-trimethyl-1,4,7-triazacyclononane) with O2. The cis-facial coordination allows both O2 and substrate to occupy the opposite face to form an intermediate that leads to the desired extradiol products. In contrast, only the intradiol-cleavage product 3,5-di-tert-butyl-1-oxocyclohepta-3,5-diene-2,7-dione (20%) was observed [32] when L in [FeL(DBC)Cl] is the meridionally coordinated 2,2′:6′,2″-terpyridine. It appears that the linear 3N ligands are involved possibly in a meridional rather than cis-facial coordination in the catecholate complex, which is unable to form the intermediate [(L)(DBSQ)Fe(II)–O2]− responsible for extradiol-cleavage to occur [33,34].
Electronic spectral, redox potentiala and kinetic data and % yield of intradiol-cleavage products of iron(III) complexes [25]
| Complex | λmax, nm (ɛ, M−1 cm−1) | DBC adduct | E1/2 (V) | (M−1 s−1) | (% Yield) | |
| CV | DPV | |||||
| [Fe(L1)Cl3] | 380 (1760) | 820 (3260) | 0.267 | 0.271 | 1.7 × 10−2 | 89.0 |
| 505 (1970) | ||||||
| [Fe(L2)Cl3] | 365 (3700) | 815 (3260) | 0.100 | 0.121 | 1.2 × 10−1 | 92.0 |
| 540 (1810) | ||||||
| [Fe(L3)Cl3] | 375 (5100) | 790 (3260) | 0.146 | 0.150 | 1.3 × 10−2 | 86.0 |
| 490 (1930) | ||||||
| [Fe(L4)Cl3] | 370 (4160) | 815 (2820) | 0.230 | 0.221 | 4.7 × 10−3 | 82.0 |
| 500 (1890) | ||||||
| [Fe(L5)Cl3] | 370 (5820) | 850 (3260) | – | 0.263 | 2.8 × 10−2 | 90.0 |
| 515 (1870) |
a Supporting electrolyte: 0.1 M NBu4ClO4 (TBAP), scan rates: 50 mV s−1 (CV), 1 mV s−1 (DPV), reference electrode: Ag/Ag+, working electrode: Pt sphere.

Oxidative cleavage products of H2DBC mediated by iron(III) complexes: 3,5-di-tert-butyl-1-oxacyclohepta-3,5-diene-2,7-dione (1), 3,5-di-tert-butyl-5-(N,N-dimethyl-amidomethyl)-2-furanone (2), 3,5-di-tert-butyl-5-(2-oxo-2-piperidin-ylethyl)-5H-furanone (3), 3,5-di-tert-butyl-5-formyl-2-furanone (4), 3,5-di-tert-butyl-2-pyrone (5), 4,6-di-tert-butyl-2-pyrone (6), 3,5-di-tert-butyl-2-hydroxy-1,4-benzo-quinone (7), 2,5-di-tert-butyl-2H-pyran-3,6-dione (8), 3-tert-butylfuran-2,5-dione (9).
The DBC2− adducts of the complexes were generated in situ in DMF solution and their reactivity towards O2 was investigated by monitoring the decay of DBC2−. The pseudo-first-order kinetics due to excess of dioxygen is judged from the linearity of the plot of [1 + log(Abs)] vs time. The rates of the reactions (Table 1) were calculated [35,28] by using the equation .
The observed values [30,32] of for the oxidative reaction catalyzed by the complexes follow the trends: Fe(L2)3+ > Fe(L1)3+; Fe(L5)3+ > Fe(L3)3+ > Fe(L4)3+. Since mainly intradiol-cleavage products are obtained, the oxidative reactions catalyzed by the complexes correspond exclusively to the intradiol-cleavage pathway; hence the trend in reactivity of these adducts may be illustrated by invoking the novel substrate activation mechanism proposed [5,14,17] for the intradiol-cleaving dioxygenase enzymes. Thus the higher Lewis acidity and lesser steric hindrance offered by the pyridine moieties in [Fe(L1)]3+ towards substrate binding illustrate the higher yield of oxygenated product observed by us earlier [19,22,23]. However, interestingly, the rate of oxygenation observed for [Fe(L2)Cl3] is almost 10 times higher than its pyridine analogue [Fe(L1)Cl3]. It is possible that the N–H groups in the benzimidazole rings in L2 may be involved in hydrogen bonding [37] to the substrate-bound peroxo group in the peroxo intermediate [(L)(DBSQ)Fe(III)O2] involved in the intradiol pathway leading to its stabilisation. Also, the release of the products from the intermediate, which is the rate-determining phase of the oxygenation reaction catalyzed by the enzymes, may be facilitated by the bulky benzimidazoles in [Fe(L2)Cl3]. Similarly, the bulky N-Ph group in [Fe(L5)Cl3], in addition to the electron sink property of the N-phenyl group, would stabilise the reaction intermediate [(L)(DBSQ)Fe(III)O2], to confer the highest rate of cleavage. Similarly, in addition to the higher Lewis acidity of the iron(III) center in [Fe(L3)Cl3], as discussed above, the hydrogen bonding involving amine N–H groups would render the catecholate adduct of the complex react much faster than the adduct of [Fe(L4)Cl3] does. Thus, the present octahedral complexes with a cis-facially coordinated tridentate linear 3N ligand tend to elicit intradiol-cleavage rather than extradiol-cleavage products, which is in remarkable contrast to the iron(III) complexes [30,31] of cis-facially coordinated macrocyclic 3N ligands. The observed rates of dioxygenase reactions of complexes could be illustrated by invoking the Lewis acidity of the iron(III) center and the hydrogen bonding potential of the ligands and by assuming the release of products from the intermediate as rate-determining step, which is facilitated by the steric bulk of the primary ligand.
2.2 Iron(III) complexes of tridentate monophenolate ligands
A series of iron(III) complexes of the linear tridentate (2NO) ligands (Scheme 6) N-(pyridin-2-ylmethyl)salicylideneamine [H(L6)], (2-hydroxy-5-nitrobenzyl)(pyridin-2-ylmethyl)amine [H(L7)], (2-hydroxy-5-nitrobenzyl)(pyridin-2-ylethyl)amine [H(L8)], N-(2-imida-zole-4-ylethyl)salicylideneamine [H(L9)], N-(benzimidazol-2-ylethyl)salicylideneamine [H(L10)], (benzimidazol-2-ylethyl)(2-hydroxybenzyl)amine [H(L11)] and (benzimidazol-2-ylethyl)(2-hydroxy-5-nitrobenzyl)amine [H(L12)] [22,38] have been employed as functional models for 1,2-CTD. The complexes with coordinated bzim and phenolate moieties provide a reasonable analogue to histidine and tyrosinate coordination in CTD enzymes. The ligands H(L6), H(L9) and H(L10) on deprotonation would be meridionally coordinated to iron(III). However, the saturation of the imine function in them to generate H(L7), H(L8), H(L11) and H(L12) would allow more flexibility in chelation leading to cis-facial coordination to metal [22,38]. As for the 3N ligand complexes, the cis-coordination sites are open in the complexes for coordination of bidentate catechols and so the complexes are convenient for investigating the effect of substrate adduct formation on spectra and redox of iron(III) complexes.
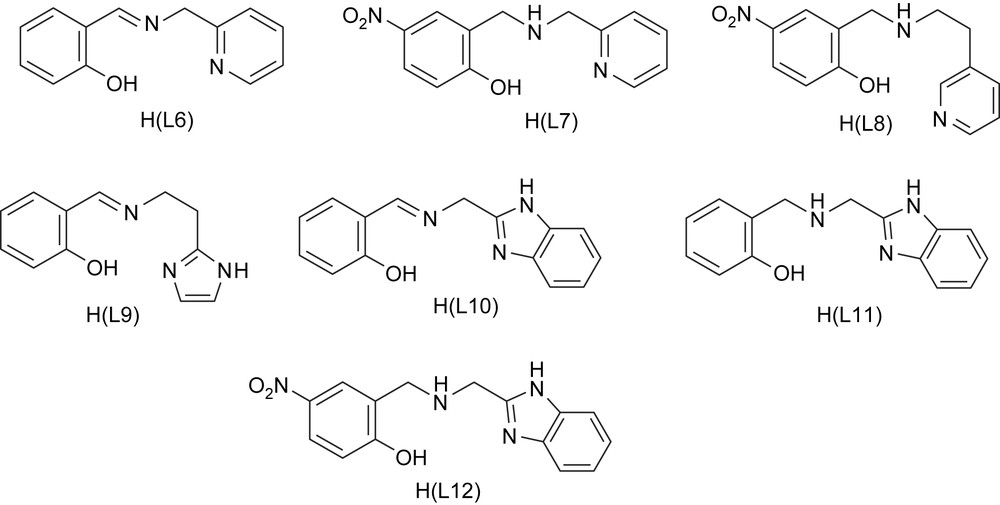
Structures of tridentate monophenolate ligands.
The electronic spectra of all the iron(III)–phenolate complexes in methanol solution (Table 2) exhibit a band in the visible region (550–470 nm), which is absent in the 3N ligand (L1–L5) complexes lacking phenolate function. This band is assigned [39,40] to phenolate (π1) → Fe(III) [dπ(dxz)] LMCT transitions. The remarkable increase in energy of this band in the order Fe(L9) < Fe(L10) < Fe(L6) < Fe(L8) < Fe(L11) < Fe(L12) < Fe(L7) represents the decreasing order of Lewis acidity of ferric center, as modified by the nature of the ligands. The two new bands (430–470, 630–890 nm) that appear on the addition of CAT2− and DBC2− to the iron(III) complexes may originate [15,41,49] from CAT2−/DBC2− → Fe(III) charge transfer transitions involving two different catecholate orbitals. The trend in the low-energy CAT2−/DBC2− → Fe(III) LMCT band energies is consistent with that in the phenolate → Fe(III) LMCT band energy. The high-energy CAT2−/DBC2− → Fe(III) charge-transfer band would have merged with the blue-shifted endogenous phenolate → Fe(III) LMCT band. The blue shift in the band indicates the conversion of coordinated catecholate to a fairly basic ligand, which would be consistent with peroxide species [50] in the proposed mechanism. The sensitivity of the band positions as well as their intensities to the iron environment is reminiscent of native CTD and PCD enzymes [42].
Electronic spectral, redox potentiala data and % yield of cleavage products of iron(III) complexes [19,22,38]
| Complex | λmax, nm (ɛ, M−1 cm−1) | DBC adduct | E1/2 (V) | (% Yield) | |
| Fe(III)/Fe(II) | DBSQ/H2DBC | ||||
| [Fe(L6)Cl2]·H2O | 543 (400) | 640 (100) | −0.169 | −0.116 | 19.8 |
| 528 (sh) | |||||
| [Fe(L7)Cl2]·H2O | 478 (450) | 693 (260) | −0.330 | −0.244 | 42.4 |
| 415 (sh) | |||||
| [Fe(L8)Cl2]·H2O | 510 (300) | – | −0.260 | −0.277 | – |
| [Fe(L9)Cl2]·H2O | 580 (1730) | – | −0.131 | −0.139 | – |
| [Fe(L10)Cl2]·H2O | 554 (1630) | 567 (3990) | −0.177 | −0.121 | 37.0 |
| [Fe(L11)Cl2]·H2O | 508 (1540) | 575 (1050) | −0.887 | −0.505 | 24.8 |
| [Fe(L12)Cl2]·H2O | 503 (1760) | 650 (sh) | −0.319 | −0.246 | 23.5 |
| [Fe(L15)Cl2] | 540 (3200) | 780 (3570) | −0.294 | – | – |
| 435 (2325) | |||||
| [Fe(L16)Cl(H2O)] | 493 (650) | – | −0.310 | – | – |
| [Fe(L17)Cl3] | 356 (4370) | 842b | −0.002 | −0.054 | 18 |
| 548 | |||||
| [Fe(L18)Cl2] | 500 (880) | 768 (360) | −0.130 | −0.130 | 59 |
| 492 (510) | |||||
| [Fe(L19)Cl(H2O)] | 492 (3120) | 780 (sh) | −0.301 | −0.240 | 50 |
a Supporting electrolyte: 0.1 M NBu4ClO4 (TBAP), scan rates: 50 mV s−1 (CV), 1 mV s−1 (DPV), reference electrode: Ag/Ag+, working electrode: Pt sphere.
b From Ref. [16].
In methanol solution all the complexes exhibit a fairly reversible to irreversible Fe(III) → Fe(II) redox behaviour [22,38]. The values of Fe(III)/Fe(II) redox potential follow the order, Fe(L9) > Fe(L10) > Fe(L6) ∼ Fe(L8) > Fe(L12) > Fe(L7) reflecting the decrease in Lewis acidity of the iron(III) center, which is consistent with the trend derived from the phenolate to iron(III) LMCT band energies and thus the present complexes exhibit a linear correlation [38] between the latter and the redox potentials. The adducts [Fe(L)(DBC)]n− generated in situ in methanol solution reveal a new reversible to irreversible wave corresponding to DBSQ/DBC2− couple of coordinated DBC2− [17], in addition to an irreversible Fe(III) → Fe(II) reduction peak located at potentials more negative than the parent complexes. The redox potential of coordinated DBSQ/DBC2− couple (0.008–0.615 V, NHE) (Table 2) is considerably more positive than that of free DBSQ/DBC2− couple (−1.096 V, NHE) [43,44], reflecting the significant stabilization of DBC2− towards oxidation by coordination to iron(III). These potentials are comparatively more positive than, and exhibit the same trend as, that of the Fe(III)/Fe(II) couple. The stabilization of DBC2− oxidation state in substrate-coordinated complex is dictated by nature of the ligand. The higher the Lewis acidity of the iron center, the larger the stabilization of the DBC2− oxidation state. Interestingly, the trend in these potentials follows that in DBC2− → Fe(III) LMCT band energies. The decreasing order of the E1/2 values Fe(L9) > Fe(L6) > Fe(L10) ∼ Fe(L7) ∼ Fe(L12 > Fe(L11) reveals the increasingly thermodynamically favourable electron transfer from chelated catecholate complex to dioxygen and hence represents a decrease in catalytic activity in this order (see below).
The experimental cleavage yields for the present DBC2− adducts follow the order of Lewis acidity, as derived from DBC2− → Fe(III) LMCT band energies and E1/2 of the Fe(III)/Fe(II) and DBSQ/DBC couples. The Lewis acidity of the iron(III) center in the complex as well as the DBC2− adducts is modulated by the nature of the donor functionalities of the phenolate ligand and the role of phenolate ligands in modulating the Lewis acidity and determining the course of the catalytic reaction is emphasized.
2.3 Iron(III) complexes of tripodal tetradentate ligands
A series of iron(III) complexes of the tetradentate tripodal ligands tris(pyrid-2-ylmethyl)amine (L13), bis(pyrid-2-ylmethyl)(pyrid-2-ylethyl]amine (L14), [2-bis(pyrid-2-ylmethyl)aminomethyl]-4-nitrophenol [H(L15)], N,N-bis(2-hydroxy-5-nitrobenzyl)-aminomethylpyridine [H2(L16)], tris(benzimidazol-2-ylmethyl)amine (L17), 2-bis(benz-imidazol-2-yl)methylaminomethyl-4-nitrophenol [H(L18)] and N,N-bis(2-hydroxy-5-nitrobenzyl)aminomethylbenzimidazole [H2(L19)] has been isolated and studied as functional models for 1,2-CTD [19,22]. These tetradentate tripodal ligands based on trimethylamine (Scheme 7) with the pendant functionalities varying from pyridine to benzimidazole to phenolate have been synthesized to provide a systematic variation of ligand Lewis basicity and charge while retaining similar coordination geometries in iron(III) complexes. The bulky bzim ring(s) seems to be a good choice to offer steric hindrance to substrates as in enzymes so as to closely approximate the active site in enzymes. The X-ray crystal structures of [Fe(L15)Cl2] and [Fe(L17)Cl2]Cl (Fig. 2) have been determined to demonstrate the availability of cis-coordination positions for the binding of bidentate catechol substrates.

Structures of the tripodal tetradentate ligands.

Structures of [Fe(L15)Cl2] and [Fe(L17)Cl2]Cl.
The electronic spectra of the iron(III)–phenolate complexes (Table 2) exhibit an intense band in the visible region (490–550 nm), which is assigned [39,40] to the phenolate(π1) → Fe(III)(dπ) LMCT transition. The increasing order of energy of this band Fe(L15) < Fe(L16) < Fe(L18) < Fe(L19) represents the decreasing order of Lewis acidity of the ferric center. Two new bands (430–470, 630–890 nm) appear on the addition of CAT2− or DBC2− to all the iron(III) complexes [15,41]. Obviously, the high-energy CAT2−/DBC2− → Fe(III) LMCT band would have merged with the endogenous phenolate → Fe(III) LMCT band, now blue-shifted on adduct formation. Further, the spectral features are similar to those observed [16] for the interaction of catechol with phenylalanine hydroxylase (PAH) (410, 700 nm) and for that of soybean lipoxygenase with 4-nitrophenol (395, 630 nm) and protocatechuic acid (455, 670 nm); this suggests that the Lewis acidity of the iron center in the present synthetic complexes with one phenolate group, rather than those with two such groups or those with three py or bzim pendants, is comparable to those in the enzyme–substrate complexes. The low-energy CAT2−/DBC2− → Fe(III) LMCT band shifts to higher energy as the softer nitrogen ligand is replaced by pendant phenolate: Fe(L13) ∼ Fe(L14) < Fe(L15) < Fe(L16); Fe(L17) < Fe(L18) < Fe(L19). All the parent chloride complexes show fairly reversible to irreversible Fe(III)/Fe(II) redox process. The values of Fe(III)/Fe(II) redox potentials of the complexes follow the orders Fe(L13) > Fe(L14) > Fe(L15) > Fe(L16); Fe(L17) > Fe(L18) > Fe(L19) reflecting the decrease in Lewis acidity of the iron(III) center as the charge on the tetradentate tripodal ligand set increases. This trend is consistent with that derived from CAT2−/DBC2− → Fe(III) and PhO− → Fe(III) LMCT band energies, and in fact, the present data fit into the straight line obtained when PhO− → Fe(III) LMCT band energies are plotted against E1/2 of Fe(III) complexes of the linear tridentate phenolate (2NO) ligands (cf. above) [19,22,23].
The oxidative intradiol-cleavage of H2DBC was observed when it was mixed with iron(III) complexes in nitromethane solution in the molar ratio 50:1 and kept for four days. All the complexes act as catalysts in the oxidative intradiol-cleavage of H2DBC (Table 2, Scheme 5) because the amounts of product 1 obtained [Fe(L13), 41%; Fe(L14), 45%; Fe(L15), 12%; Fe(L16), 56%; Fe(L17), 18%; Fe(L18), 59%; Fe(L19), 50%] are more than that of the complex used. The yields show a dependence on the nature of the tetradentate ligand, revealing that the coordination chemistry of the iron(III) center plays a role in determining the course of the reaction. Thus the yields for Fe(L13) and Fe(L14) are higher than that for Fe(L17), possibly due to a decrease in Lewis acidity and an increase in steric hindrance to substrate binding on replacing py by the more basic bzim pendant. It has been rationalized [5] that a decrease in Lewis acidity of the iron center decreases the covalency of the iron–catecholate interaction and decreases the semiquinone character of the bound catecholate. The present observation also supports the proposal [49] that the increased yield of the desired cleavage product with increased Lewis acidity of the metal center reflects the coordination [45] of the intermediate peroxide to the metal center. A decrease in Lewis acidity on introducing one phenolate donor, as in Fe(L18), diminishes the Lewis acidity of the iron center and hence the yield of the cleavage product. On the other hand, the introduction of two phenolate donors as in Fe(L16) and Fe(L19) tends to increase the yield. This is consistent with the observation [46] that the rate-determining phase of the enzyme reaction is the product release and not the oxygen attack or the ring opening. If the dissociation of the Fe–O(substrate) bond in the oxygenated product in the novel substrate activation mechanism [5,14,17] proposed by Que and co-workers is the rate-determining step, then it would be facilitated by the increased negative charge built on the iron centers in Fe(L16) and Fe(L19) complexes. Thus, though the observed yields of desired cleavage product could not be illustrated solely on the basis of the Lewis acidity of complexes, all these observations are consistent with the substrate activation mechanism proposed for intradiol-cleaving dioxygenases.
2.4 Iron(III) complexes of sterically hindered tripodal monophenolate ligands
The iron(III) complexes [Fe(L15)Cl2]·CH3CN, [Fe(L20)Cl2], [Fe(L21)Cl2] and [Fe(L22)Cl2], where L20 = N,N-dimethyl-N′-(pyrid-2-yl-methyl)-N′-(2-hydroxy-4-nitrobenzyl)ethylenediamine, L21 = N,N-dimethyl-N′-(6-methylpyrid-2-ylmethyl)-N′-(2-hydroxy-4-nitrobenzyl)ethylenediamine and L22 = N,N-dimethyl-N′-(1-methylimidazole-2-ylmethyl)-N′-(2-hydroxy-4-nitro-benzyl)ethylenediamine (Scheme 8) have been isolated [24]. The pendant pyridine/imidazole functionality has been incorporated into the ligand to provide a systematic variation in the Lewis acidity of the iron(III) center. The bulky N,N-dimethyl group, the pyridine ring nitrogen sterically hindered by 6-methyl group and the more basic N-methylimidazole moiety [pKa (BH+): imidazole, 6.0; pyridine, 5.2] in the ligands are expected to influence the iron(III) coordination structures as well as the electronic properties of the complexes and offer steric hindrance to the substrates so as to closely resemble the active site in enzyme–substrate complexes. The tetradentate ligands provide a reasonable analogue to histidine and tyrosinate coordination in CTD and PCD enzymes via the heterocyclic nitrogen donors and phenolate moieties, respectively.
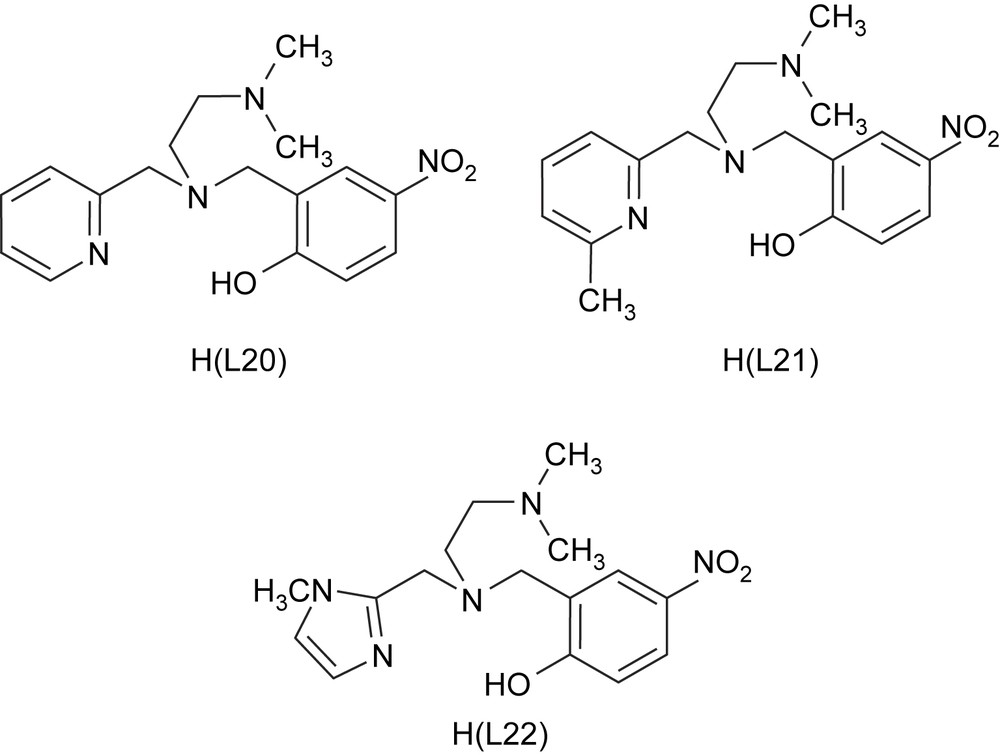
Structures of the sterically hindered monophenolate ligands.
The X-ray crystal structures of [Fe(L15)Cl2]·CH3CN and [Fe(L20)Cl2] reveal the presence of two chloride ions in cis-positions, which can be replaced by bidentate catecholate ions. In [Fe(L20)Cl2], the Fe–N3 bond is significantly longer than the Fe–N2 bond, due to the inability of the sterically hindering –NMe2 group to strongly coordinate to iron. The replacement of the pyridine moiety in [Fe(L15)Cl2]·CH3CN by the sterically demanding –NMe2 group enhances the Fe–O–C bond angle from 128.5° to 136.1°. This angle is higher than the ideal value of 120° for sp2 hybridized phenolate oxygen atom, indicating that the latter (in-plane pπ orbital) interacts less strongly with a half-filled dπ∗ orbital on iron(III), and also that the angle is higher than the average Fe–O–C bond angle of ∼128.5° observed in other octahedral iron(III)–phenolate complexes [5,18,20,47,48]. This illustrates the importance of extended π-delocalisation involving p-nitrophenolate and iron(III) d-orbitals.
The iron(III) monophenolate complexes [Fe(L20)Cl2]–[Fe(L22)Cl2] display a relatively intense band in the 535–550 nm region, which is assigned to phenolate (π) → Fe(III) (dπ∗) LMCT transition [39,40]. On the other hand, the high-energy band observed in the 425–435 nm range is assigned to phenolate (σ) → Fe(III) LMCT transition. The band energy of the lowest energy LMCT band decreases in the order [Fe(L22)Cl2] > [Fe(L20)Cl2] > [Fe(L15)Cl2] > [Fe(L21)Cl2], reflecting the increase in Lewis acidity of the iron(III) center in this order. Thus the Lewis acidity of the iron(III) center is fine-tuned by modifying the ligand environment through the replacement of a pyridine moiety by an imidazole moiety and suitable incorporation of methyl groups on the heterocyclic rings. Two new visible bands (480–490, 635–800 nm, Table 3) appear on adding catecholate dianions. The position of the low- rather than high-energy LMCT band of the catecholate adducts shows remarkable dependence on the nature of the primary ligand [15–17,35,50] and, in fact, the magnitude of energy of this band represents the Lewis acidity of the iron(III) center as modified by the phenolate ligands.
Electronic spectral, redox potentiala, kinetic data and % yield of cleavage products of iron(III) complexes [20,24]
| Complex | λmax, nm (ɛ, M−1 cm−1) | E1/2 (V) | (M−1 s−1) | (% Yield) | |
| CV | DPV | ||||
| [Fe(L20)Cl2] | 535 (3570) | −0.262 | −0.266 | ||
| 430 (3195) | |||||
| [Fe(L20)(DBC)] | 790 (3260) | – | – | 4.6 × 10−3 | 60.3 |
| 490 (3325) | |||||
| [Fe(L21)Cl2] | 550 (3190) | −0.176 | −0.152 | ||
| 365 (13 890) | |||||
| [Fe(L21)(DBC)] | 800 (3465) | – | – | – | – |
| 495 (3265) | |||||
| [Fe(L22)Cl2] | 535 (3570) | −0.278 | −0.280 | ||
| 425 (3645) | |||||
| [Fe(L22)(DBC)] | 760 (2630) | – | – | 2.9 × 10−2 | 83.0 |
| 490 (3085) | |||||
| [Fe(L23)Cl] | 475 (4530) | −0.495 | −0.483 | ||
| [Fe(L23)(DBC)] | 540 (4530) | −0.320 | −0.313 | – | |
| [Fe(L24)Cl(H2O)] | 515 (3530) | −0.202 | −0.205 | ||
| 430 (5510) | |||||
| [Fe(L24)(DBC)] | 700 (15 970) | −0.495 | −0.525 | 2.8 × 10−3 | 100 |
| 410 (14 780) | −0.101 | ||||
| [Fe(L25)Cl] | 550 (2820) | −0.581 | |||
| 340 (4360) | |||||
| [Fe(L25)(DBC)] | 545 (2670) | −0.394 | −0.883 | – | – |
| 470 (2480) | −0.395 | ||||
| [Fe(L26)Cl(H2O)] | 515 (4860) | −0.354 | −0.345 | ||
| 425 (8250) | |||||
| [Fe(L26)(DBC)] | 690 (2015) | −0.763 | Very fast | – | |
| 410 (25 000) | −0.191 | −0.189 |
a Supporting electrolyte: 0.1 M NBu4ClO4 (TBAP), scan rates: 50 mV s−1 (CV), 1 mV s−1 (DPV), reference electrode: Ag/Ag+, working electrode: Pt sphere.
In methanol solution, all the monophenolato complexes exhibit a completely irreversible redox behavior (Table 3). The E1/2 values of FeIII/FeII redox potentials of the monophenolato complexes follow the trend [Fe(L21)Cl2] > [Fe(L20)Cl2] > [Fe(L22)Cl2] > [Fe(L15) Cl2], which represents the decrease in Lewis acidity of the iron(III) center, consistent with the above spectral results. On replacing the pyridyl arm in [Fe(L15)Cl2] by –NMe2 group to obtain [Fe(L20)Cl2], the iron(III) center is destabilized due to weak σ-bonding interaction by the sterically demanding –NMe2 group. Similarly, on introducing the sterically hindering 6-methyl group on the pyridyl ring in [Fe(L20)Cl2] to give [Fe(L21)Cl2], the iron(III) oxidation state is destabilized. The Lewis basicity of N-methylimidazole moiety in H(L22), which is higher than that of pyridine moiety in H(L20), leads to enhanced stabilization of iron(III) oxidation state in [Fe(L22)Cl2] rendering its FeIII/FeII redox potential more negative.
The complexes [Fe(L20)(DBC)] and [Fe(L22)(DBC)] were found to catalyze the cleavage of H2DBC. The oxidative intradiol (2, 3, Table 3, Scheme 5) and very small amounts of extradiol (4–6) and side (7–9) products are formed. The products 2 and 3 are derived [17] from the nucleophilic attack, respectively, of Me2NH as impurity in DMF and piperidine on cis,cis-muconic anhydride, which is the immediate product of oxidative cleavage. The very low amounts of the extradiol products are expected because the six-coordinate catecholate adducts of [Fe(L20)Cl2], [Fe(L22)Cl2] have no vacant coordination site for O2 to attack [32]. The incorporation of a coordinated phenolic hydroxyl group into [Fe(TPA)(DBC)]+ (, 1.5 × 103 M−1 s−1) [17] as in [Fe(HDP)(DBC)] [H(HDP) = 2-[(bis(2-pyridylmethyl)-aminomethyl]-4,6-dimethylphenol] [12] decreases the Lewis acidity of the iron(III) center and hence lowers the rate of dioxygenation enormously (, 3.3 × 10−3 M−1 s−1). On the other hand, the incorporation of p-nitro-phenolate moiety as in [Fe(L15)Cl2] leads to lack of reactivity towards dioxygen and H2O2 as well [18]. The p-nitrophenolate donor, as it is weakly σ-bonding, does not appear to facilitate the decomposition of the reaction intermediate into products (cf. above). Also, on replacing one of the pyridyl moieties in [Fe(L15)Cl2] by the –NMe2 pendant to obtain [Fe(L20)Cl2], the dioxygenase activity is restored; obviously, the weak coordination of the sterically hindering –NMe2 group (cf. above) enhances the Lewis acidity of the iron(III) center sufficiently, thereby increasing the reaction rate. Further, as previously demonstrated by Que [15–17,49] and Palaniandavar [19,20,22–25], the higher Lewis acidity of the iron(III) center in [Fe(L20)Cl2] (cf. above) would be expected to confer a higher rate of oxygenation on [Fe(L20)(DBC)]. But, interestingly, the latter reacts (, 2.9 × 10−2 M−1 s−1) approximately six times slower than [Fe(L22)(DBC)] does (, 4.6 × 10−3 M−1 s−1), which is consistent with the former converting the substrate into intradiol products more efficiently than the latter {[Fe(L22)Cl2]: 2, 1.0; 3, 52.6%; [Fe(L20)Cl2]: 2, 1.5; 3, 75.6%}. The inactivity of [Fe(L21)Cl2] towards catechol cleavage, in spite of enhanced Lewis acidity of its iron(III) center, is intriguing. The sterically high-demanding 6-methyl group in this adduct appears to hinder the approach of dioxygen. A similar dioxygenation activity of [Fe(MeTPA)Cl2] [51] [MeTPA = (6-methylpyrid-2-ylmethyl)bis(pyrid-2-ylmethyl)amine], extremely lower than [Fe(TPA)Cl2]Cl, has been noted by Que et al.
2.5 Iron(III) complexes of sterically hindered tripodal bis(phenolate) ligands
The iron(III) complexes of the bis-phenolate ligands [N,N-dimethyl-N′,N′-bis(2-hydroxy-3,5-dimethylbenzyl)ethylenediamine [H2(L23)], N,N-dimethyl-N′,N′-bis(2-hydroxy-4-nitro-benzyl)ethylenediamine [H2(L24)], N,N′-dimethyl-N,N′-bis(2-hydroxy-3,5-dimethylbenzyl)ethylenediamine [H2(L25)] and N,N′-dimethyl-N,N′-bis(2-hydroxy-4-nitrobenzyl)ethylenediamine [H2(L26)] have been constructed (Scheme 9) and studied as models for the 3,4-PCD enzymes [20].

Structures of the bis(phenolate) ligands.
The coordination environment around iron atom in [Fe(L23)Cl] is described as distorted trigonal bipyramidal (trigonality index τ, 0.79) (Fig. 3) [20]. Interestingly, this coordination environment is closely related to the trigonal bipyramidal metal core (τ, 0.44) in the substrate-free 3,4-PCD enzyme; however, both the phenolates are equatorial in [Fe(L23)Cl], while they are mixed axial–equatorial in the enzyme active site. On the other hand, the analogues complex [Fe(L24)-(H2O)Cl] exhibits a distorted octahedral coordination geometry (Fig. 4). The trans-disposed Fe–O(phenolate) bonds are significantly different (Fe–O1, 1.890; Fe–O4, 1.993 Å) and are interesting because the difference in the two Fe-tyrosinate bonds in the 3,4-PCD enzyme is thought to influence the asymmetric binding of the chelated substrate moiety. The Fe–O–C bond angles (Fe–O1–C1, 135.3°; Fe–O4–C14, 135.6°) are similar to those in [Fe(L20)Cl2], but greater than those in [Fe(L15)Cl2]·CH3CN and other octahedral iron(III) complexes of phenolate ligands (∼128.5°), obviously due to the difference in phenolate substitution in the ligands.

Structures of [Fe(L15)Cl2]·CH3CN and [Fe(L20)Cl2].
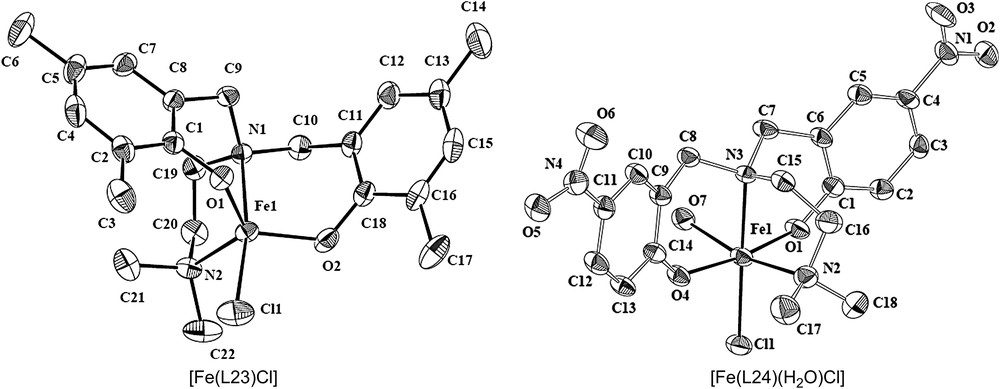
Structures of [Fe(L23)Cl] and [Fe(L24)(H2O)Cl].
The iron(III) complexes of bis-phenolate ligands show that the charge transfer transition from the out-of-plane pπ orbital (HOMO) of the phenolate oxygen to the half-filled orbital of iron(III) would arise from the charge-transfer transition [39,40] from the phenolate to iron(III) LMCT band (475–550 nm). The shift of this LMCT band to lower energy, on replacing methyl group in [Fe(L23)Cl] by the electron withdrawing p-nitro group [47] to obtain [Fe(L24)Cl(H2O)], reflects the lower Lewis acidity of the iron center in the former. This is supported by the shorter and hence stronger Fe–O bonds found in [Fe(L23)Cl]. A similar shift has been observed for [Fe(L25)Cl] and [Fe(L26)Cl(H2O)]. Further, the tripodal ligand complexes [Fe(L23)Cl] and [Fe(L24)Cl(H2O)] exhibit the LMCT band at energies higher than their respective linear ligand complexes [Fe(L25)Cl] and [Fe(L26)Cl(H2O)]. On adding catecholate anions to [Fe(L24)Cl(H2O)], [Fe(L25)Cl] and [Fe(L26)Cl(H2O)], two new catecholate → Fe(III) CT bands are observed [15,41,49]. On the other hand, interestingly, only one catecholate-to-Fe(III) LMCT band (475–540 nm) is observed for [Fe(L23)Cl].
The E1/2 values for the FeIII/FeII couple exhibit the following trends: Fe(L23) < Fe(L24), Fe(L25) < Fe(L26). This reflects the increase in Lewis acidity of the iron(III) center as the electron-releasing methyl groups on the phenolate donors are replaced by p-nitro group, which is consistent with the trend in PhO− → Fe(III) LMCT band energies. Interestingly, the redox potentials of linear ligand complexes [Fe(L25)Cl] and [Fe(L26)Cl(H2O)] are more negative than the respective tripodal ligand complexes [Fe(L23)Cl] and [Fe(L24)Cl(H2O)]. This is consistent with the trend observed in the energies of LMCT bands and suggests that linear ligands are more suitable than tripodal ligands to strongly bind to and hence confer decreased Lewis acidity on iron(III) center.
From the reaction mixture of complexes with H2DBC two intradiol (1, 61; 2, 25%) and one extradiol (5, 14%), cleavage products (Scheme 5) were identified for [Fe(L24)Cl(H2O)]. In contrast, interestingly, only one intradiol-cleavage product (1) (Scheme 5) was identified for [Fe(L26)Cl(H2O)]. The complexes [Fe(L23)Cl] and [Fe(L25)Cl] were found to show no activity and the rate of reaction calculated for [Fe(L24)Cl(H2O)] is 3.76 × 10−3 M−1 s−1. The latter with Lewis acidity lesser than the former (cf. above) would be expected to exhibit a lower rate of dioxygenase reaction. However, interestingly, [Fe(L26)Cl(H2O)] reacts much faster [14] than [Fe(L24)Cl2)]. Unlike the tripodal ligand complex [Fe(L24)Cl(H2O)], the linear tetradentate ligand in [Fe(L26)Cl(H2O)] would rearrange itself to provide cis-coordination positions for bidentate coordination of the catechol substrate. The increased steric congestion and enhanced negative charge built on iron(III) in [Fe(L26)(DBC)]− compared to [Fe(L24)(DBC)]− adduct would facilitate the rate-determining product releasing phase in the reaction mechanism proposed by Que et al. for intradiol-cleaving dioxygenases.
3 Conclusions and relevance to iron oxygenases
This review comprehensively collects the mononuclear iron(III) complexes of phenolate and non-phenolate ligands, which have been isolated and studied by our research group as structural, spectral and functional models for the intradiol-cleaving catechol 1,2-dioxygenase enzymes. It is remarkable that the complexes of suitably tailored ligands can closely mimic the interesting structural, spectral and chemical properties of the intradiol-cleaving enzymes. The cis-facially coordinating linear 3N ligands form octahedral iron(III) complexes and catalyze the cleavage of 3,5-di-tert-butylcatechol into intradiol-cleavage rather than extradiol-cleavage products. All the tripodal and linear tetradentate mono- and bis-phenolate ligands also form octahedral iron(III) complexes with two cis-coordination positions vacant and elicit intradiol-cleavage. However, interestingly, one of the complexes possess a trigonal bipyramidal coordination geometry, which bears a close resemblance to that of 3,4-PCD enzyme active site, but fails to elicit catechol cleavage activity. The Fe–O–C bond angle of 136.1° in two complexes with a coordinated –NMe2 group is similar to that in the enzyme, which leads to an enhanced rate of catechol cleavage. Also, it is remarkable that the imidazole-based complex confers an enhanced reaction rate with efficient conversion of substrate into intradiol-cleavage products. The substituents on the phenolate ligands lead to tune the Lewis acidity of the iron(III) center and hence determine the course and products of dioxygenase activity of the complexes. Thus the ligand donor functionalities –NMe2, (6-methyl)pyridine and N-methylimidazole exhibit different stereoelectronic effects in clearly influencing the shielding of the iron(III) center and hence its Lewis acidity, as demonstrated by the spectral and electrochemical properties. The rates of dioxygenase reactions catalyzed by the complexes could be illustrated, not only on the basis of the Lewis acidity of the iron(III) center alone, but also by assuming that product release is the rate-determining phase of the catalytic reaction.
Acknowledgments
We sincerely thank the Department of Science and Technology, New Delhi [Scheme No. SP/S1/F-20/2000 and SR/S1/IC-45/2003] and Council of Scientific and Industrial Research, New Delhi [Scheme No. 9(299)/90 EMR-II] for supporting this research and the latter for a Senior Research Fellowship to R.M.