

1 Introduction
Metalloproteins and metalloenzymes that contain dinuclear active sites are prevalent in nature. The active sites of a majority of these dinuclear proteins have been characterized by spectroscopic and magnetic methods as well as X-ray crystallography in several cases; they contain two metal atoms connected by at least one endogenous carboxylate bridge and usually one or two oxo-, hydroxo- or aqua-bridges derived from exogenous water [1,2]. Despite their common structural characteristics, the functional diversity is one of the most striking aspects of this class of proteins which incorporates diiron proteins such as hemerythrin [3], a non-heme dioxygen carrier, and oxidative enzymes such as ribonucleotide reductase [3] and soluble methane monooxygenase [4], dimanganese proteins such as catalase [5], arginase [6], and the dinickel enzyme urease [7,8]. The dicopper monooxygenases are structurally closely related to the above-mentioned enzymes.
The structure–function relationships that are of importance for these metal sites include the larger protein surrounding of the active site, where specific amino acids may regulate substrate accessibility and orientation, and also the direct coordination environment of the metals, where subtle changes may have profound influence on the reactivity of the metal site. There are often pronounced asymmetries in catalytically active metallobiosites, and these asymmetries are very likely essential for the catalytic properties [1,2].
Adequate structural modeling of dinuclear metallobiosites has long posed a formidable challenge to the synthetic chemist. Because of the above-mentioned structural considerations, it has become increasingly clear that an essential part of biomimetic model chemistry is the development of new ligands that may reproduce the important irregularities of the protein environment. In attempts to model the asymmetries that often occur in dinuclear active sites of enzymes, a number of asymmetric dinucleating ligands have been developed. The use of such ligands in bioinorganic model chemistry began with the development of models for the active sites of Cu [9] and Fe proteins [10,11]. In this review, we aim to highlight some important recent investigations on the use of such ligands in biomimetic chemistry.
In this context, the term “asymmetry” is not easily defined. If the ligand/complex possesses C1 symmetry, then it is indeed asymmetric. We have chosen a relatively liberal interpretation of asymmetry; thus several ligands/complexes that possess C2 or Cs (or higher) symmetries are included in this review. Also, a few complexes that are symmetric in all respects but the fact that they are heterodinuclear have been included. Dinucleating macrocyclic ligands that impart differing coordination numbers on the two metals have been reviewed recently [12] and are, in general, not discussed here.
This review is not comprehensive; it focuses on studies that have taken place during the past decade. As a consequence, this review covers complexes that have been prepared as models for hydrolases or oxygenases; relevant active sites in metalloenzymes are briefly introduced. A number of complexes that are not explicit models for metallobiosites, but are nevertheless relevant, have also been included. Any faults and omissions are entirely the fault of the authors. The reader may also wish to refer to previous reviews [2,12–15] that partially cover the topic(s) of this article.
2 Models for hydrolases containing dinuclear active sites
2.1 Models for purple acid phosphatases
The purple acid phosphatases (PAPs) are enzymes that have an intense pink/purple colour and catalyze the hydrolysis of a diverse group of phosphomonoesters in vitro with a pH optimum around 4.9–6 [16]. They have been isolated from mammals, plants and fungi but their function(s) in vivo is still uncertain. Proposed functions include transport of iron in fetal pigs [17,18], control of bone resorption [19,20], and phosphate homeostasis in plants [21]. It has also been proposed that PAPs play a part in the pathological development of Alzheimer's disease [22]. Mammalian PAPs are often termed TRAPs (tartrate resistant acid phosphatases) due to their resistance towards tartrate inhibition compared to other mammalian acid phosphatases [23].
The structures of several enzymes from different sources (rat [23], rat bone [24], porcine uterus (UfPAP) [25], kidney bean (kbPAP, Fig. 1) [26,27] and sweet potato (spPAP) [28]) have been determined by X-ray crystallography. They all have very similar active sites except that kbPAP and spPAP have heterodinuclear Fe(III)Zn(II) and Fe(III)Mn(II) cores, respectively, (an FeZn isoform of spPAP has also been isolated [29]), and the mammalian enzymes have heterovalent Fe(II)Fe(III) cores in the active enzyme although they are in the oxidized and inactive Fe(III)2 state in the determined structures. Two quite different coordination pockets are present, giving very selective binding of the trivalent and the divalent metal ions. Fe(III) is coordinated by an aspartate, a histidine and a deprotonated tyrosine residue (Fig. 1). The latter stabilizes the ferric oxidation state and gives rise to the intense LMCT band around 560 nm which is responsible for the colour of these enzymes [30]. In the mammalian and kidney bean PAPs, a hydroxide and a 1,1-μ-aspartate residue bridges to the other metal ion that is further coordinated by two histidines and one asparagine. Although not resolved in the crystal structures, the octahedral environments are generally believed to be fulfilled by one water and one hydroxide coordinating to the divalent and the trivalent ion, respectively. There are also a few other non-coordinating amino acid residues in the active site that are thought to be involved in the binding and activation of the substrate by hydrogen bonding [26,31]. The FeMn isoform of sweet potato PAP was crystallized with phosphate which coordinates in a μ–η2–η2 bridging mode with one of the phosphoryl oxygens forming a 1,1-bridge while two other oxygens bridge in a 1,3 manner, displacing the exogenous water molecules (Fig. 2) [28]. In the native spPAP enzyme, an oxido group is thought to bridge the two metal ions as indicated by the very strong antiferromagnetic coupling (−J > >70 cm−1, H = −2JS1·S2) observed by magnetic susceptibility measurements and indicated by absence of EPR signal [32]. It should be mentioned that there are no previous examples of μ-oxido-bridged dinuclear enzymes or complexes containing Mn(II) and the highly ionic character usually observed for Mn(II) compounds contradicts the presence of a strong exchange coupling. The true identity of the divalent metal ion of spPAP, and also other PAPs, is still debated since it is easily lost or substituted during isolation or preparation.

Schematic structure of the active site of kidney bean purple acid phosphatase (kbPAP).
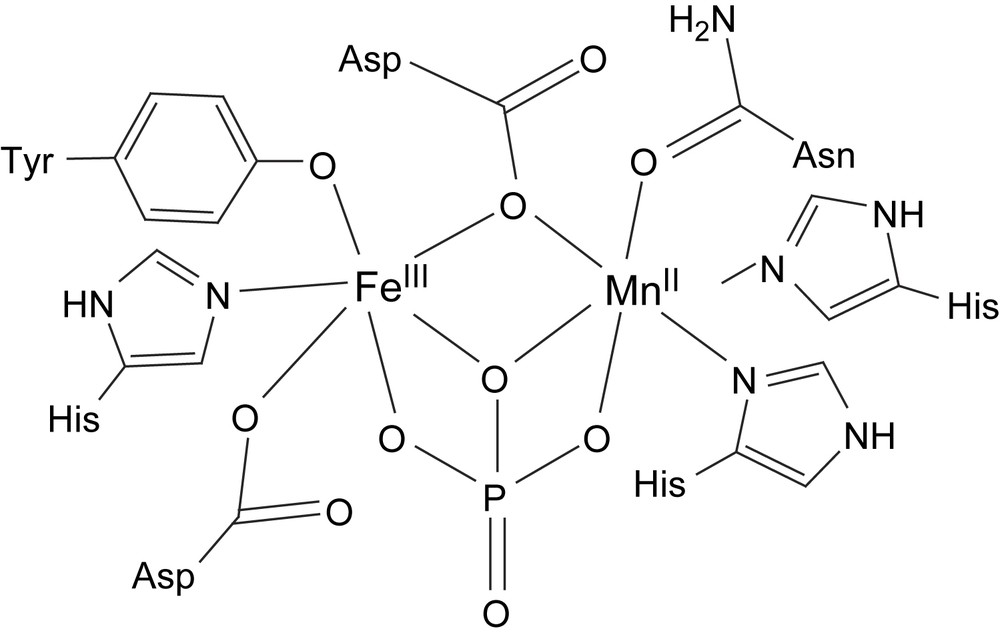
Schematic structure of the active site of sweet potato PAP with the coordinated phosphate group.
The kbPAP has an EPR signal of g = 4.3 corresponding to the isolated Fe high-spin (S = 5/2) ground state of the FeZn core [33]. The mammalian PAPs show weak antiferromagnetic exchange couplings (J = −4.4 to −13 cm−1, H = −2JS1·S2) [34,35]. The oxidized form is EPR silent, while the active Fe(III)Fe(II) form displays a pH-dependent rhombic EPR signal (gav ≈ 1.75) [36].
The narrow pH range for activity stems from the requirement of a deprotonated and a protonated species (pKa,1: 4–5.4 and pKa,2: 5–7.5, respectively) [37,38] where the acid dissociation constants are somewhat dependent on the specific form of enzyme [31,39]. The former (pKa,1) was shown to be a hydroxide coordinated to the ferric ion (terminally or bridging) [36] and the latter might be an amino acid residue near or in the active site. It has been proposed that a histidine residue (His92 in recombinant human PAP) may fulfill this role but recent results are not in agreement with this proposal [40].
It is now widely accepted that the hydrolysis of phosphomonoesters by the PAP enzymes occurs through a direct nucleophilic substitution on the phosphorus but neither the identity of the nucleophile nor the binding mode of the substrate in the active site have been unambiguously determined. The three most common proposals for the active nucleophile are those depicted in Scheme 1: (a) [pathway 1] the terminally Fe(III)-coordinated hydroxide, (b) [pathway 2] the bridging hydroxide or (c) [pathway 3] a hydroxide generated from a water molecule in the second coordination sphere through deprotonation by the terminal hydroxide. Both (a) and (b) were originally based on the crystal structures of kbPAP [26] and similar enzymes [41] with tetraoxidoanions (e.g. phosphate, tungstate) coordinated in 1,3-bridging mode because in the case of pathway 1 the attack would yield this structure as an intermediate and in pathway 2 the coordinated phosphate (product) is believed to mimic the coordination mode of the organophosphoester substrate, aligning it perfectly for a nucleophilic attack by the hydroxy bridge with inversion at the phosphorus, as observed experimentally [42].

Three mechanistic proposals for the PAP catalyzed hydrolysis of phosphomonoesters (adapted from Ref. [43]).
2.1.1 Diiron complexes as models for purple acid phosphatases
The ligands H21 and. H22 have been prepared to provide a central bridging phenolate moiety and a terminal phenolate that may mimic the binding of a single (terminal) tyrosine to the diiron site in purple acid phosphatases. The diferric and mixed valent Fe(III)Fe(II) complexes of the general formula [Fe2(L)X2]n+ (L = 1 or 2; X2 = 1,3-m-benzene dipropionate (mpdp), (OAc)2, (OBz)2, (H2PO4)2; n = 1 or 2) have been prepared and fully characterized by mass spectrometry, NMR, UV–vis, and Raman spectroscopies and cyclic voltammetry [44]. Mössbauer and NMR spectroscopic measurements on the mixed valence species indicate that they are valence localized at room temperature; a weak and broad intervalence transition can be observed around 1100 nm. The electrochemical studies established that the fully reduced Fe(II)2 and the mixed valence oxidation states are thermodynamically stabilized relative to related complexes while the oxidized Fe(III)2 state is destabilized. The mixed valence complexes were found to be oxidized to the diFe(III) species by reaction with iodine.
The analogous complexes of 1 and 2 with two diphenylphosphates as bridging ligands have also been prepared [45]. These complexes mimic the inactivated form of purple acid phosphatases, which is formed when phosphate is added to the active form of the enzyme under aerobic conditions. This inactivated form is believed to contain a bridging phosphate, as seen in the crystal structure of the FeMn isoform of sweet potato PAP (vide supra). The exchange of the diphenylphosphate ligands with mpdp, and of the corresponding mpdp complexes with diphenylphosphate (dpp), has been studied by NMR and electrochemistry (Scheme 2). When Na2mpdp is added to the mixed valence complexes [Fe(III)Fe(II)(L)(dpp)2]+ (L = 1, 2) in acetonitrile, the phosphate/carboxylate exchange is rapid. However, the reverse reaction could not be effected, even when a tenfold excess of dpp was added. Addition of three equivalents of Na2mpdp to the corresponding diferric diphenylphosphate complex leads to ligand exchange and reduction of the complex to form the mixed valence species. The reducing agent was indirectly identified as the ligand, as the phenylacetate analogue of the Fe(III)2 complex was prepared and was shown to generate dibenzyl upon reaction with dpp – this reaction indicates that the carboxylates act as reducing agents by a one-electron oxidation of the carboxylate to form the corresponding radical, which subsequently undergoes decarbonylation and dimerization.
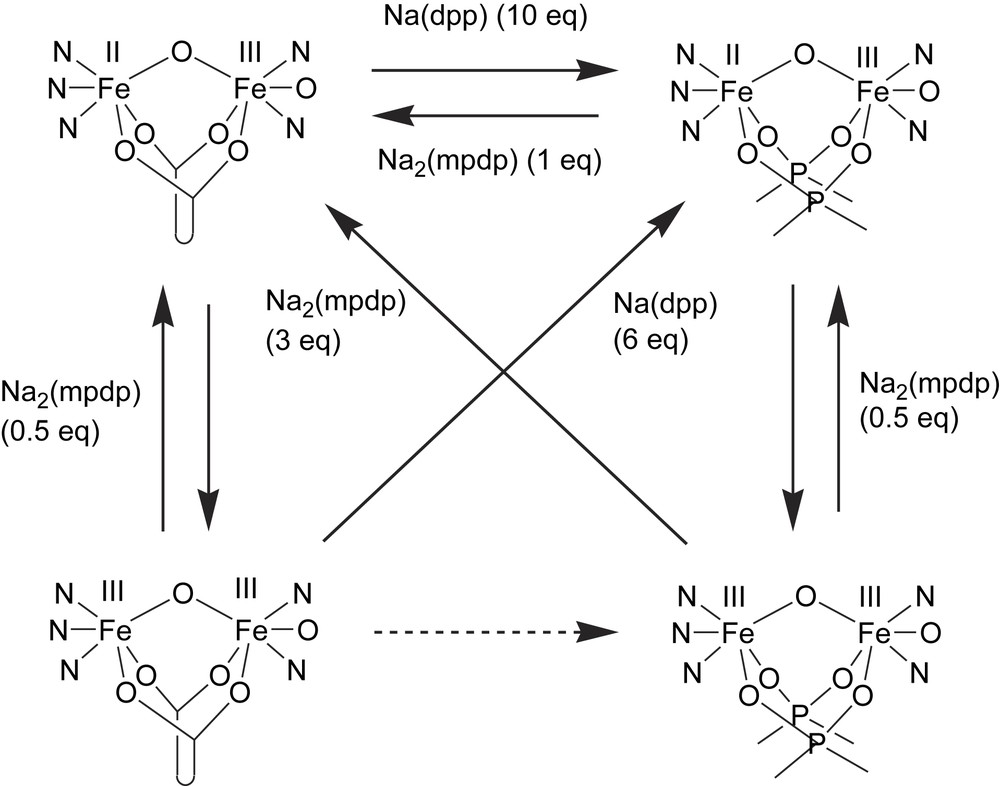
Exchange of diphenylphosphate (dpp) and 1,3-m-benzene dipropionate (mpdp) ligands of diFe(III) and mixed-valent (Fe(II)Fe(III)) complexes of ligands 1 and 2 (adapted from Ref. [45]).
The ligands H33 and H4 are closely related to H21 and H22 and have also been used to prepare model complexes for purple acid phosphatases. The diferric complex [Fe2(3)(μ-OMe)(OMe)]+ (5) was synthesized by reaction of two equivalents of Fe(ClO4)2·9H2O with H33 in the presence of triethylamine [46]. The crystal structure of 5 reveals that the Fe(III) ion that is coordinated by two pyridyl moieties is hexacoordinate with a terminal methoxide ligand while the other Fe(III) ion is pentacoordinate with a distorted trigonal bipyramidal coordination geometry. Cyclic voltammetry revealed that the diiron(III) state was stabilized by 0.85 V when compared to equivalent complexes with four pyridyl moieties based on ligand H8 (vide infra), while the diiron(II) state was destabilized by 0.9 V, meaning that the mixed valence complex had a similar stability domain (ΔE1/2 = 0.7−0.75 V) as the equivalent symmetrical pyridyl complexes. Variable temperature magnetic measurements revealed a weak antiferromagnetic coupling between the two Fe(III) ions (J = −13 cm−1; H = −2JS1·S2).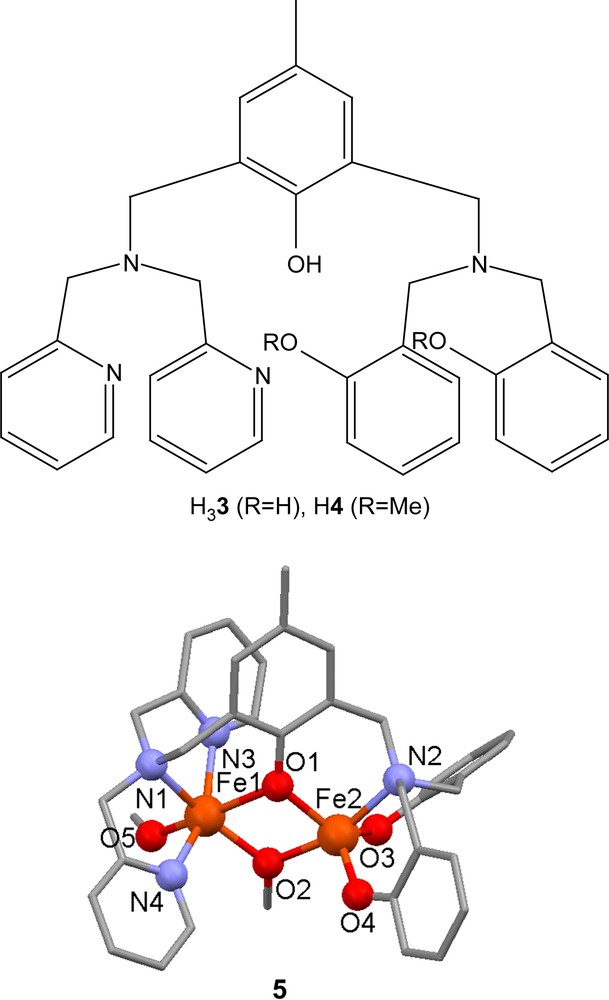
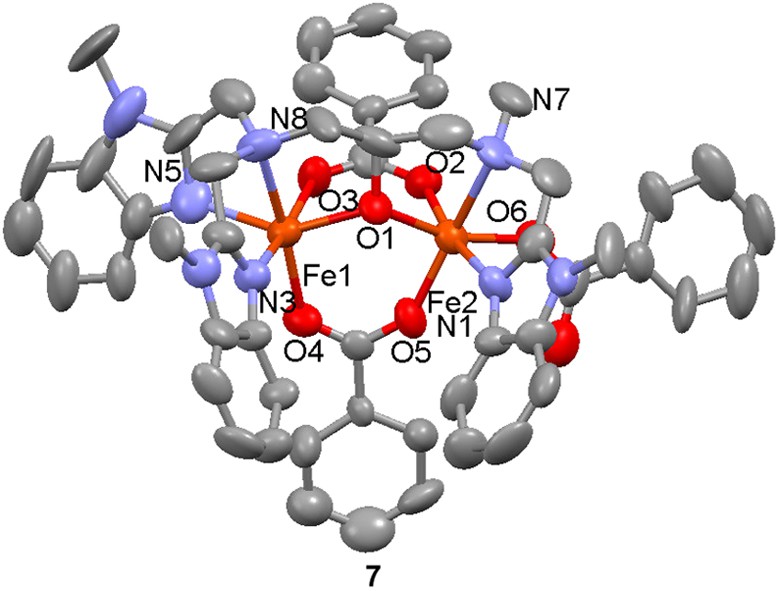
The mixed-valent dinuclear complex [(Fe(II)Fe(III)(6)(O2CPh)3]+ (7) has been prepared by reaction of the diFe(II) complex [Fe2(6)(O2CPh)(MeOH)1.5(H2O)0.5]2+ with five equivalents of sodium benzoate under a dinitrogen atmosphere followed by bubbling of air through the solution [47]. In addition to two bridging benzoate ligands, the third benzoate is bound in a terminal (monodentate) coordination mode. On the basis of metal–ligand donor atom distances, the metal valences were assigned as +2 for the iron coordinated by the N3O3 donor set and +3 for the iron coordinated by the N2O4 donor set. A magnetic study showed a weak antiferromagnetic interaction between the two metal centers (J = −4.4 cm−1, H = −2JS1·S2). In the absence of air, the above-mentioned reaction leads to the formation of [Fe(II)2(6)(O2CPh)2]+ with one bridging and one terminal benzoate ligand.
2.1.2 Heterodinuclear Fe(III)M(II) complexes
The heterodinuclear complexes [FeF2M(H2O)n(8)]2+ (M = Zn, Cu; n = 1; M = Co, n = 2) have been prepared by reaction of the dinucleating ligand H8 with one equivalent of Fe(BF4)2·6H2O, followed by addition of the appropriate hydrated M(BF4)2 salt [48]. The crystal structure of [FeF2Cu(H2O)(8)]2+ (9) reveals that the iron ion is octahedral with two terminal fluoride ligands, while the copper is in a square pyramidal coordination environment. It is suggested that the coordination mode of the fluoride(s) in fluoride-inhibited purple acid phosphatases may be similar to that detected in these model complexes, i.e. terminal rather than bridging. Cyclic and differential pulse voltammetry demonstrate that the heterodinuclear complexes exhibit a reversible Fe(III)/Fe(II) redox couple.

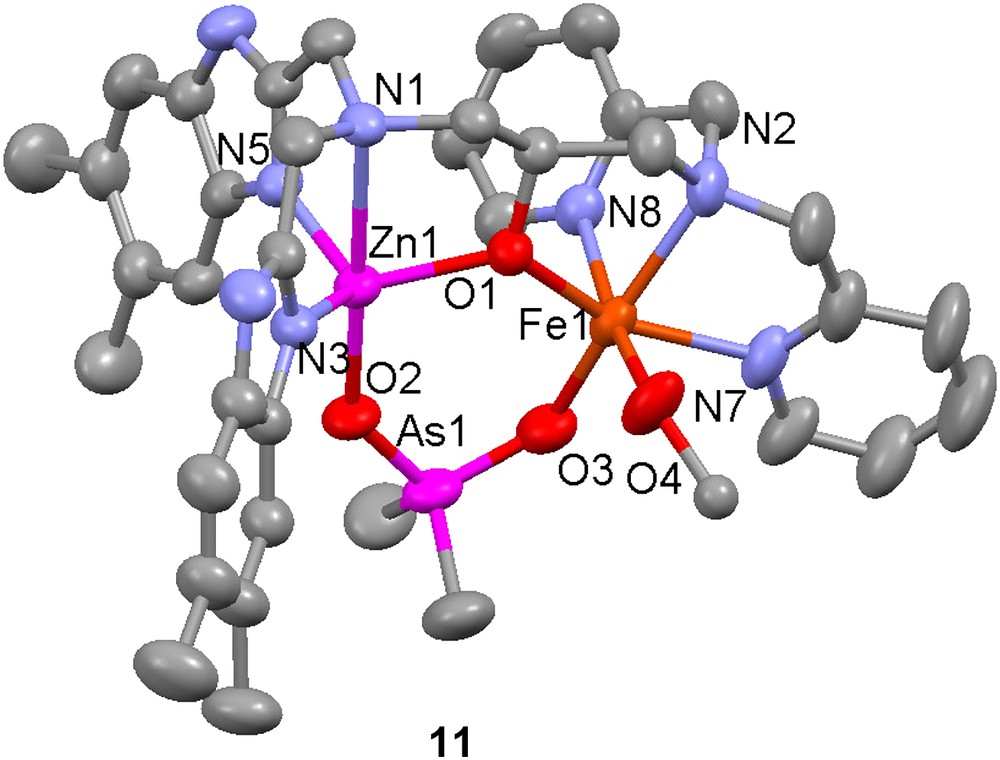
Several heterodinuclear Fe(III)Zn(II) complexes with asymmetric ligands have been prepared as structural models for the active site of kidney bean purple acid phosphatases and the hydrolytic activity towards organophosphoesters has been investigated for some of these complexes. The complex [FeZn(10)-(O2AsMe2)(MeOH)]3+ (11) was synthesized by sequential addition of zinc(II) perchlorate and iron(III) perchlorate to the dinucleating ligand H10 [49]. Its crystal structure reveals that the complex is a good structural model for the active site of kidney bean purple acid phosphatase, with the bound cacodylate (dimethylarsenate) mimicking a bound phosphoester substrate (or an inhibitor). The zinc atom has a trigonal bipyramidal coordination geometry, while the iron is octahedral with the sixth coordination site occupied by a solvent (methanol) molecule.
Regioselective coordination of zinc to the ligands H33 and H4 (vide supra) has been utilized to prepare a dinuclear Zn(II)Fe(III) complex [50]. NMR and mass spectrometry measurements indicate that reaction of either ligand with Zn(ClO4)2·6H2O leads to mononuclear Zn complexes in which the Zn ion coordinates preferentially in the bispyridyl pocket of the ligand. Utilizing this fact, a heterodinuclear complex was prepared by initial addition of 1 equiv of Zn(II) to H33 followed by NEt3 and Fe(ClO4)2·9H2O. Finally, addition of diphenylphosphate led to the formation of the complex [ZnFe(3){O2P(OPh)2}2] (12), which is a good structural model for the phosphate adduct of kbPAP.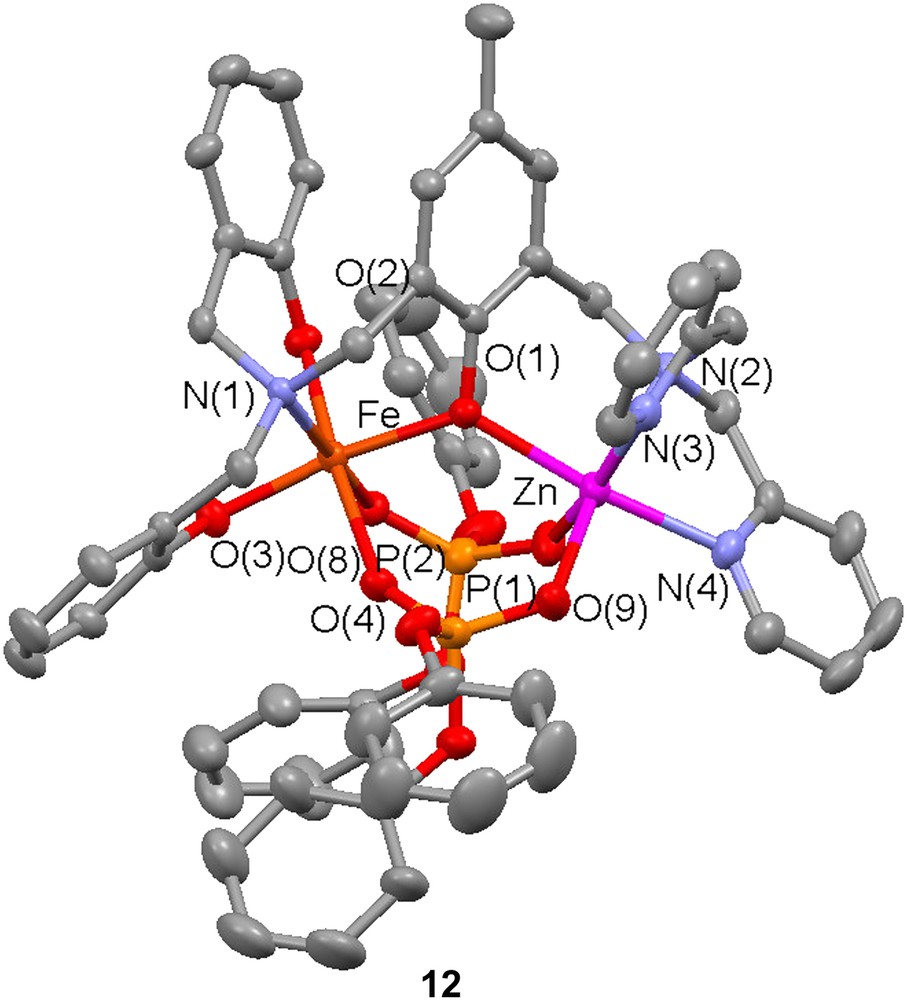
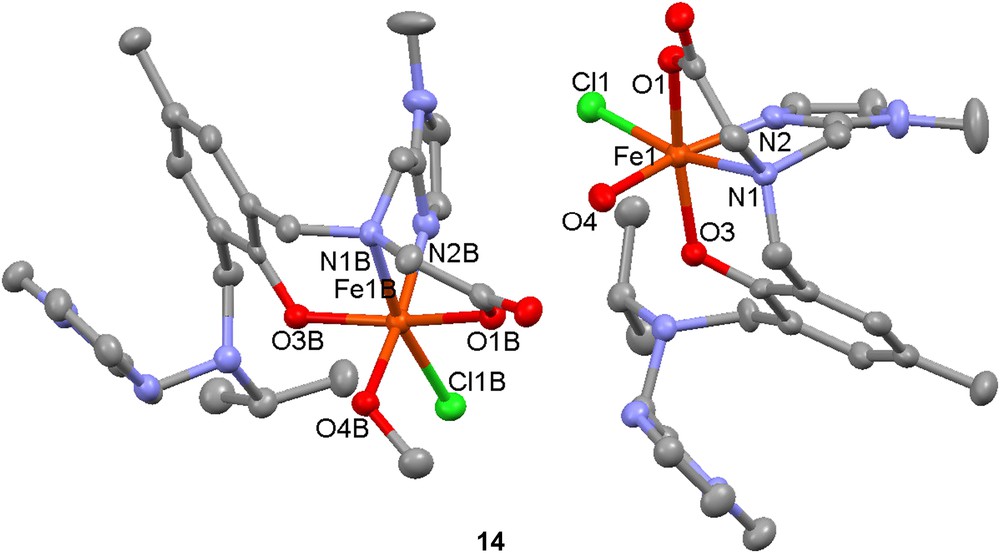
Synthesis of a FeZn complex based on the polydentate ligand H313 has also been performed [51]. A mononuclear complex that crystallizes as [Fe(H213)(H2O)Cl][Fe(H213)(MeOH)Cl][ClO4]4 (14) was used as a starting material/synthon. Reaction of this mononuclear synthon with zinc acetate in methanol/acetonitrile led to the formation of a heterodinuclear complex that could be crystallized as [FeZn(13)(OAc)2][ClO4]·2CH3OH (15). Reactivity studies were performed by investigating the ability of 15 to catalyze the hydrolysis/transesterification of the substrate 2-(hydroxypropyl)-4-nitrophenyl phosphate (hpnp, Scheme 3). Hydrolytic activity was detected but due to the low stability of 15 in water, this activity was mainly attributed to a homodinuclear zinc complex generated in situ.

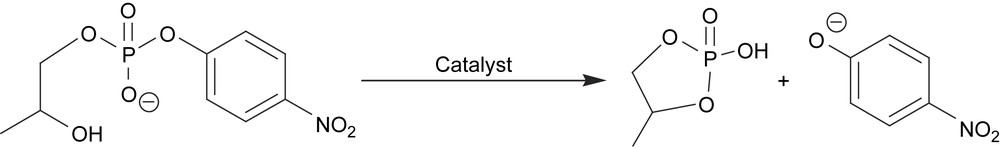
Hydrolysis/transesterification of 2-(hydroxypropyl)-4-nitrophenyl phosphate (hpnp).
Ligand H21 was used to prepare the complex [Fe(III)Zn(II)(1)(μ-OAc)2]+ (16) [52]. The crystal structure of the complex reveals an Fe–Zn distance of 3.490(9) Å, which is close to that observed for kbPAP in the presence of phosphate (3.33 Å). Three protonation equilibria for the Fe–Zn complex in water/methanol solutions could be detected and studied by UV–vis spectrophotometry. These equilibria were proposed to involve terminal and bridging water molecules that are coordinated upon the dissociation of the bridging acetates in aqueous solution. The Fe–Zn complex was found to catalyze the hydrolysis of 2,4-bis(dinitrophenyl)phosphate (Scheme 4) with an optimum activity at pH ca. 6.5.

Hydrolysis of 2,4-bis(nitrophenyl)phosphate (bdnpp).
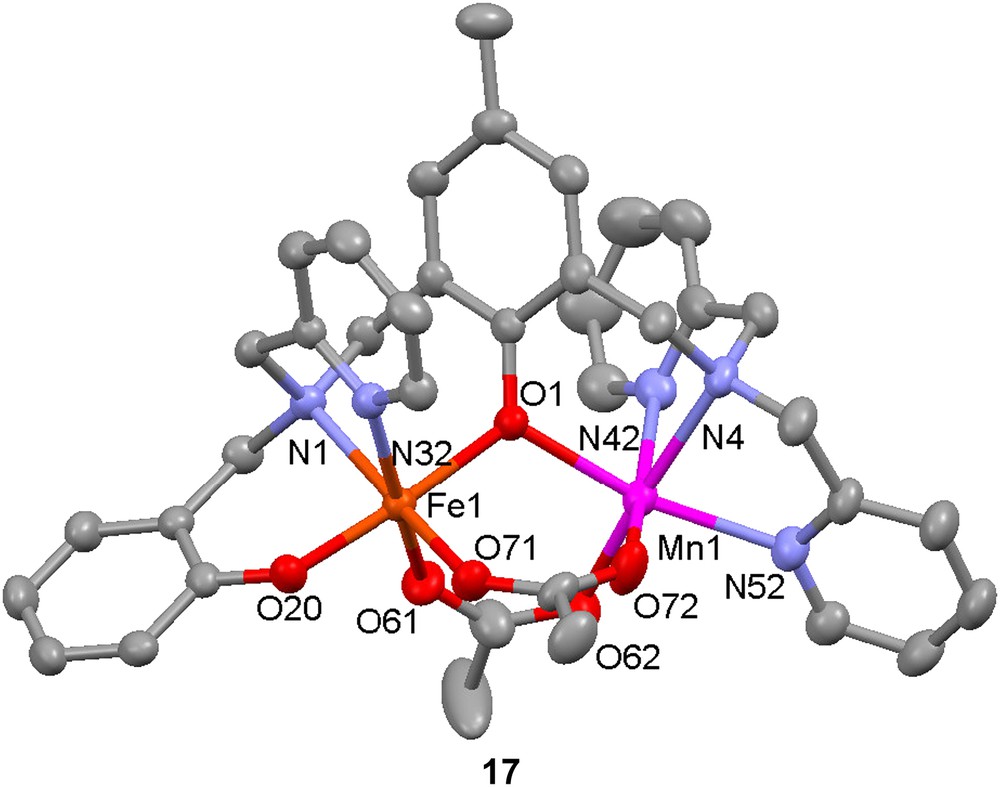
The same ligand has been used to prepare a model for the active site in the purple acid phosphatase from sweet potato. Reaction of H21 with 1 equiv each of Mn(OAc)2·6H2O and FeCl2·4H2O in methanol leads to the formation of the Fe(III)Mn(II) complex [FeMn(1)(OAc)2]+ (17) [53]. The crystal structure of the complex reveals that the metal–metal distance is 3.5 Å, to be compared with 2.9 Å in the PAP enzyme. Low-temperature Mössbauer spectroscopy confirms the presence of a high-spin ferric ion (ΔEQ = 1.04 mm−1, δ = 0.48 s−1). Variable-temperature magnetic measurements indicate that the two metal ions are weakly antiferromagnetically coupled (J = −6.8 cm−1, H = −2JS1·S2), as opposed to the strong antiferromagnetic coupling found in the biological site. The complex was found to catalyze the hydrolysis of 2,4-bis(dinitrophenyl)phosphate with an activity maximum at pH = 6.7; Michaelis–Menten-like saturation behaviour was observed for this reaction.
2.2 Models for manganese catalase
Catalases are enzymes that catalyze the disproprotionation of hydrogen peroxide into dioxygen and water. Heme-containing catalases are prevalent in nature and effect the catalase reaction by the same kind of mechanism that is used by heme peroxidases to oxidize substrates. The first manganese-containing catalase was isolated from Lactobacillus plantarum in 1983 [54], and crystal structures of manganese catalases from L. plantarum (in oxidized form) and Thermus thermophilus (in reduced Mn(II)2 form) have been determined [55,56]. The structure of the active site of the L. plantarum enzyme reveals that the two Mn ions are hexacoordinate and bridged by a glutamate residue and two solvent-derived (oxido, hydroxido or water) bridges, probably an oxido and a hydroxido bridge. Each manganese is coordinated by one histidine; on one Mn ion the remaining coordination sites are filled by a chelating glutamate residue (in the reduced T. thermophilus structure, this glutamate is monodentate, rendering the Mn pentacoordinate), while the second ion is coordinated by a terminal glutamate and a coordinated water molecule (Fig. 3).
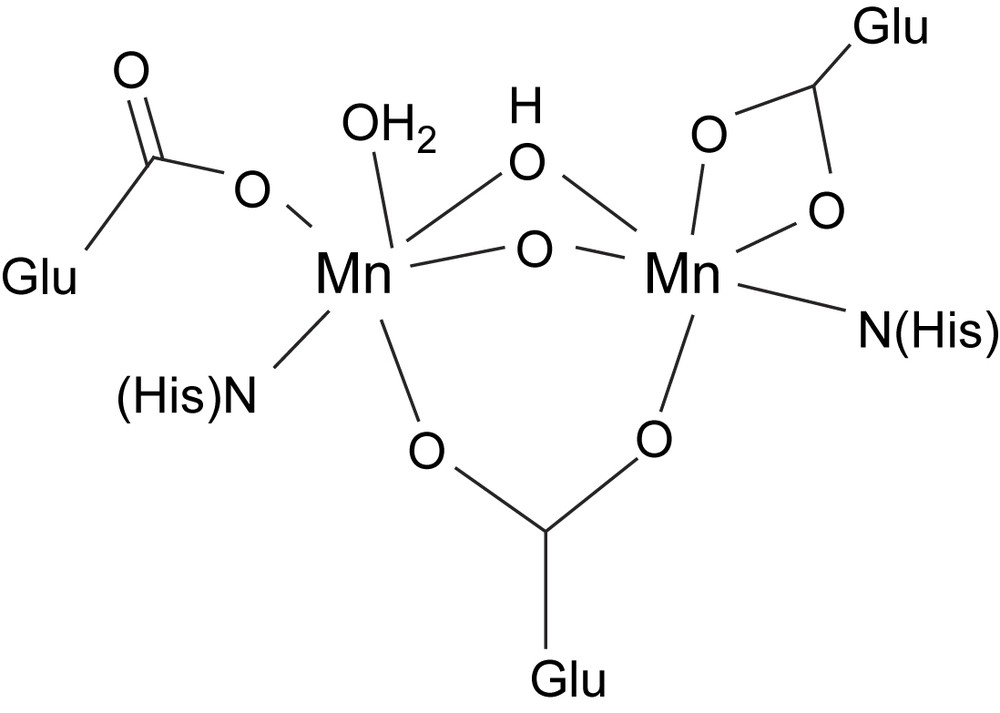
Schematic structure of the active site of Mn catalase.
The mechanism(s) of the catalase reaction that is effected by the Mn catalases has (have) not been determined, but the essence of most proposals is summarized in Scheme 5. It is proposed that initial coordination of hydrogen peroxide in the catalytic cycle occurs as a terminal hydroperoxide, and that a protic amino acid residue close to the dimanganese site is involved in protonation/deprotonation steps.
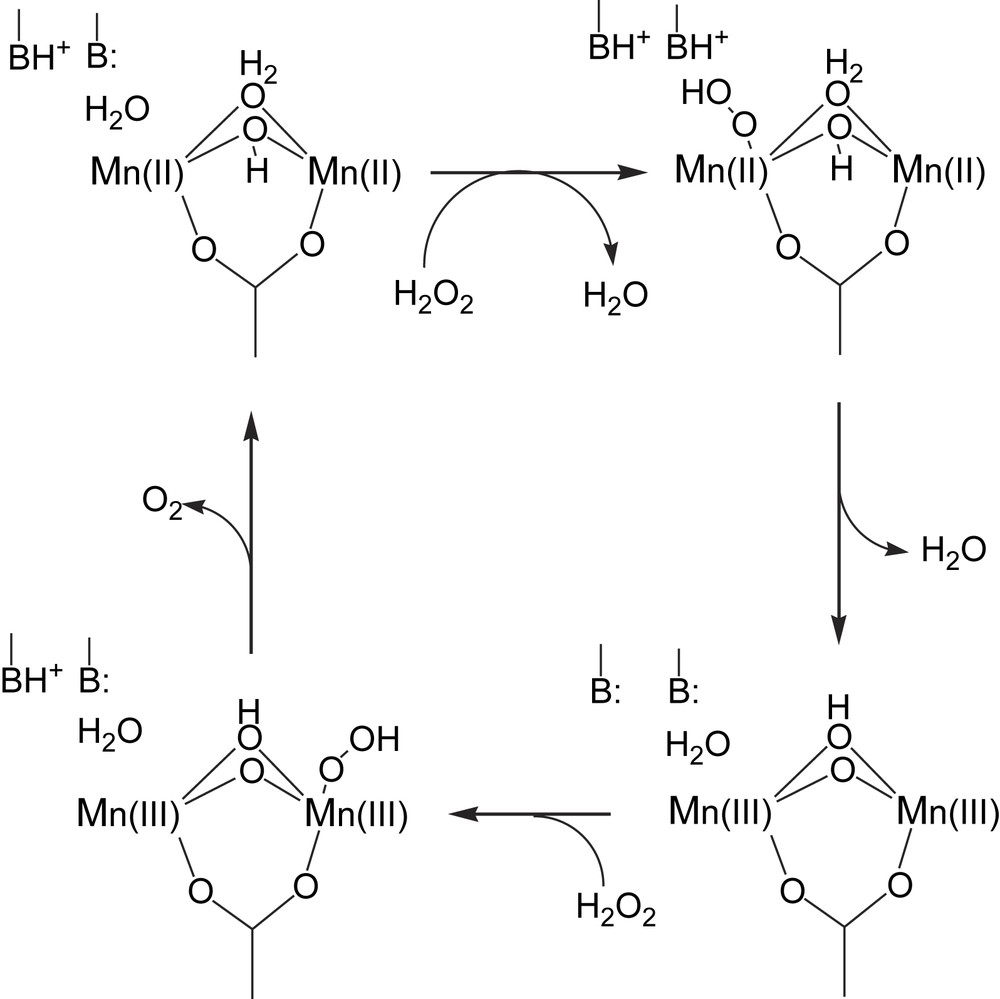
A proposed mechanism for the manganese catalase reaction (adapted from Ref. [5]).
Dinuclear manganese complexes of the unsymmetrical ligand H18 have been shown to catalyze the disproportionation of H2O2 [57,58]. Two dimanganese(II) complexes, [Mn(II)2(18)(OAc)2(CH3OH)](ClO4) (19) and [Mn(II)2(18)(OBz)2(H2O)](ClO4) (20), were produced when Mn(II) salts were used as starting materials, but with manganese(III) acetate, a mixed valent dinuclear manganese complex, [Mn(II)Mn(III)(18)(OAc)2(H2O)](ClO4) (21) was formed. Depending on reaction and crystallization procedures, the identity of the exogenous ligand was either methanol or water. The crystal structures of complexes 19–21 show that in all complexes the Mn ions are in distorted octahedral environments with one phenolate bridge and two carboxylate groups bridging in a syn-μ-1,3-fashion. The mixed-valent complex shows decreased bond lengths for one of the manganese ions, as expected for a valence localized dinuclear complex. The somewhat shorter bond between the exogenous coordinating molecule and the Mn(II) ion is explained by the weaker trans effect exerted by the phenolate moiety, which has a longer metal–oxygen bond to this Mn ion than what is usually observed. This latter elongation depends on the displacement of electron density towards the stronger Mn(III)–phenolate bond. The bond between the solvent molecule and the Mn ion with less ligand-derived coordinating groups is shorter for the heterovalent complex, which may be explained by the decreased trans effect caused by the increased Mn(II)–phenolate bond length. This increase is due to the displacement of the electron density to the stronger Mn(III)–phenolate bond, which is shortened because of Jahn–Teller distortion.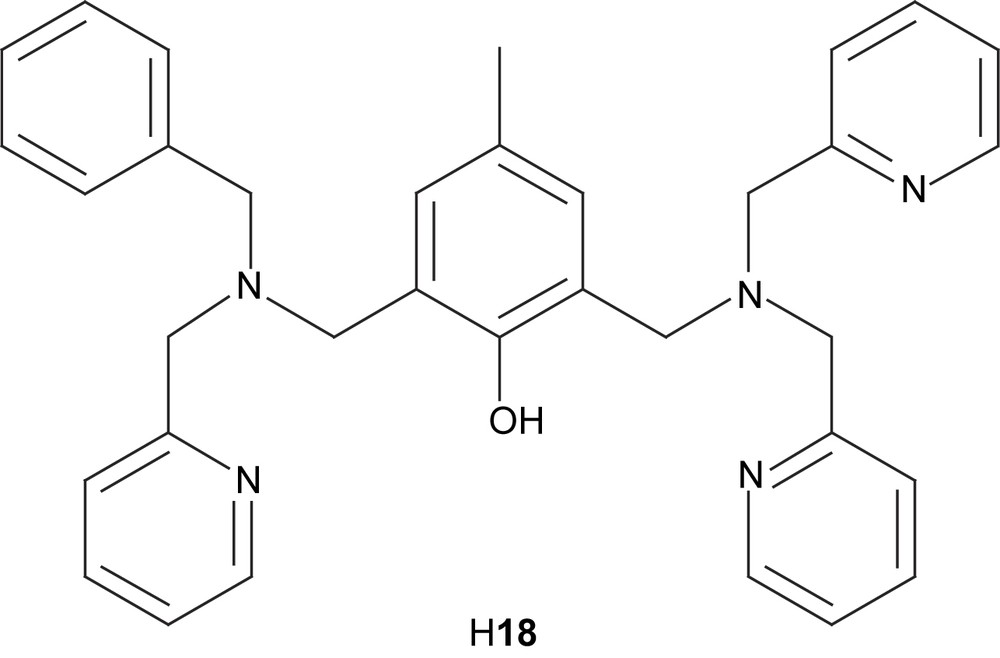
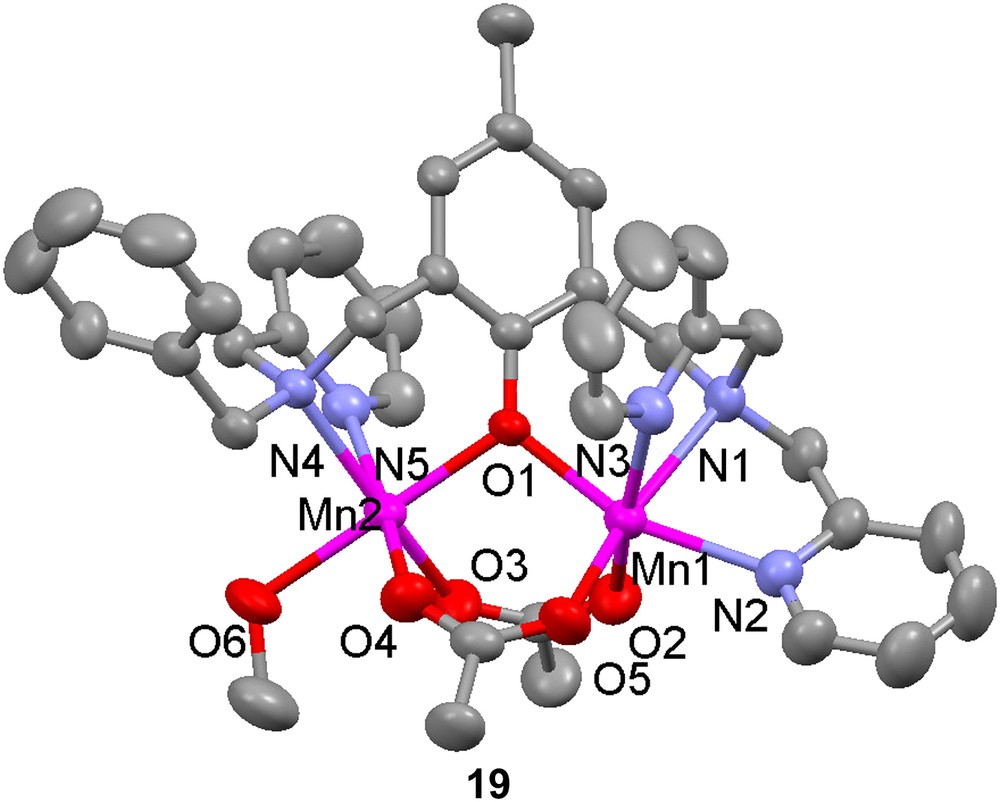
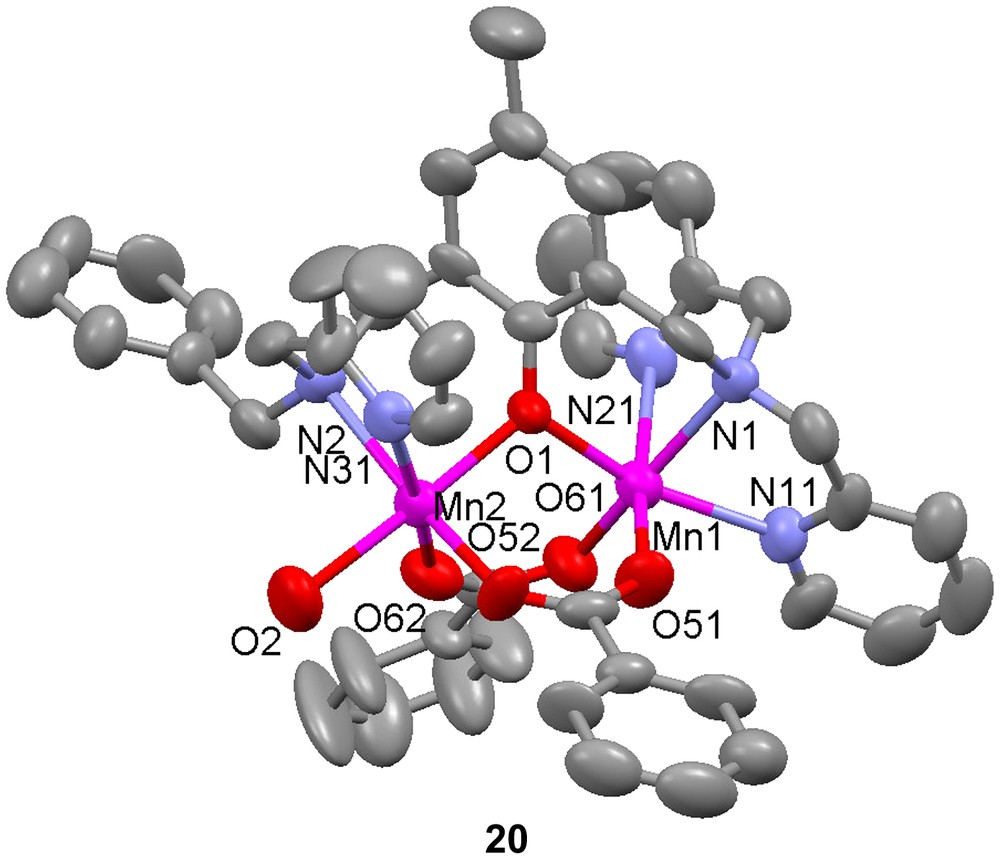

Magnetic susceptibility measurements show very similar magnetic behaviours for both the homo- and heterovalent complexes, indicating moderately antiferromagnetically coupled high-spin manganese ions with magnetic exchange interactions at J = −4.3(1) cm−1 (H = −2JS1·S2). A comparison between these and several other (μ-phenoxido)bis(μ-carboxylato) dimanganese complexes show that −J increases when the Mn–phenoxido distances decrease. Only rough correlations are observed when the direct Mn–O bond distances are used, but using the difference in bond length between Mn(II)–O and Mn(III)–O in the mixed valent complexes gives a strict relationship. This correlation shows the somewhat surprising feature that the exchange interaction increases with increasing the Mn(III)–O distance. This phenomenon is attributed to the fact that increased Mn(III)–O distance decreases orbital overlap and lowers the energy of the Mn(III) centered orbital, which is one of the orbitals involved in the exchange interaction. This lowering of orbital energy leads to an increased population of and therefore an increased magnetic exchange.
Electrochemical studies on 19–21 revealed a reversible Mn(II)2/Mn(II)Mn(III) redox couple, but an irreversible Mn(II)Mn(III)/Mn(III)2 oxidation at higher potentials. It was suggested that a substitution of the coordinated exogenous ligand(s) could be the cause of this, which is supported by the observation by X-ray crystallography that different solvent molecules can bind in the complexes. An increased stability domain of the mixed valence complex in acetonitrile was observed compared to symmetrical tetrapyridyl derivatives of the ligands (H8). It was explained by the coordination to the Mn(II) ion of an acetonitrile molecule, which is less able to stabilize higher oxidation states.
The homovalent dimanganese complexes catalyzed the disproportionation reaction of hydrogen peroxide, and with combinations of UV–vis, EPR, ESI-MS, IR spectroscopy, electrochemistry and X-ray absorption experiments, three intermediates in the reduction process of this reaction could be identified [58]. An early and very short-lived Mn(II)Mn(III) species forms, which has the phenoxido bridge displaced to yield an uncoupled dinuclear species, probably due to concomitant oxidation of one manganese ion and protonation of the phenoxide. This species is suggested to bind hydrogen peroxide, thus forming a compound that is observed during the whole reaction. Thorough investigations identified this compound to have the formula [Mn(III)Mn(IV)(L)(O)2(OAc)]+ with both oxido bridges derived from the same H2O2 molecule, as indicated by isotope-labeling experiments. In this complex, the phenolate appears to be deprotonated and the acetate most likely binds in a bridging mode. The complex was directly synthesized by reaction of the diMn(II) complexes with tBuOOH (TBHP) and despite its instability it could be characterized at −20 °C by UV–vis, EPR, ESI-MS and EXAFS.
As the reaction proceeds, another complex is formed and becomes the dominant species towards the end of the reaction. This complex was formulated as [Mn(II)Mn(III)(18)(O)(OAc)]+ on the basis of EPR, ESI-MS, and XANES spectroscopic measurements. By isotopic labeling it could be concluded that it is formed from the μ-dioxido complex. The catalytic cycle is suggested to shuttle between this and the Mn(III)Mn(IV) complex. The fact that the latter species dominates the reaction except in the end suggests that the oxidation is slower than the reduction process studied here.
2.3 Models for dinuclear zinc phosphoesterases
In the middle of the 1970s, it was shown that two different strains of soil bacteria (Pseudomonas diminuta and Flavobacterium sphaeroides) were able to hydrolyze organophosphates [59]. The reaction is catalyzed by the enzyme zinc phosphotriesterase, which contains a dinuclear zinc moiety in its active site (vide infra). Since organic phosphotriesters are fairly uncommon in nature, it is still not completely clear if there is a natural substrate for the enzyme [60]. Organophosphates were not widely used as insecticides until after World War II, and it is unlikely that the ability to break down the poison instead of being destroyed, as other organisms are, has developed during such a short time span. Until a natural substrate is found, the insecticide paraoxon (Scheme 6) is often used as an example of a wide range of phosphotriesters that have been identified as substrates.
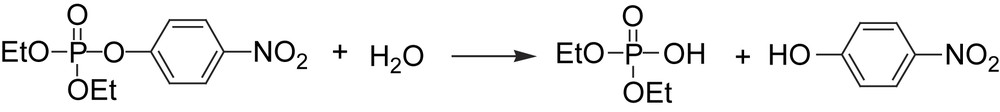
Hydrolysis of the insecticide paraoxon.
A number of crystal structures of the enzyme zinc triphosphoesterase have been published [61,62]. They also include some metal-exchanged proteins. Among others, a Cd2 substituted phosphotriesterase has been used for a number of spectroscopic studies [63]. The two zinc ions in the native enzyme are bridged by a carbamylated lysine residue (Fig. 4), a feature which zinc phophotriesterase shares with the dinuclear nickel enzyme urease (vide infra). A hydroxido group bridges the zinc ions and each ion has two histidine residues in its coordination sphere. In addition, one zinc (Zn2) coordinated by a monodentate aspartate. In the crystal structure, the other zinc ion (Zn1) has a labile oxygen donor coordinated, which completes the pentacoordinate geometry for both centers. The metal–metal distance is 3.4 Å.
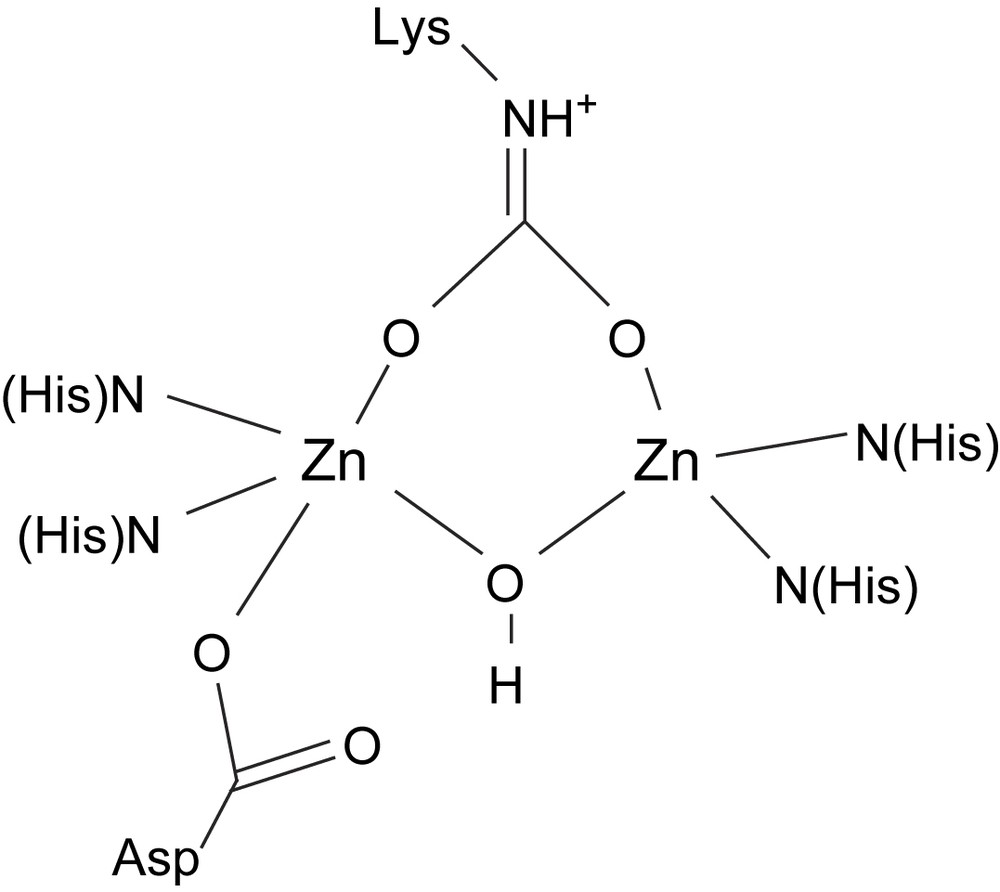
Schematic structure of the active site of zinc phosphotriesterase.
With regard to catalytic mechanisms, three main proposals have been made [64]. The first step is similar in all cases, viz. the coordination of the free phosphate oxygen to the coordinatively unsaturated Zn ion (Zn1), replacing the labile oxygen donor. It demonstrates the need for a vacant/labile coordination site, but not necessarily a symmetric coordination sphere. In the next step, either a terminal hydroxido group on Zn2, or a water molecule activated by the same metal, attacks the phosphorus atom of the phosphate group and releases the product alcohol. This leads to a bridging or terminal phosphate group, which is subsequently released. A similar mechanism involving the bridging hydroxido group as nucleophile has been discussed [65].
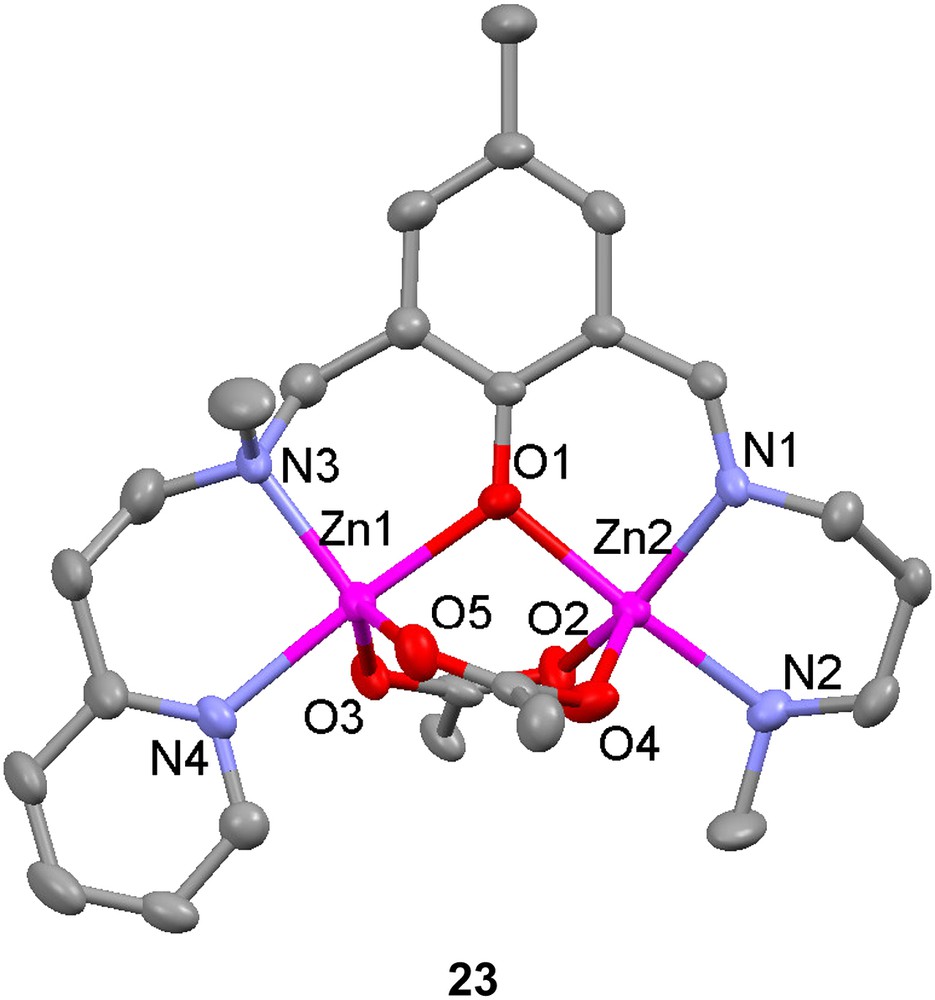
The unsymmetrical ligand H22 has been used to prepare two phenolate-bridged dinuclear zinc complexes and one phenolate-bridged dinuclear nickel complex with acetates as bridging exogenous ligands [66]. The bridging mode of the acetates in solid state for the two zinc complexes differed depending on whether sodium thiocyanate was added or not. Without SCN− present, two syn–syn bidentate bridging acetates were found in complex 23, but with thiocyanate ions present and coordinating to the zinc ions, only one acetate bridges in the uncommon monodentate mode (24). The distance between the zinc ions in these complexes are 3.309 Å and 3.254 Å, respectively.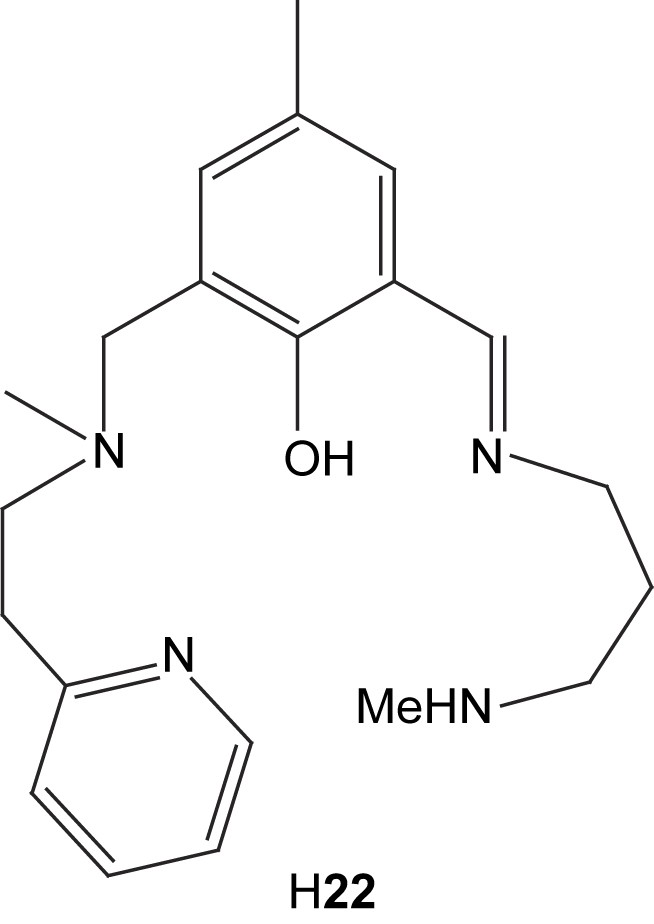

Several new 3,5-substituted pyrazole-containing ligands have been synthesized, where the pyrazole moiety is expected to act as the bridge between two metal centers [67]. To avoid dimerization and other unwanted reactions associated with the nucleophilic pyrazole moiety, the tetrahydropyran (thp) protecting group was used. An enantiomerically pure C2-symmetric ligand was prepared and used to synthesize a dinuclear zinc complex 25, the structure of which has been determined by X-ray crystallography. The proton originally on the pyrazole is probably transferred to the tertiary amine and participates in hydrogen bonding between the amine, the ether oxygen and one of the chlorides, because of the short distances between these atoms. This prevents one of the side arms from coordinating to the metal center, resulting in an unsymmetrical dinuclear zinc complex.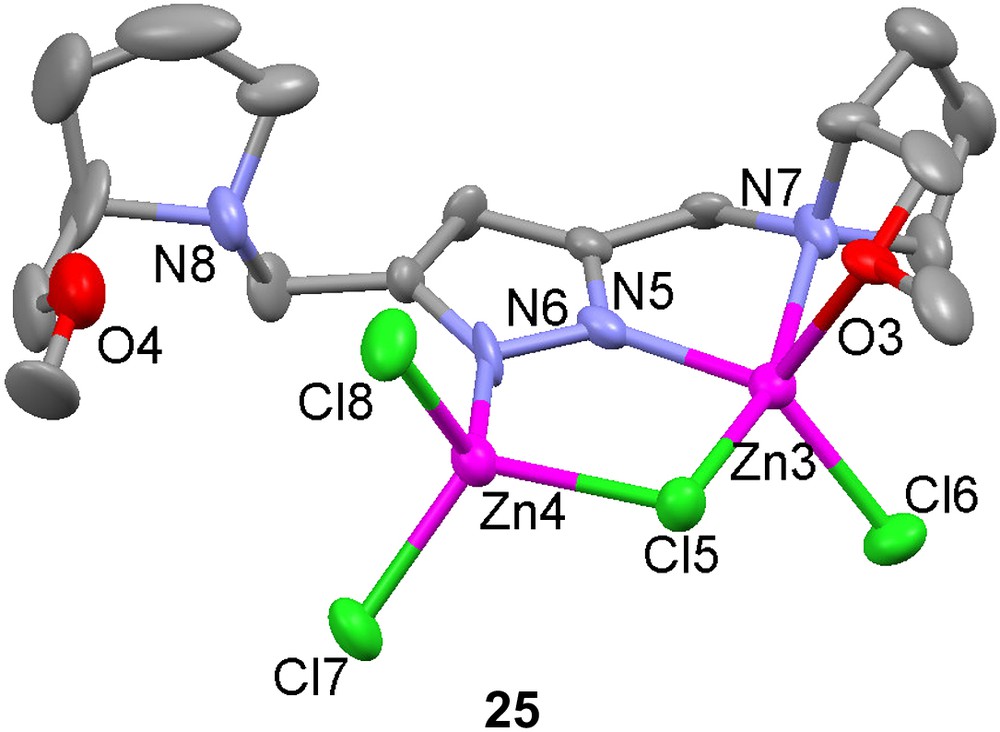
The homodinuclear complex [Zn2(10)(O2AsMe2)]2+ (26) has been prepared [49]. The dizinc complex 26 is a structural and functional model for phosphoesterases with dinuclear active sites, such as alkaline phosphatase, phospholipase C, nuclease P1 and phosphotriesterase. The hydrolytic activity of the dizinc complex was studied by monitoring the transesterification of hpnp (cf. Scheme 3). The complex was found to significantly promote the transesterification at pH > 6, and solution speciation studies indicated that the species [Zn2(10)(OH)]2+ (and [Zn2(10)(OH)2]+) predominate in this pH range. It is proposed that these species bind hpnp and that the metal bound hydroxide deprotonates the alcohol moiety of hpnp to initiate the transesterification.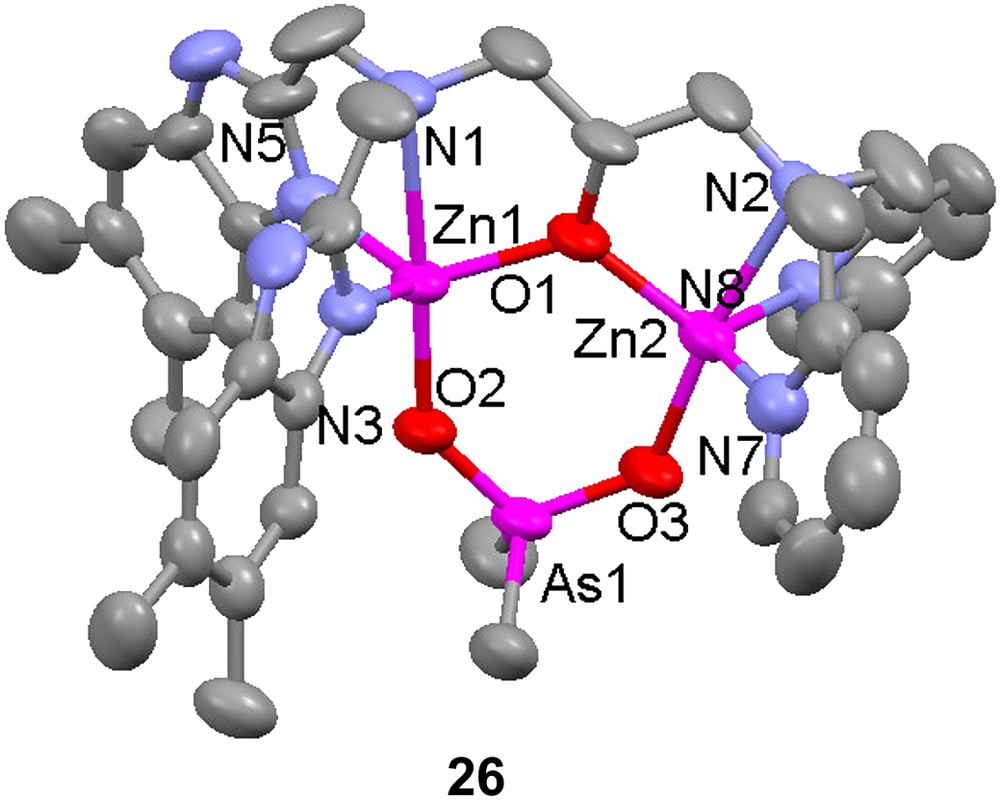
2.4 Models for the active site of urease
Urease is a hydrolytic enzyme that catalyzes the breakdown of urea to ammonia and carbamate, which spontaneously decomposes to ammonia and carbon dioxide [68,69]. Early data predicted that the active site of urease contained two nickel ions [70]. Several crystal structures have been published [71–74]. They show that the catalytic site consists of a dinuclear nickel unit bridged by an oxygen donor (hydroxide) and a carbamylated lysine, i.e. a lysine residue that has reacted with one molecule of carbon dioxide (Fig. 5). Each nickel ion is also coordinated by two histidine residues and one water molecule, while one of the ions (normally termed Ni2) in addition has an aspartate side chain in its coordination sphere. This means that the nickel centers are non-equivalent, which may be important for the catalysis. The nickel–nickel distance is about 3.5 Å.

Schematic structure of the active site of urease.
A number of mechanisms for the catalytic urea hydrolysis have been suggested [72,75–77]. They are all in agreement when it comes to the first part of the catalytic cycle, in which the carbonyl oxygen of urea is coordinated to the open coordination site on Ni1. The origin of the attacking oxygen donor is being debated. A terminal water/hydroxide on Ni2 or a bridging hydroxide have been suggested. The latter hypothesis has been inferred from the fact that urea analogues which function as inhibitors bind at the active site in a tetrahedral fashion, bridging the two nickel atoms [72]. Other suggestions include, for example, a cyanate intermediate [77].
2.4.1 Pyrazolate-bridged dinickel model complexes
A number of symmetric and asymmetric ligands based on pyrazolate with amino and thioether-containing pendant arms have been synthesized and their Ni(II) complexes have been prepared [78–80]. In reaction with nickel chloride, the most common architecture of the resulting complexes could be described as ClNi(μ-Cl)(μ-L)NiCl, where L is either of the ligands 27, 28 or 29 [78,80]. In the case of the hexadentate ligand 27, a tendency to dimerization or polymerization of the dimeric units was seen, in which the chlorine atoms, only bound to one of the nickel atoms in the dimer, act as bridge-points to coordinatively unsaturated nickel ions in neighboring dimers [80]. This is to the most part avoided when the octadentate ligand 28 is used, where the ligand can provide a saturated distorted octahedral coordination sphere together with the chloride ions.
In the case of the asymmetric ligand H29, two different modes can be seen. The straightforward reaction with nickel chloride results in a structure (31) in which one of N-donating pendant arms of 29 is “dangling”, leaving one nickel atom in a square-pyramidal orientation [79]. Furthermore, it has been shown that the dangling side chain can be made to re-coordinate by the exchange of the terminal chloride for BPh4− as a counter ion, as seen in complex 32 [79]. The internuclear distance varies between 3.82 Å (31) and 3.90 Å (32).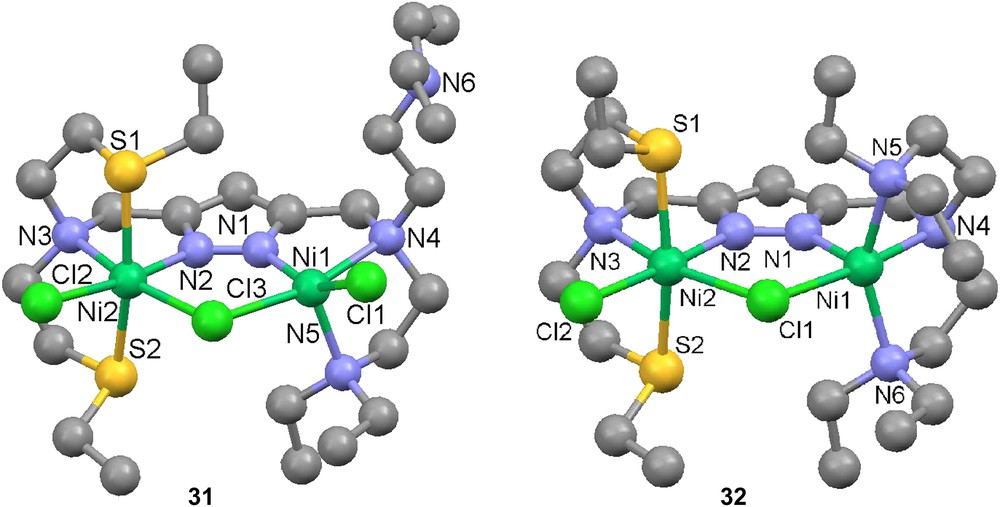
In the case of the hexadentate ligand 30 with all nitrogen donors, a number of new complexes based on [Ni2(30)(O2H3)](BPh4)2(acetone)2 (33(BPh4)2(acetone)2) were synthesized [79]. These include μ-amidato-N,O-bridged complexes based on the corresponding nitrile compound, presumably by oxygen attack from a terminally bound hydroxyl group on the nitrile carbon. Complex 33 can also generate a urea complex (34) [81]. This structure features a bridged urea molecule binding through the oxygen atom to one nickel center and through one nitrogen atom to the other. Also the urea complex can generate an amidato-bridged complex, which is the same as the one originating from dimethylcyanamide.
With the intention to mimic the dinuclear Ni active site of urease, a number of complexes with urea or urea-like ligands were prepared, [Ni2(27)(μ-OAc)(N,N′-dimethylurea)2(MeOH)2](ClO4)2 (35), [Ni2(28)(μ-OAc)(benzylurea)2](ClO4)2 (36), [Ni2(28)(μ-OAc)(acetone)2](ClO4)2 (37) and [Ni2(29)(μ-OAc)(urea)](ClO4)2 (38) [82]. All the urea molecules feature coordination through their oxygen atom to nickel and in available instances hydrogen bonding through an amine hydrogen to the neighboring acetate oxygen atom. No secondary metal interaction is observed. Due to the additional carboxylate ligation, the internuclear distance is longer, 4.16–4.31 Å.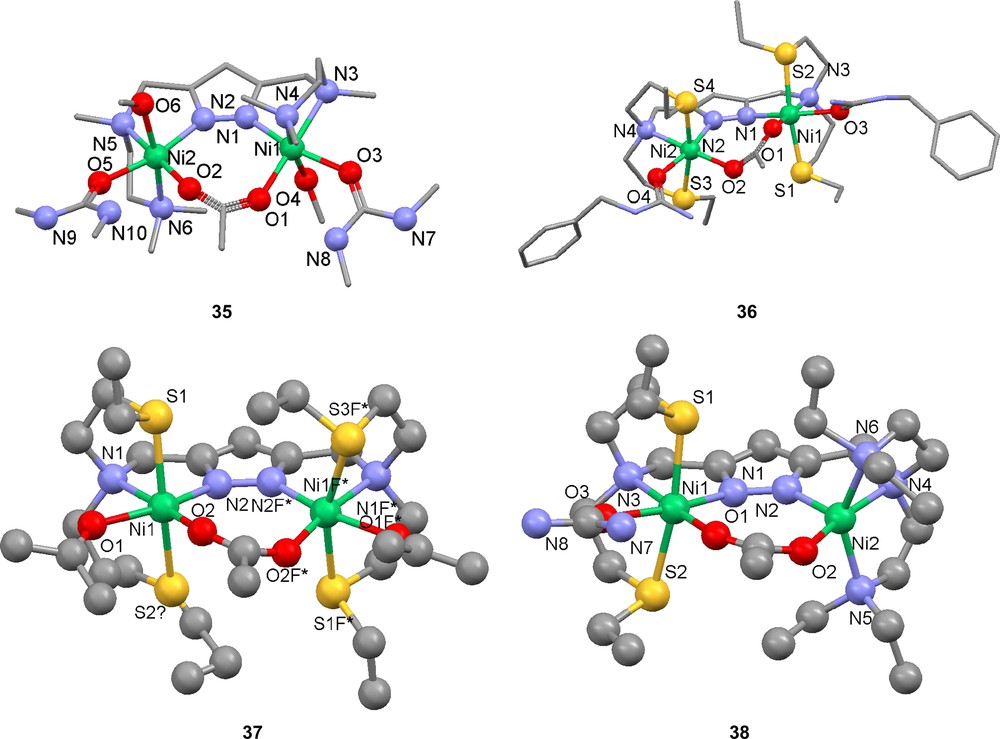
In order to investigate the catalytic activity of the generated complexes, ethanolysis of urea to ethyl carbamate was studied [82]. Most of the results show low to medium stoichiometric activity with the exception of complex 37. However, in the case of 37, the presence of a mononuclear catalyst cannot be excluded. Complex 38, which includes the asymmetric ligand 29, shows no measurable activity. Coordination of cyanide or cyanamide (NCNH2) to compound 37 yields complexes with the additive in a bent bridging coordination (39 and 40) [83,84]. The cyano and hydrogencyanoamido ligands are coordinated in between the two nickel atoms, which in turn are held together by the pyrazolate in the backbone. If the cyano-compound is extended further, as in the case of 2-cyanoguanidine (NCNC(NH2)2) and ortho-hydroxybenzonitrile, the ligand stays bridging, but is forced out of the internuclear space, leaving a vacant coordination site at the second nickel that may be filled by an exterior ligand [84]. In the case of 2-cyanoguanidine, IR spectroscopy and computational modeling indicate a weak interaction between the second nickel center and atoms of the cyanide group. This also drastically shortens the internuclear distance to 3.99 Å [83].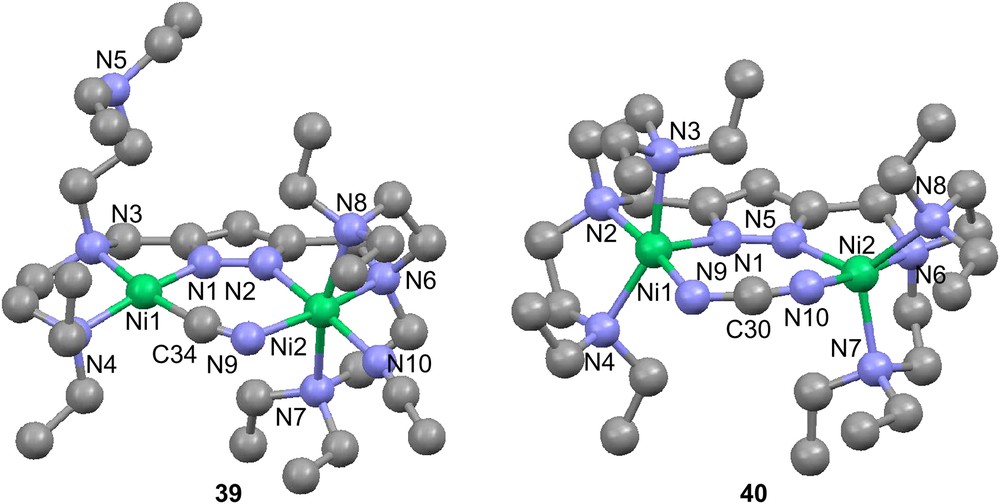
2.4.2 Phenolate-bridged dinickel complexes
A large number of methylphenolate-based ligands have shown to generate interesting nickel complexes and biomimetic models [85–95]. A number of these structures has previously been reviewed [13,96]. The general structure of the asymmetric ligands can be described as a 2,6-substituted 4-methylphenol. The side arms normally contain a bridging amine or a Schiff base and additional arms (with donors) that make the ligand denticity vary from tetradentate to heptadentate (Fig. 6). Among the recently published structures, side arms containing amines only [86–88,95], pyridines [85–88,90,93,94], benzimidazole [88], imidazoles [91,92], thioethers [88,90], aldehydes [85], and carboxylic acids [91,92] have been noted.
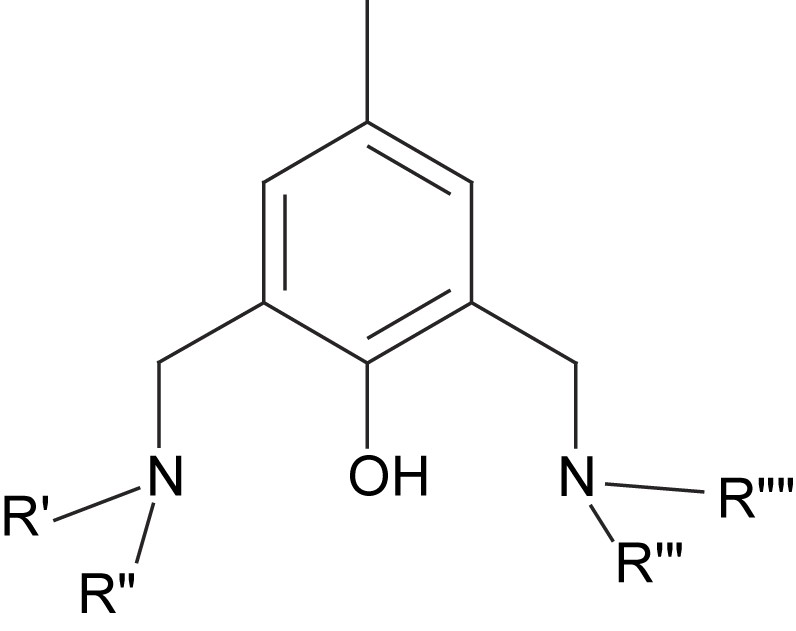
General structure of a phenolate-based dinucleating ligand.
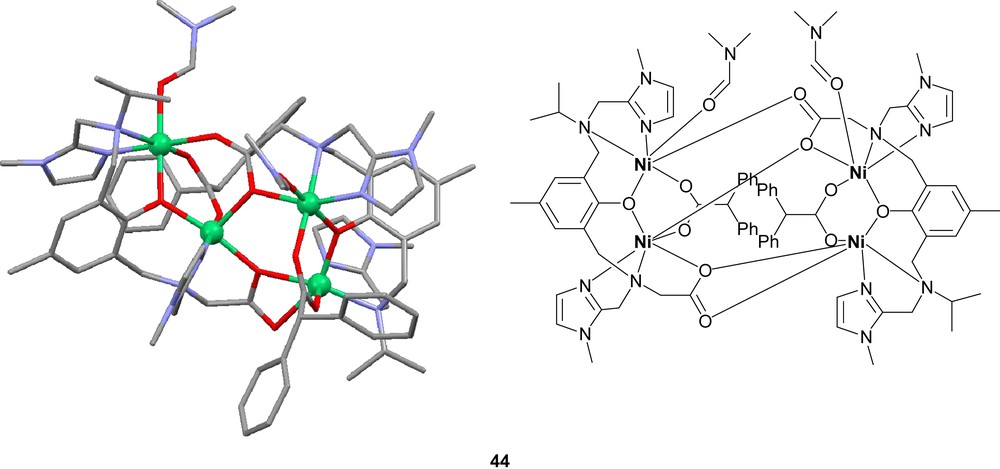
The nickel complexes formed from these ligands can in most cases be described as dinuclear complexes with phenolate-bridging ligands in an “end-off” configuration (Fig. 7). The additional side arm donor moieties then fill up the coordination sites on each nickel center. Diversions from this general concept are common. Ligands with low denticity tend to generate complexes with more than one ligand per two nickel ions. The tetradentate ligands H41 and H42 are examples of this phenomenon. Ligand H41 form Ni2L2 complexes with nickel [86], while the bulkiness of ligand H42 only permits Ni3L2 complexes [88]. Also pentadentate ligands can generate Ni3L2 coordination [90]. Dimer of dimer type structures are also common [85,88,91–93]. These are normally generated by bridging oxygen atoms, which stems either from the solvent or from one of the ligand arms. In the case of the pentanuclear product formed via reaction of ligand H43 with nickel acetate, two dimers are bridged by acetate and hydroxyl bridges that are partly kept together by a fifth metal center [88]. In the case of the mixed imidazole/carboxylate ligand H313, the dimer of dimer structure is generated by bridging carboxylates from the ligand itself (44) [91,92].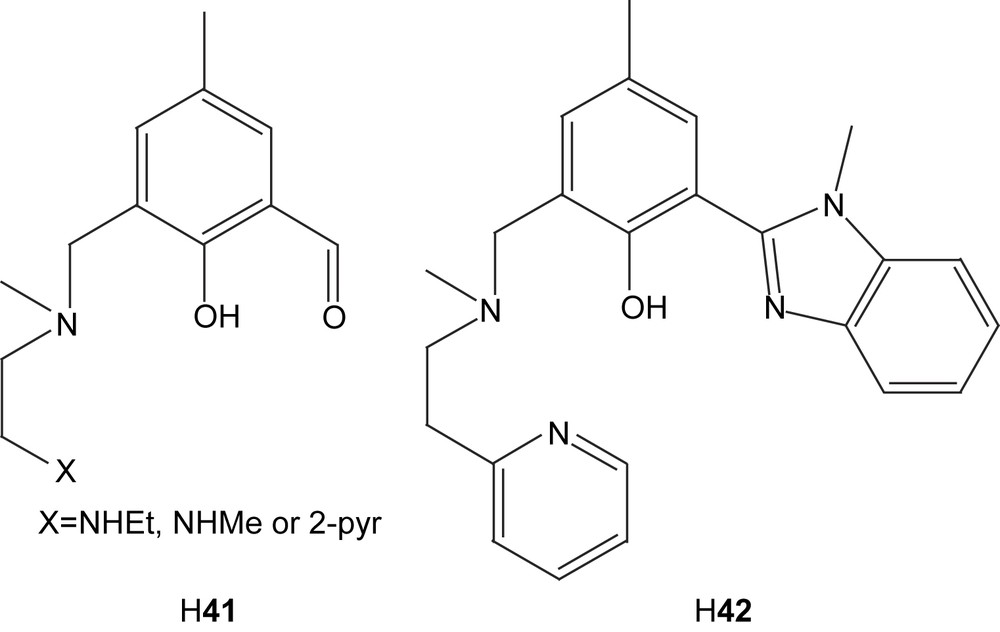
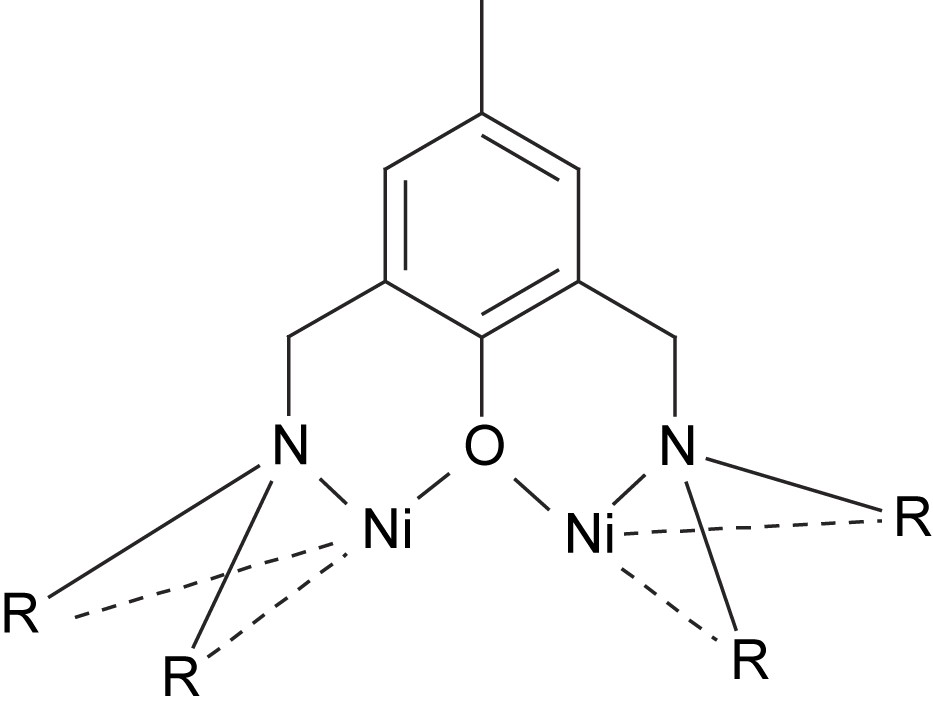
General structure of phenolate-bridged Ni2 complexes.
The phenolate-bridged structures result in closer internuclear distances, which tend to vary from 3.1 to 3.6 Å. In several cases this yields structures very similar to the asymmetric dinuclear site found in the active site in urease [87,91,92]. The common phenolate bridge is not directly modeling an amino acid side chain, but rather the hydroxide ligand found in the resting structure of the enzyme. The similarities found between the complexes and the active site(s) of urease make the complexes potent structural models for the enzyme. In some cases, urea has been found to coordinate to these structures. In the case of the pentadentate amine-rich ligand H45, urea was found to bind in the presence of nickel perchlorate to form [Ni2(45)(OH)(OH2)2(urea)][BPh4]2 (46). Urea binds to the nickel ion with the least steric hindrance and hence avoids the amino group with an ethyl substituent. Another pentadentate amine-rich ligand, H47, also shows signs of interaction with urea. When dinickel complexes of the ligand are stirred in ethanol and urea, isocyanate complexes can be crystallized after reflux in 15–20% yields [85]. The isocyanate nitrogen atom is found to bridge the nickel centers.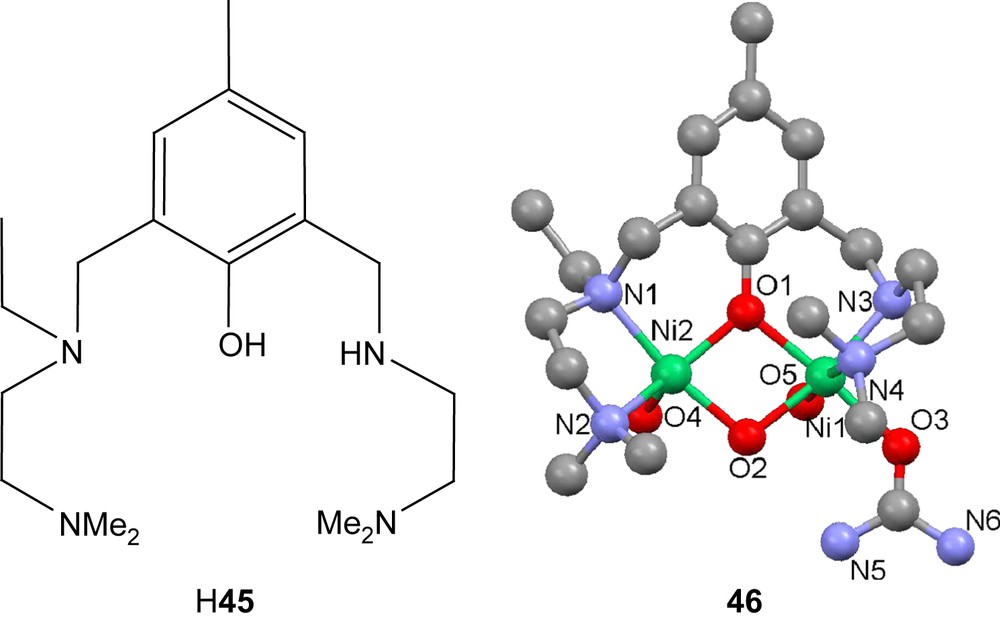
The tetranuclear complex 44 also coordinates urea [92]. The slightly asymmetric configuration of the two dimers leads to two different models for the active site of urease, both with internuclear distances very close to the crystal structure of urease (3.47 and 3.51 Å vs. about 3.5 Å in urease) and very similar coordination spheres. One of the dimers has a solvent molecule coordinated on the same nickel center as the ligand carboxylate, while the other has the open site on the other nickel. If urea is added to complex 44, crystals of complex 48 are generated with urea bound through its oxygen atom to the latter dimer. This would indicate that urea would prefer associating with the nickel center not carrying the carboxylate, which would mean Ni1 in the case of urease. Evidence for an alternative mechanism progressing via a cyanate intermediate has been seen in nickel complexes containing the ligand 49 [77]. Both urea- and intermediate cyanate-bound complexes have been generated. Complexes with coordinated urea have also been shown to form with nickel and the ligand 50 [95]. The equivalent structure with two imine arms does not show this ability.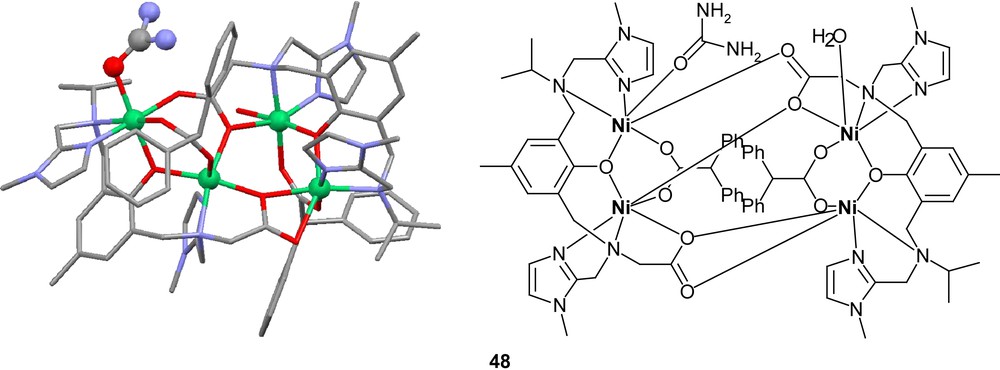

Another nickel complex based on ligand H313 also shows evidence of catalytic activity [92]. A functional study using hydrolysis of hpnp as a substrate shows comparative rates to several previously published dinuclear catalysts. However, the applicability of observations on the model complex to the active site of urease is debatable since the otherwise relatively fine-tuned structural model blocks out the important bridging site by the phenolato ligand.
2.4.3 Other asymmetric dinickel complexes of biological relevance
A number of new ligands which provide an alkoxy unit as a bridging entity rather than a phenolate group have been synthesized and their nickel complexes have been studied [97,98]. As examples can be mentioned ligands H51 (cf. H6) and H52, which both generate well-behaved dinuclear complexes with nickel. Also here, both coordination sphere architecture and internuclear distances are reminiscent of the urease structure. Also these complexes show hydrolytic properties [97].
When ligands H53 and H54 were reacted with Ni(ClO4)2·6H2O, hydrolysis of the Schiff base moiety in each ligand was observed, and two hydrolyzed ligands were found to coordinate in head-to-tail fashion to form the complexes [NiX(OH2)2]2+ (X = hydrolysed 53 or 54) [99]. In the case of 52, the hydrolysis was not restricted to the Schiff base but also led to partial hydrolysis of the terminal amine to form a secondary amine. The nickel ions were found to be antiferromagnetically coupled in both complexes; J = −31 cm−1 for the 53-based complex and J = −17 cm−1 for the 54-based.
A dinickel complex of ligand H22, [Ni2(22)(OAc)2(MeOH)]+ (55) has two acetates bridging in different manners [66], one in a syn–syn bidentate and another in a monodentate fashion. In the analogous zinc complex 23 (vide supra), the latter acetate is bidentate for one of the zinc ions. This bridging mode is very common for acetate-bridged dinickel complexes when ligands forming six-membered chelating rings with the imine coordinating side arms are used. The distance between the two nickel ions in the complex is 3.096 Å, which also lies in the range of previously synthesized complexes of this kind.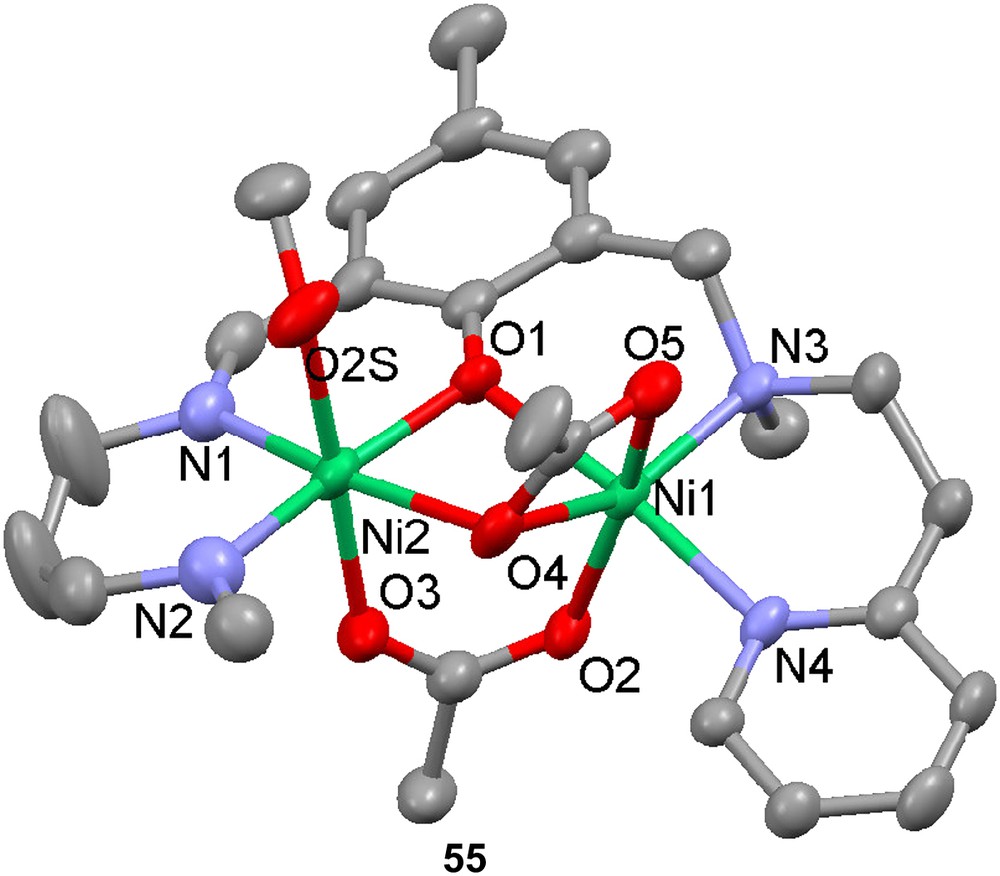
3 Models for dismutases and monooxygenases containing dinuclear active sites
3.1 Models for superoxide dismutase
Superoxide dismutases catalyze the disproportionation of the superoxide anion into dioxygen and hydrogen peroxide. There are mononuclear superoxide dismutases (SODs) that contain iron or manganese in their active sites. In eukarytic erythrocytes, the disproportionation is catalyzed by SODs containing a binuclear Cu, Zn active site. The crystal structure of bovine erythrocyte Cu–Zn SOD reveals that the two metal ions are linked via a deprotonated imidazolyl moiety of a histidine. In addition, the copper ion is coordinated by three histidines, while the Zn ion is coordinated by two histidines and a monodentate aspartate (Fig. 8). The mechanism by which the enzyme disproportionates the superoxide anion is not completely determined, but it is recognized that an arginine residue in close proximity to the dinuclear site plays an essential role in the catalysis and that the copper is the redox active metal that oxidizes/reduces the superoxide ion. The essence of the proposed catalytic mechanism is summarized in Scheme 7.
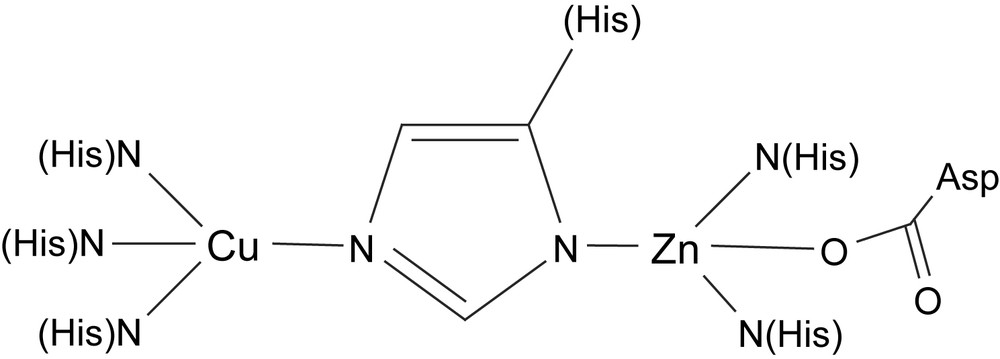
Schematic structure of the active site of Cu, Zn superoxide dismutase.
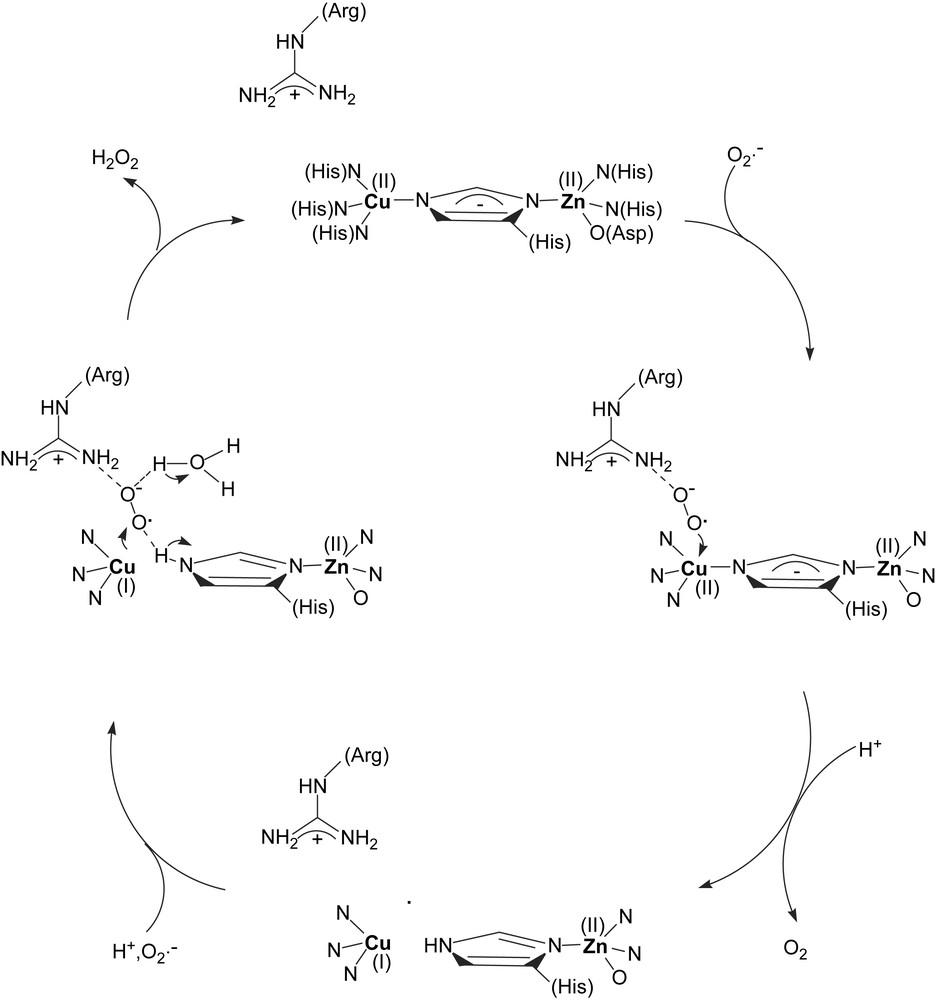
Schematic depiction of the Cu, Zn superoxide dismutase mechanism (adapted from Ref. [100]).
Two dinuclear copper complexes containing the asymmetric ligand H56, viz. [Cu2(56)Cl3] (57) and [Cu2(H56)Cl4] (58) [101], have been prepared and structurally characterized by X-ray crystallography. The first complex is formed when the metal-to-ligand ratio is 2 or higher, while the second is formed when the same ratio is less than 2. The crystal structures reveal that in both complexes, the copper that is coordinated by pyrazole moieties is in a distorted square pyramidal coordination environment, while the second copper is in a distorted tetrahedral environment. The first complex, in which the metals are bridged by an imidazole moiety, is a good structural model for Cu–Zn superoxide dismutase (it was not possible to synthesize an analogous Cu–Zn complex). pH-dependent EPR and UV–vis spectroscopy of the complex shows that the imidazolate bridge begins to form at pH 5 and is fully formed at pH 7–8, demonstrating that the solid-state structure is maintained in aqueous solution at biologically relevant pHs. Recent measurements of kinetic isotope effects support inner-sphere electron transfer between the copper and the superoxide ion [102]. The complex is a good catalyst for superoxide dismutation, on par with bovine erythrocyte superoxide dismutase as well as the best model complexes for superoxide dismutase.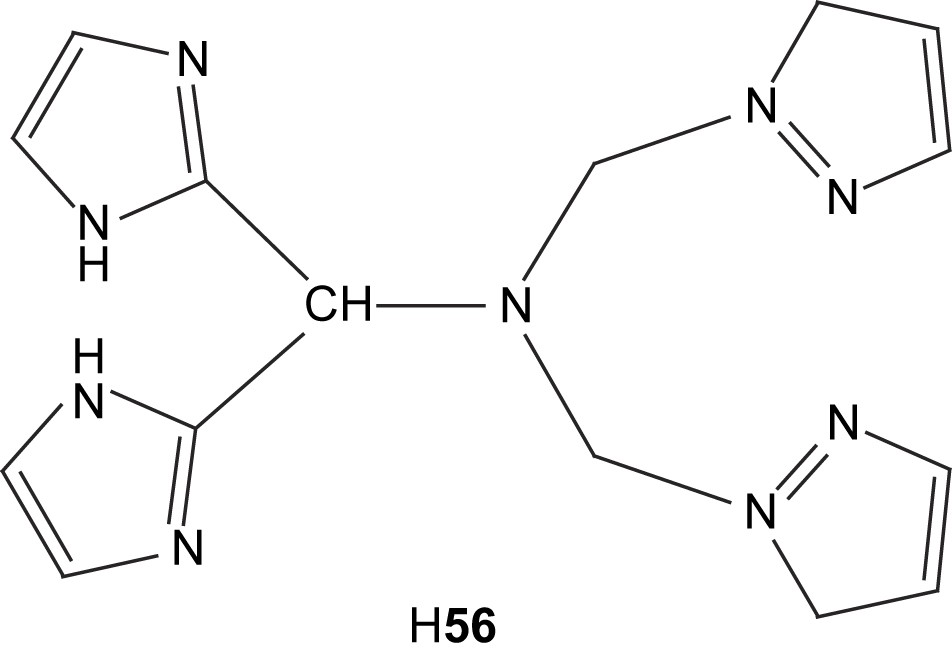

3.2 Models for tyrosinase
Tyrosinase is both a monooxygenase that ortho-hydroxylates tyrosine and an oxidase that can oxidize the resulting catechol to an orthoquinone which is used in the biosynthesis of melanin. The enzyme is found in all organisms, including man. The first crystal structure of a tyrosinase (from the bacterium Streptomyces castaneoglobisporus) was determined recently [103] and this structure confirmed that the active site contains a dinuclear type-3 copper site with each copper coordinated by three histidines. The active site is thus very similar to those of the oxygen-carrying protein hemocyanin and the enzyme catechol oxidase (vide infra). The reactivity/substrate selectivity of these three types of proteins is probably regulated by the specific protein structure/amino acids surrounding the active site [104,105].
The proposed tyrosinase mechanism (Scheme 8) involves the initial coordination of dioxygen to the fully reduced Cu(I)2 site resulting in the formation of a Cu(II)2 peroxide complex with the peroxide bound in the μ–η2–η2 coordination mode. Subsequent coordination of the substrate phenol (tyrosine) to one of the copper ions leads to the substrate being suitably oriented for the ortho-hydroxylation reaction. The exact coordination mode of the resulting catecholate has not been determined. Subsequent electron transfer leads to reduction of the two coppers and oxidation of the bound catecholate to an orthoquinone.
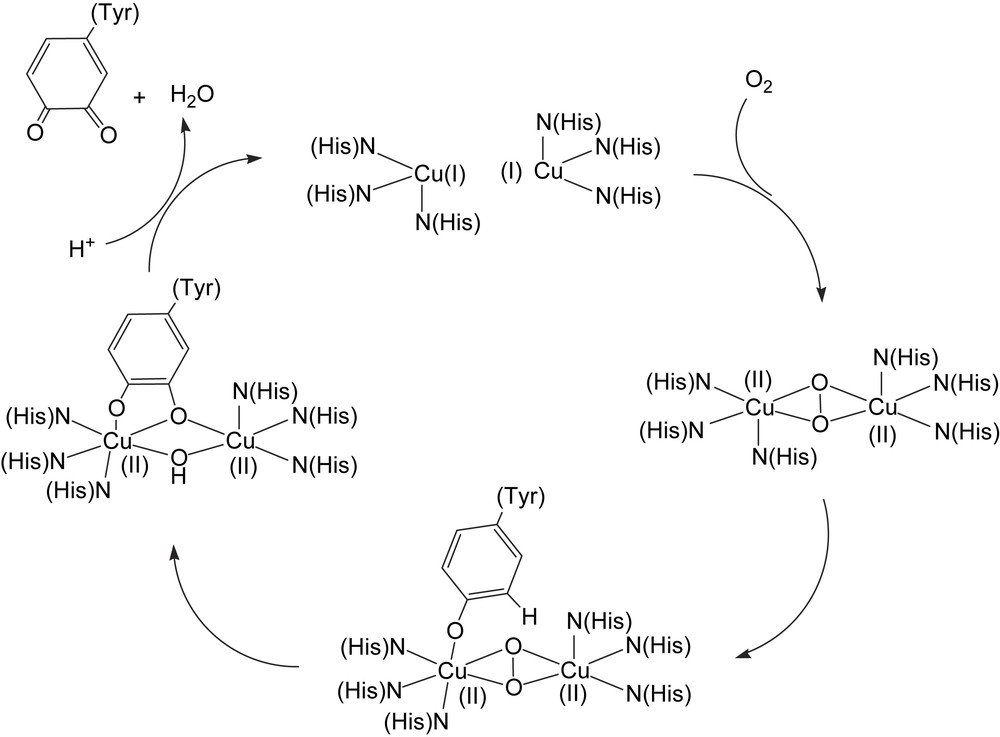
Schematic depiction of the proposed tyrosinase mechanism (adapted from Ref. [105]).
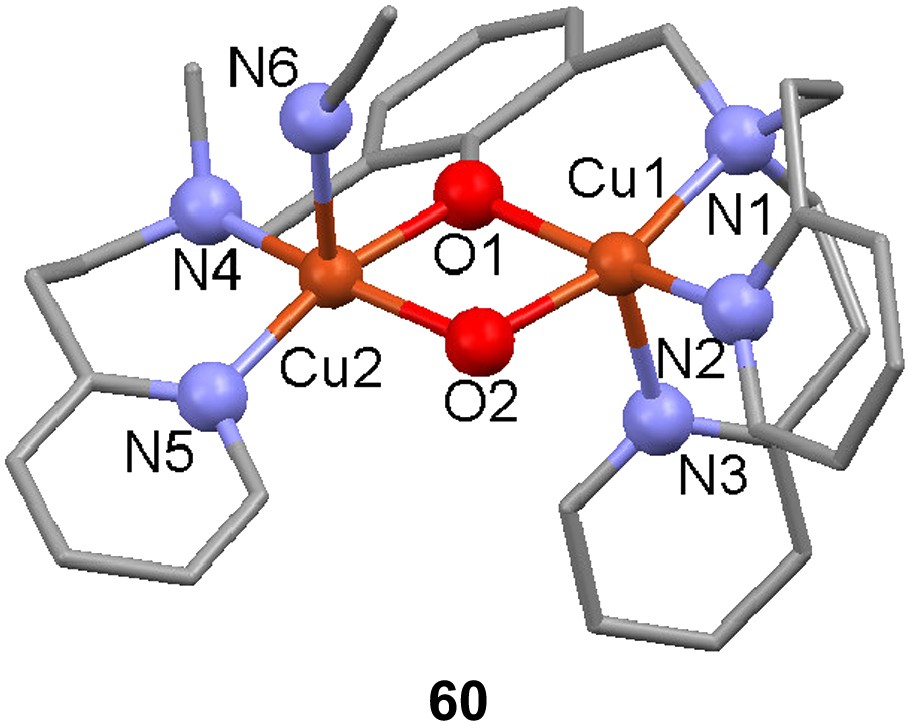
The new unsymmetric dinucleating ligand 59-H with a m-xylyl spacer moiety was used to prepare a dicopper(I) complex that upon reaction with dioxygen hydroxylates the ortho position between the side arms of the spacer, yielding a phenolate and hydroxido-bridged dicopper(II) complex ([Cu(II)2(59-O−)(OH)(CH3CN)](PF6)2) in 90% yield [106]. NMR and elemental analysis of the initial dicopper(I) complex both concur to it being formulated as [Cu2(59-H)]X2 (X = ClO4− or PF6−) without coordinated solvent (in the solid state) leaving one of the metal ions coordinated by only two nitrogen donors, although a Cu–Cu interaction cannot be excluded. The [Cu(II)2(59-O)(OH)]2+ complex formed (60) could be crystallized however and its X-ray structure displayed a slightly distorted square pyramidal coordination of the two metal centers that, due to ligand asymmetry, resides in different environments where the less ligand-coordinated copper also has a acetonitrile molecule bound to it with the rather unusual Cu–N–C angle of 149°. The structure does not differ significantly from the complexes made from the symmetrical version of this ligand. ESI-MS in acetonitrile shows that the formula is maintained except for a considerable loss of coordinated acetonitrile, and IR confirmed the presence of an O–H group in the complex. Antiferromagnetic coupling between the copper ions results in a low magnetic moment of 0.95 μB per copper ion and no signal in EPR measurements. As observed previously for the symmetric derivative, the cyclic voltammogram do not show two sequential reversible one-electron redox reactions, but instead one cathodic and two anodic waves at −0.49, −0.37 and −0.25 V, respectively, probably because the bridging hydroxyl group does not readily support two different oxidation states for the copper ions. The similar reactivity towards dioxygen between the complexes derived from the symmetric and unsymmetric ligands may be due to coordination of solvent at the available site in the unsymmetric complex, which levels out the differences in electronic properties.
The new phenolate ligand produced by reaction of the initial complex with dioxygen could be extracted by removing the Cu(II) ions with ammonia, and a new dicopper(I) complex could be formed with the ligand. This new copper(I) complex, which also could be produced by reduction with diphenyl hydrazine, appears to have an acetonitrile molecule coordinated at the copper ion coordinated by only two ligand-derived nitrogen moieties according to spectroscopic and analytical data. Coordination of the phenolate to either copper could not be experimentally determined.
The new copper(I) complex also shows reactivity towards uptake of dioxygen (Cu:O2 2:1) forming a new complex with a strong absorption at 390 nm (LMCT HOO− → Cu(II) and/or PhO− → Cu(II)), a shoulder at 450 nm and a weaker absoption at ∼630 and 1000 nm (d–d transitions) observed at −78 °C by UV–vis spectroscopy. The product is formulated as [Cu(II)2(59-O−)(OOH−)]2+ because this is in agreement with two previously well-characterized complexes with symmetrical and unsymmetrical hydroxylated ligands, respectively, that have similar absorptions in UV–vis but also have been characterized by X-ray structure and EXAFS measurements. The peroxido complex synthesized here oxidizes PPh3 to OPPh3 in 85% yield, a reaction also characteristic of hydroperoxido complexes.
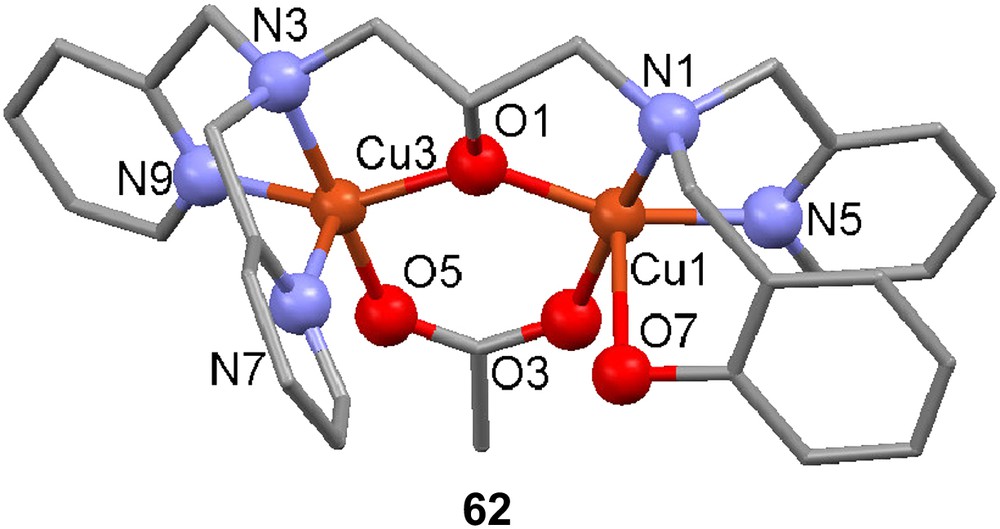
The ligand H261, containing a terminal phenolate donor moiety (cf. H21), was reacted with two equivalents of Cu(II) acetate in methanol in the presence of NaClO4 to afford [Cu2(H61)(OAc)]2+ (62) [107]. The crystal structure of 62 contains two crystallographically independent molecules that have similar geometric parameters. Both five-coordinate copper ions are in distorted square pyramidal geometries. The phenol moiety remains protonated, and the complex may be a relevant structural model for initial tyrosine coordination to the active site of tyrosinase. The EPR spectrum of the complex reveals two signals, one broad signal at g = approx. 4 attributed to a ΔMs = 2 transition and a transition at g = approx. 2 that has the typical characteristics of a square pyramidal Cu(II) geometry. Cyclic voltammetry reveals a reduction at −0.89 V (vs. Fc+/Fc) and a quasi-reversible wave at −1.11 V; the first reduction is attributed to Cu(II)2/Cu(II)Cu(I), with the Cu in the N3O2 coordination sphere being reduced, and the second reduction is attributed to reduction of the mixed valence state to the all cuprous oxidation state.
3.3 Models for catechol oxidase
As mentioned above, the enzyme catechol oxidase is very closely related to tyrosinase. The crystal structure of catechol oxidase from sweet potato [108] reveals that its type-3 Cu site is very similar to that of tyrosinase and hemocyanin. The functional difference between catechol oxidase and tyrosinase is that the former enzyme only catalyzes oxidation of orthocatechols to orthoquinones (presumably via a mechanism similar to that of tyrosinase), while tyrosinase catalyzes both hydroxylation of phenols to orthocatechols and subsequent further oxidation (vide supra). It has been proposed that the difference in reactivity may be due to the immediate environment of the active site in catechol oxidase, preventing its catechol substrates from coordinating to a copper in the same way as tyrosine (phenol substrates) coordinates in tyrosinase [105]. A review on synthetic modeling of catechol oxidase has been published recently [109].
The unsymmetrical compartmental ligand H63 was used to synthesize both a mono- and a dinuclear copper(II) complex without and with acetate as exogenous ligand, respectively [110]. The crystal structure of the dinuclear complex 64 revealed that the alkoxy group and one acetate were bridging the two metal ions, the latter in a syn,syn-μ-1,3 bidentate manner, and that the copper ions were coordinated in square planar and trigonal bipyramidal geometry, leaving an open coordination site on the less coordinated ion. In aqueous solution this site seems to be occupied by a water molecule, as indicated by potentiometric titration.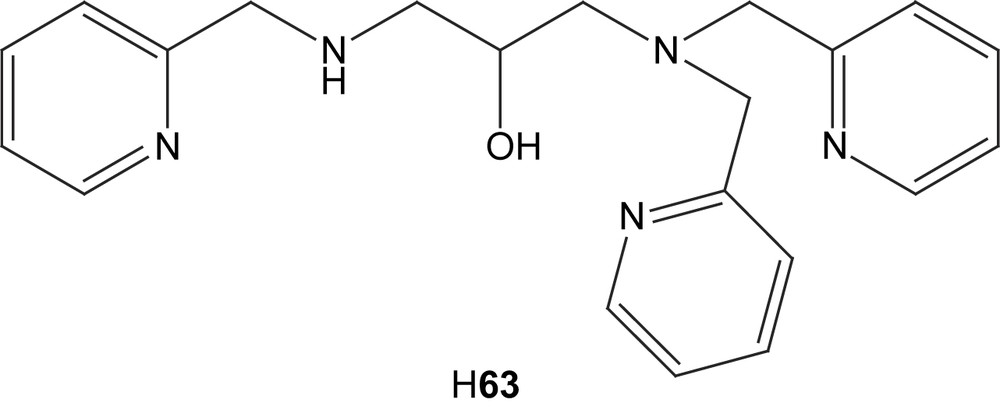

Magnetic susceptibility measurements at variable temperature show that the dinuclear complex exhibits a ferromagnetic coupling between the Cu(II) centers and the values g = 2.08 and J = +25.41 cm−1 (H = −2JS1·S2) could be extracted. The mononuclear complex has a UV–vis absorption peak at 758 nm and a shoulder at 843 nm that corresponds to d–d transitions for square pyramidal copper complexes. The dinuclear complex shows two well-defined absorption bands at 667 and 889 nm and this is probably due to the existence of two different geometries for the copper ions. EPR spectra of the complexes are in agreement with the square pyramidal geometry for the mononuclear complex, as suggested by the electronic spectra and a triplet state for 64, resulting from the ferromagnetic coupling of the metal ions. The electrochemical behaviour of the complexes are as expected, displaying one reversible reduction at −0.19 V for the mononuclear and two irreversible ones at −0.38 V and −0.56 V, all attributed to one-electron reductions of the copper centers.
Oxidation of 3,5-di-tert-butyl catechol was investigated by UV–vis spectroscopy, showing that 64 catalyzes the reaction. The mononuclear complex is inactive despite its higher reduction potential, indicating that the electrochemical properties play a secondary role and that the possibility to coordinate both oxygen atoms of the catechol is more important. The rate of the oxidation by the dinuclear complex 64 increased rapidly at pH = 8.05, which was explained by the fact that the water molecule that is coordinated in solution gets deprotonated at this pH and in turn deprotonates the substrate, which increases the rate of the reaction.
Dinuclear copper(II) complexes containing the ligands H365 and H366 were synthesized with either hydroxides, acetates or nitrates as exogenous ligands [111]. Elemental analysis (not performed on the hydroxide complexes) supports the formulation of the complexes as [Cu2L(X)]·H2O (X = OAc or NO3) and the molar conductivities also indicate neutral complexes. Coordinated or lattice water is evidenced in IR spectra as is the coordination of the nitrate and the acetates in the corresponding complexes. The IR absorption for the acetates is in the range of that for bridging acetate groups. The UV–vis spectra show the normal absorptions of d–d transitions and LMCT observed for this kind of complexes. It is worth noting that the d–d band for the acetate-bridged complexes are redshifted compared to the others, which could be explained by the distortion of the coordination geometry introduced by the more flexible bridging acetates. Antiferromagnetic coupling is inferred by both EPR spectra that show broad signals without hyperfine splittings and magnetic susceptibility measurements that gave values substantially lower than in the spin-only case. The catecholase activity for the complexes was also investigated by studying the oxidation of catechol by UV–vis spectroscopy. The acetate-bridged complexes all show higher activity than the hydroxido and nitrate derivatives. This was explained by an easier substitution of the acetate for the substrate, which is attributed to the higher flexibility of the acetates and the higher stability of the hydroxide and nitrate complexes.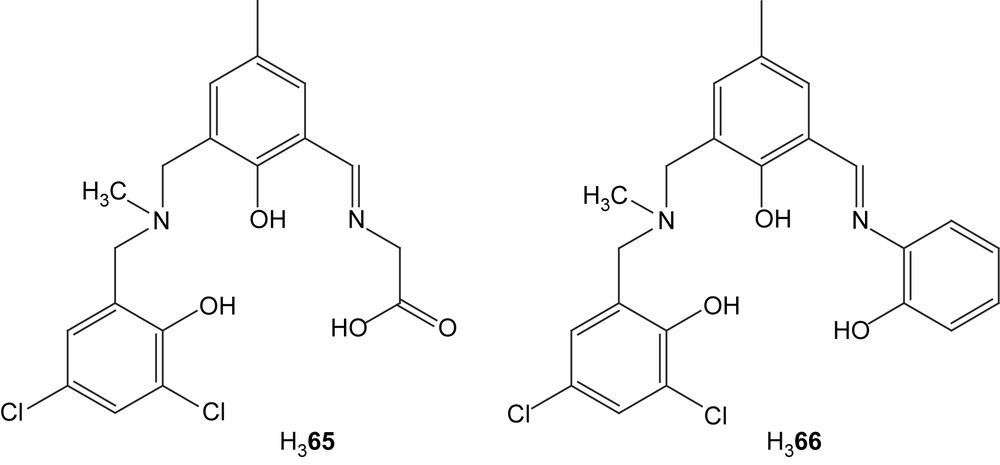
Symmetric and unsymmetric dinuclear copper(II) complexes were prepared from the new ligands H67 and H68 [112]. Three different exogenously coordinating anionic ligands were used (chloride, hydroxide and acetate) to yield six different complexes. The complexation of the ligand was confirmed by IR which showed that the imino groups were coordinating. Absorptions in the UV–vis spectra around 430 nm (ɛ = 1000–3000 M−1 cm−1) indicated a charge-transfer interaction either involving the phenolate or the benzimidazole functionalities.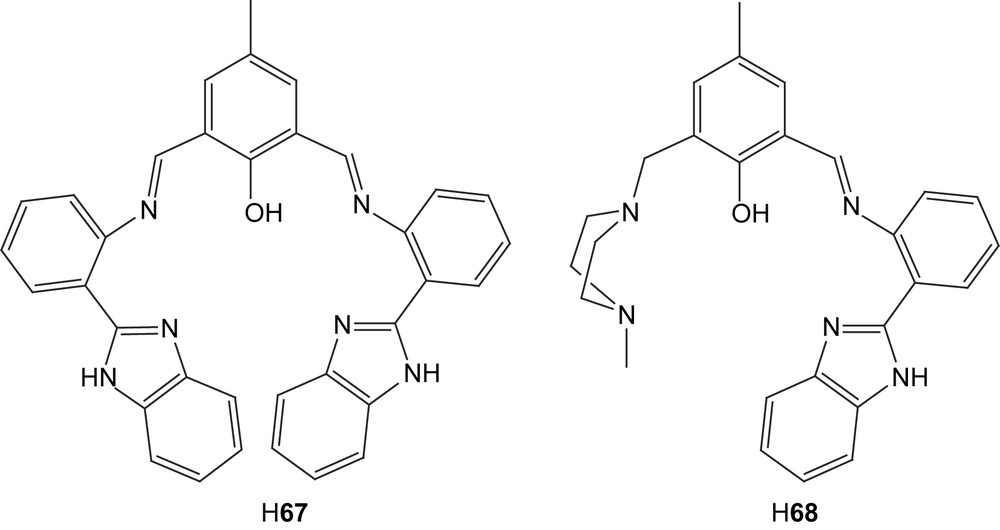
Cyclic voltammetry displayed two quasi-reversible reductions corresponding to two successive one-electron transfers also supported by coulometric measurements. Comparisons of the CVs for the complexes formed from the different ligands showed that the side arms containing the imine and benzimidazole caused a shift of the reduction potential to more negative values. This was explained by the fact that reduction of metals is easier with more flexible coordinating groups. In this case the piperazine moiety is less rigid than the imine–benzimidazole group due to the extended conjugation provided by the overlapping π-orbitals in the latter. For both ligands the reduction potentials are also dependent on the exogenous ligands; somewhat surprisingly, the more electron-donating acetate appears to make the first reduction easier than the hydroxide and chloride groups. This may be due to the higher flexibility of the acetate compared to the other exogenous ligands, if the acetate is bridging in a μ-1,3 mode.
Magnetic susceptibility measurements indicate an antiferromagnetic coupling, which is considerably stronger for the symmetric complex based on 67 than for the unsymmetric based on 68, which could be explained by the rigid planarity of the imine–benzimidazole groups which forces the orbitals of the metal ions to align themselves for a better exchange interaction over the phenolate. An exchange pathway over the extended conjugation in the symmetrical complexes is also possible. This difference in antiferromagnetic coupling between the ligands was also observable in EPR spectra of the complexes where the symmetrical complexes gave no EPR signal but the unsymmetrical ones gave broad peaks without any hyperfine signals.
The unsymmetric dinuclear active site in catechol oxidase was modeled with the three new structurally related ligands (H69–H71) that contain piperidine, morpholine and thiomorpholine moieties, respectively [113]. Except for these groups the ligands also contain a central phenolate, piperazine and one secondary amine as coordinating groups. When reacted with mixtures of copper(II) tetrafluoroborate and copper(II) acetate, all three ligands formed similar complexes [Cu(L)(OAc)2]+ (L = 69 (72), 70 (73), 71 (74)] with one bridging phenolate-based ligand and two μ-1,3 acetate bridges according to their crystal structures. These structures also reveal that the piperidine, morpholine and thiomorpholine rings are all in chair conformations with the nitrogen coordinating the copper ion in an equatorial position.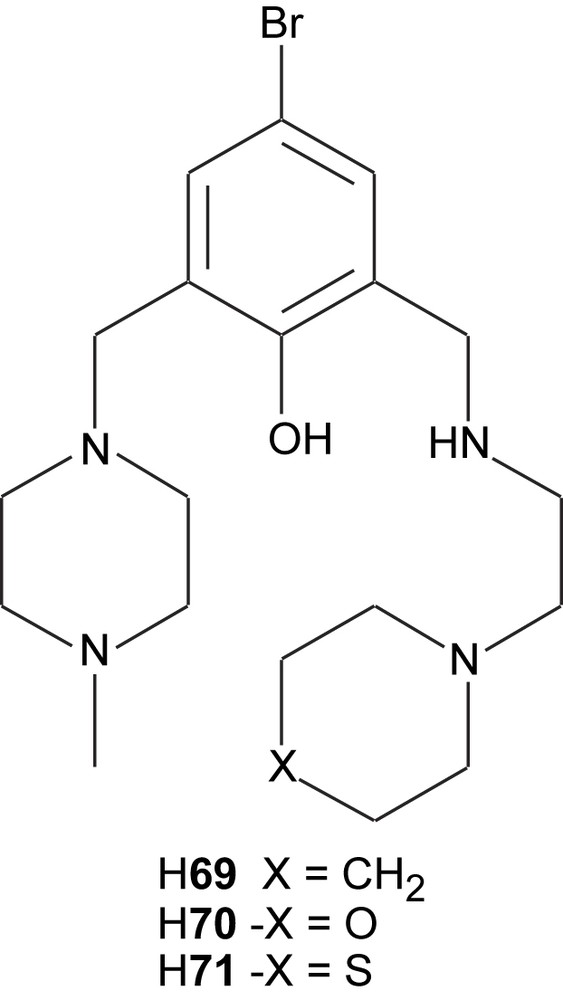
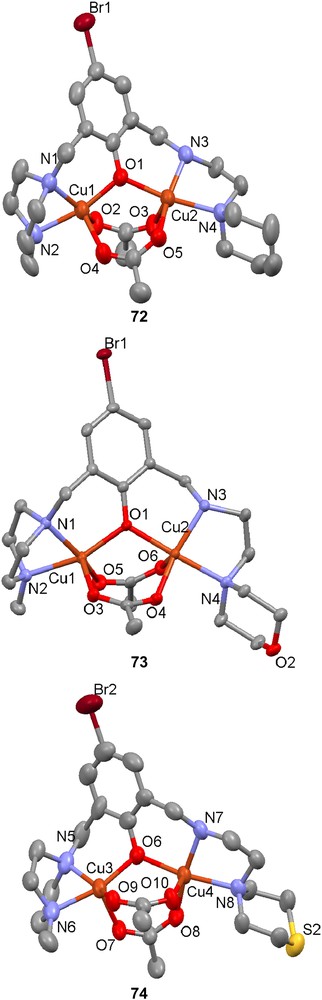
The electronic spectra of the complexes show similar behaviour, with one LMCT band below 400 nm and a d–d transition at 675–680 nm, as usually found for dinuclear copper complexes with phenolate and carboxylate bridges in square pyramidal coordination spheres. The only difference is that the thiomorpholine derivative 71 has a blueshifted LMCT band compared to the other complexes, which could be due to a displacement of one or both acetates by methanolates or hydroxides.
When 72–74 were reacted with 3,5-di-tert-butylcatechol, a significant oxidase activity was observed by UV–vis spectroscopy. All complexes showed saturation kinetics at high substrate concentrations but the thiomorpholino derivative (74) had the highest turnover number, followed by the morpholino (73) and then the piperidine (72) derivatives. This was explained by the higher propensity of the heteroatoms, especially sulfur, to coordinate and thereby weaken the acetate coordination, which results in a higher reactivity and turnover number. ESI-MS experiments showing an increased intensity for the peak corresponding to the monocarboxylate-bridged complex when the coordinating ability of the atom in position 4 of the piperidine rings increases (S > O > CH2) also conform to these results. The conclusion was further supported by DFT calculations that showed that the boat conformation necessary for the coordination of the heteroatom had 5.5 kcal mol−1 lower energy than the chair for the thiomorpholino complex and the sulfur atom formed a bond to the closest copper ion. This difference was reversed but small for the morpholino complex (1.4 kcal mol−1) and reversed and big for the piperidine complexes (10.4 kcal mol−1).
Ligand H75 was synthesized and reacted with Cu(NO3)2 to give a dinuclear copper complex of formula [Cu2(75)(H2O)1.5(NO3)2](NO3) (76) as a model for the active site of catechol oxidase [114]. X-ray crystallography revealed that both metal ions reside in distorted octahedral environments but are non-equivalent due to donor atom asymmetry (N3O3 vs. N2O4). They are only bridged by the phenolate oxygen, and there is thus a relatively large metal–metal separation (3.9003 Å) compared to complexes with two or more bridges. Both nitrate ions bind to the copper ion in the tridentate ligand pocket, while the water molecules coordinate to the other copper.

The UV–vis spectra both in the solid state and in acetonitrile solution show the same absorptions around 456 and 640 nm corresponding to LMCT between the phenolate and the copper ions and Cu(II) d–d transitions, respectively. As the spectra do not differ to any significant extent, this indicates that the complex persists in solution. An ESI-MS spectrum performed in acetonitrile indicates that the complex during the measurement loses the coordinated water molecules observed in the solid state. Cyclic voltammetry displays two irreversible reductions at −0.13 and −0.33 V, corresponding to Cu(II)2/Cu(II)Cu(I) and Cu(II)Cu(I)/Cu(I)2, respectively. At −0.7 V both copper ions are reduced to Cu(0), which is deposited on the electrode surface. Magnetic susceptibility measurements indicate a weak ferromagnetic coupling with an exchange integral of J = −4.6 cm−1 and a Curie–Weiss temperature of 0.87 K.
The unsymmetrical ligand H77 was synthesized and reacted in situ with copper(II) acetate monohydrate to yield a phenoxido and monohydroxido-bridged dinuclear complex [Cu2(77)(μ-OH)(OAc)]+ (78) with the copper ions coordinated with different geometries – trigonal bipyramidal or nearly so on the dipicolyl side of the ligand and almost square pyramidal on the imine side, including a monodentate acetate in the apical position [115]. The UV–vis spectrum of the complex in DMF shows the normally encountered d–d transitions around 653 nm. Cyclic voltammetry displayed two quasi-reversible couples at −0.49 and −0.98 V, where the latter one corresponds to reduction of the copper ion with trigonal bipyramidal coordination geometry. Magnetic susceptibility measurements showed typical antiferromagnetic coupling between two S = 1/2 ions with an exchange integral of J = −85 cm−1 (H = −2JS1·S2), suggesting a relatively ineffective superexchange pathway realized through an antibonding molecular orbital formed by the of the square pyramidal Cu(II), a filled p orbital of the hydroxy bridge and the orbital of the trigonal bipyramidal Cu(II) ion.

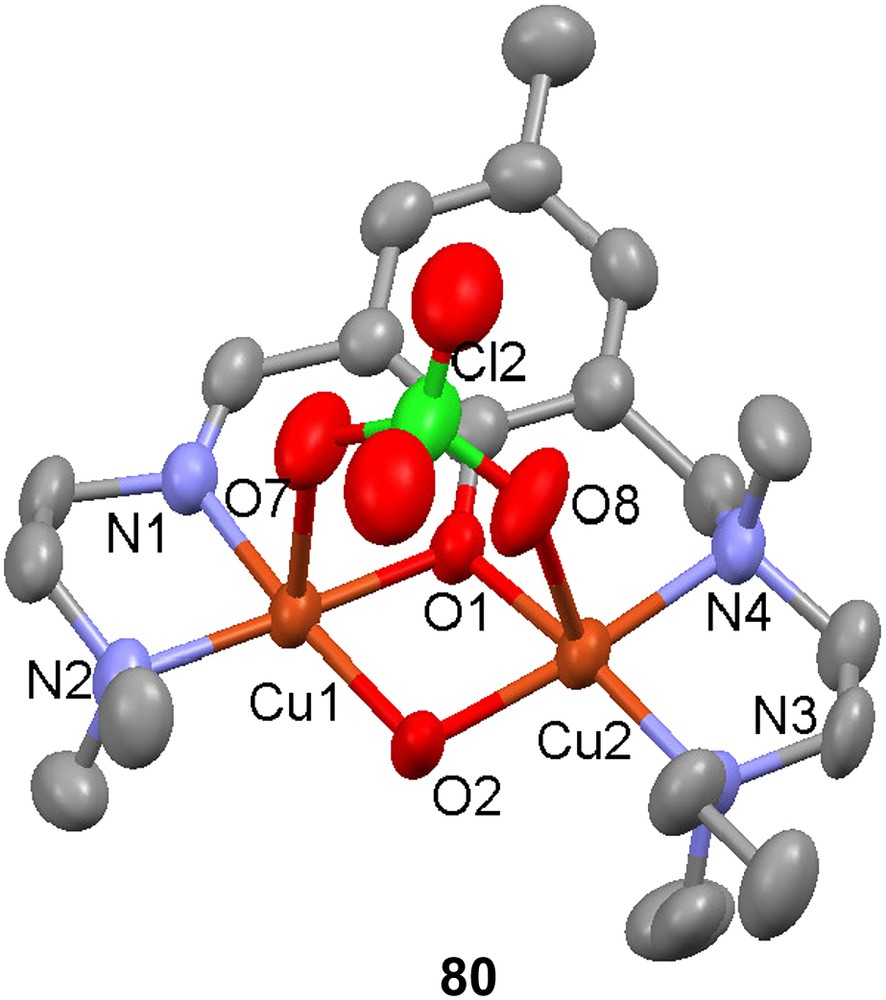
Reaction of H53 with CuBr2 in methanol gave [Cu2(53)Br3] (79) [99], the crystal structure of which reveals that each Cu(II) ion is in a square pyramidal environment with τ indices [116] of 0.34 and 0.054 for the coppers coordinated by the amine and the imine moieties, respectively. The corresponding Cu–N(amine) and Cu–N(imine) distances are significantly different, with the latter being 0.77 Å shorter than the former. This may be a consequence of the conformational flexibility and the hybridizations of the nitrogen donors, but may also reflect an inherent coordination preference of Cu(II) for an imine rather than an amine. The reaction of H54 with Cu(ClO4)2·6H2O resulted in the formation of the phenoxido/hydroxido-bridged dinuclear complex [Cu2(54)(OH)(ClO4)]+ (80) where each Cu ion again is in a square pyramidal coordination environment (τ values of 0.13 and 0.03); each copper is bonded to an apical oxygen atom that is a part of the coordinated perchlorate ion.
4 Summary and conclusions
The synthesis of asymmetric ligands for the preparation of model complexes for active sites in metalloenzymes has advanced significantly and has become much more common during the past decade. The asymmetry of the model complexes is often essential to impart specific coordination geometry and reactivity to the two metals in a dinuclear complex, and is thus necessary for adequate structural and functional modeling of metallobiosites. There is also very active development of mono- and polynuclear coordination complexes as catalysts or stoichiometric reagents for organic reactions [117–119] and the future is likely to bring many new and increasingly sophisticated ligands and metal complexes that may be used in bioinorganic model chemistry, bioinspired catalysis and asymmetric organic reactions.
Acknowledgements
The authors' research on asymmetric model complexes for biological metal sites has been sponsored by the Swedish Research Council (VR).