

1 Introduction
The Pd-catalysed asymmetric allylic substitution (AAS) is one of the most versatile routes for preparing optically active compounds through carbon–carbon or carbon–heteroatom bond formation [1–3]. The catalytic cycle for AAS is well established and involves olefin coordination to form a η2-olefin complex 2, subsequent intramolecular oxidative addition with the loss of the leaving group to produce η3-allyl complex 3, nucleophilic addition to allylic moiety to generate 4 and finally, the substituted olefin decoordination to give the initial catalytic species 1 (Scheme 1).

Accepted mechanism for Pd-catalysed allylic substitutions.
Except for the last step (olefin decoordination), the rest of the cycle reactions offers possibilities for enantiodiscrimination [2]. In particular, when the allylic substrate generates a meso π-allyl intermediate (3, Scheme 1), the enantioselectivity is controlled by the external nucleophilic attack on the more electrophilic terminal allylic carbon of 3. However, two diastereomeric complexes can be formed (W-type/M-type or endo/exo isomers, depending on the relative disposition of the allyl fragment with respect to the chiral ligand L,L′), and it can suffer an interchange through π–σ–π mechanism. Thus, the enantioselection is determined for both the regiochemistry of the nucleophilic addition and the diastereomeric composition of π-allyl intermediates. Two typical models of Pd-catalysed allylic substitutions are shown in Scheme 2, rac-3-acetoxy-1,3-diphenyl-1-propene (5) is widely used as substrate for alkylation reactions with dimethyl malonate (Scheme 2a) or for amination reactions with benzylamine (Scheme 2b).
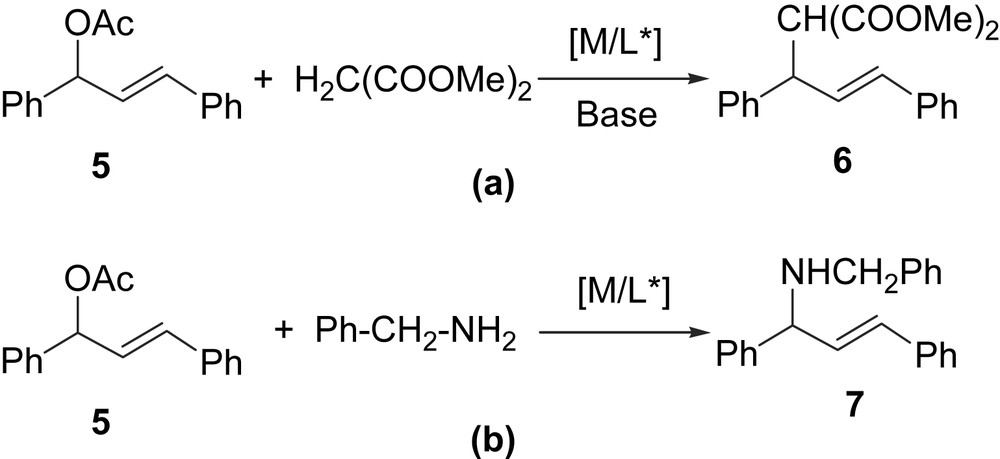
Model allylic substitution reactions of 5 with soft nucleophiles: (a) dimethyl malonate (alkylation) and (b) benzylamine (amination).
Since the first enantioselective catalytic process described by Trost in 1977, many catalytic systems have been tested, affording excellent enantiomeric excesses [3]. Whereas several chiral ligands bearing nitrogen and phosphorus donors (either homo or heterodonors) are widely used in AAS [3,4], other donor atoms, such as sulphur, have been much less studied [5]. In relation to thioether containing ligands, structural data of the allylic palladium intermediates, both in solution and solid state, have been reported for several types of ligands, mainly for heterodonors, but also for sulphur homodonors. Because a mixture of diastereomers may exist in solution due to the configuration inversion at sulphur atom [6], a decrease of stereoselection may occur if the relative rates of the intermediates are similar. In spite of this feature, excellent enantiomeric excesses have been achieved in asymmetric allylic substitutions as well as in other metal-catalysed asymmetric reactions [7].
Thus, this review will cover reports in the literature on the use of thioether ligands in Pd-catalysed asymmetric allylic substitution reactions in the most emerging period for this research area (1996–June 2006). In Section 2, we will cover the results using (S,S)-homo and (S,S)-heterodonor ligands. In Section 3, we will present the results using heterodonor (S,X)-ligands, emphasizing the results obtained with (S,N)- and (S,P)-donor ligands. We also discuss any reported mechanistic aspects according to the allylic substitution results.
2 (S,S)-donor ligands
2.1 (S,S)-homodonor
The first successful result using chiral dithioethers in palladium-catalysed allylic alkylation of 5 (Scheme 2a) was obtained with DEGUS-type ligands (up to 81% e.e.) (Fig. 1) [8]. Other chiral dithioether ligands, such as 9–11, have been developed and applied in the same reaction, yielding modest-to-good asymmetric inductions [9]. Recently, the efficiency of the cyclic DIOS-S type ligands in AAS was improved by introduction of two new stereogenic centres in the propyl chain (DIOS–DMPS, 12) [10].
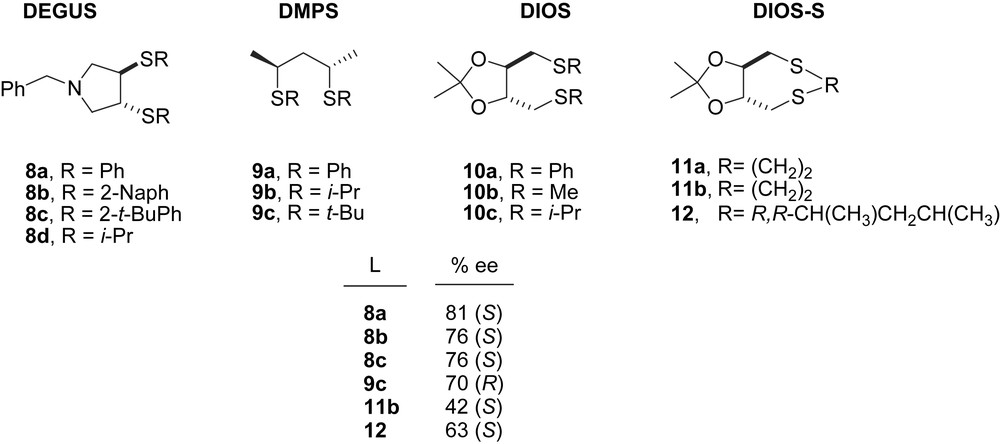
Chiral dithioether ligands 8–12. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
The structural study of the reaction intermediates of dithioethers 8–12 showed that the coordination modes to palladium occur through both sulphur donor atoms. NMR spectra of Pd allylic complexes [Pd(η3-1,3-PhC3H3Ph)(L)]PF6 (L = 8, 9, 11, 12) show different diastereomeric species in solution. In several cases, the diastereomeric excesses are in good agreement with the enantiomeric excesses obtained in the catalytic reactions [8–10].
The ligands 8, 9 and 11 were also evaluated in palladium-catalysed allylic alkylation of rac-3-acetoxy-1-cyclohexene, obtaining 20–34% of e.e. The Pd/9 systems were more active than Pd/8 systems and the butterfly-type palladium complexes (with cyclic ligands 11) gave the best enantioselectivities. The cyclohexenylallyl palladium complex [Pd(η3-cyclohexenyl)(11b)]PF6 has also been prepared and studied in solution and in solid state [9].
The allylic alkylation of (E)-3-acetoxy-1-phenyl-1-propene (13) and (E/Z)-1-acetoxy-2-butene (15) catalysed by palladium complexes was studied using 8, 9 and 11 chiral dithioether ligands (Fig. 2). Pd/11 systems were very active and the most selective to linear isomers in the reaction with substrate 13, whereas Pd/9 systems gave the best enantioselectivities for the branched product. In the case of the later systems, a strong dependence on activity and selectivity of the nature of the S-substituent was observed. Using substrate 15, excellent enantioselectivity was achieved with Pd/DMPS-tBu (9c) [9].
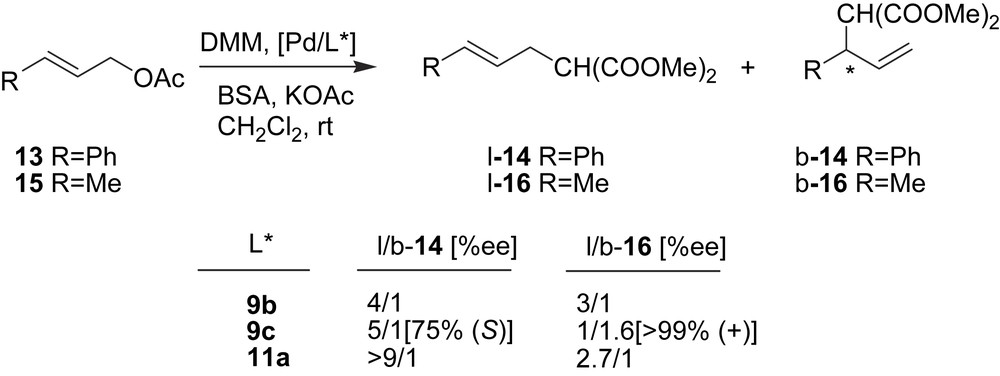
Selected data of allylic alkylation of unsymmetrical substrates 13 and 15 using Pd/dithioether catalytic systems.
The complexes [Pd(η3-1-PhC3H4)(9b)]PF6, [Pd(η3-1-MeC3H4)(9a)]PF6, and [Pd(η3-1-MeC3H4)(9c)]PF6 were prepared and both the solution and solid-state species were analysed in order to understand the catalytic performance. For the latter complex, the most important catalytic fact was the preference to yield the branched product, and also there was good agreement between the diastereomeric ratio of the two Pd isomers (1.3/1) and the regioselectivity of the process (b/l-16 = 1.6/1).
A series of chiral dithioethers containing rigid cyclopentane and heterocyclic analogues (17,18) were prepared and tested in the allylic alkylation of 5 (Fig. 3). The most efficient systems were the ligands containing pyrrolidine moieties (17), achieving enantiomeric excesses up to 90%. The authors, however, suggest that alkyl-3,4-bis-(arylthio)pyrrolidine derivatives behave as (N,S)-donating ligands more than (S,S)-donating ligands. Monothioethers 19–21 bearing benzylpyrrolidine groups were also considered as (N,S)-ligands because of the high e.e.’s obtained (79–90%). Only ligands 18 were considered (S,S)-homodonor ligands, but low-to-modest enantioselectivities were attained in the alkylation reaction [11].
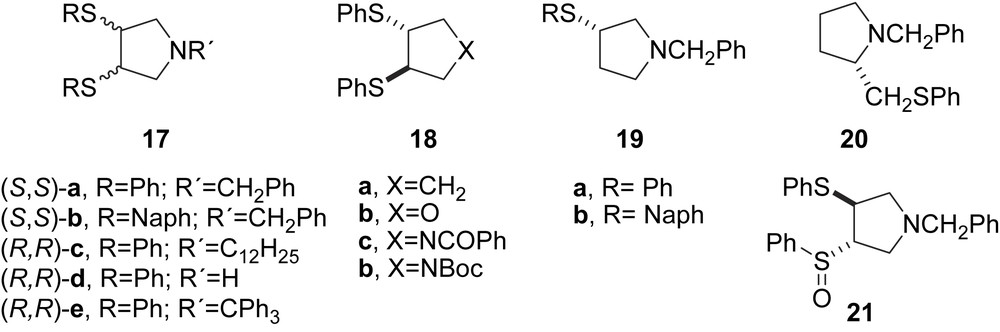
Chiral heterocyclopentanyl thioethers.
Chiral sulfideoxathianes 22–23 have proved to be effective asymmetric inductors for palladium-catalysed allylic alkylation of substrate 5 with dimethyl malonate (Fig. 4).
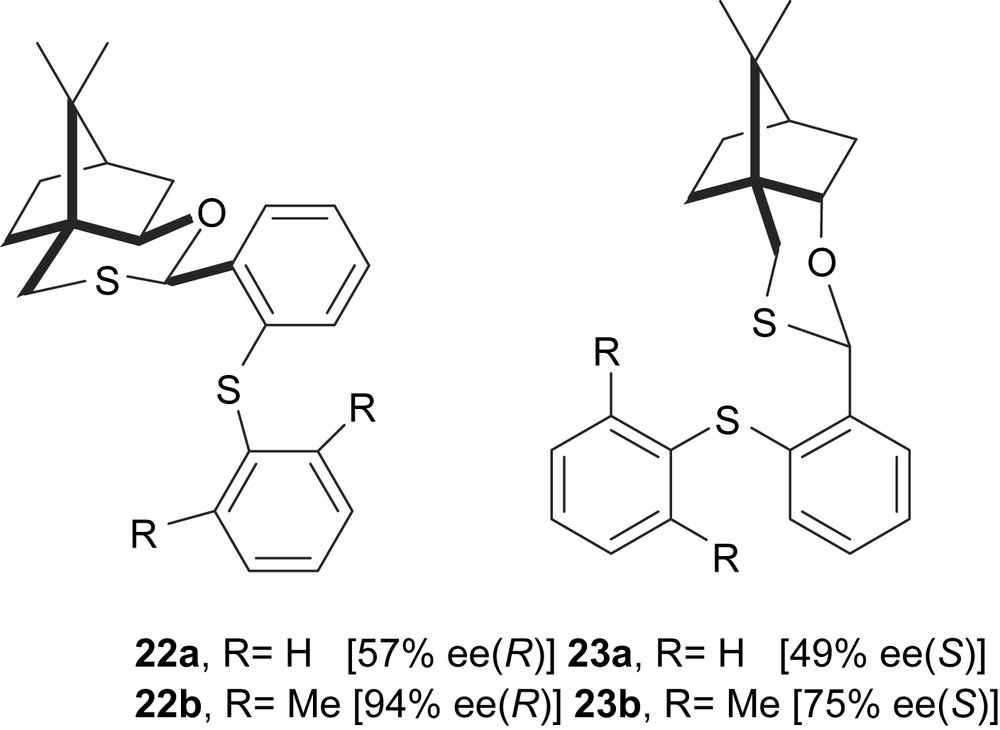
Chiral sulfideoxathianes 22–23. In brackets, the enantiomeric excesses of alkylated product 6 are shown.
Enantomeric excesses up to 99% were achieved with ligand 22b, using the same substrate and dimethyl and diethyl methylmalonates as nucleophiles. MO calculations give a rational explanation for the different catalytic behaviour between Pd/22a and Pd/22b. The palladium allyl optimised structure indicates that the steric hindrance of the methyl group on the phenyl ring in [Pd(η3-1,3-PhC3H3Ph)(22b)] favours one of the four possible isomers (Scheme 3). This result was related to the high enantioselectivity obtained using 22b [12].

Possible isomers for [Pd(η3-1,3-PhC3H3Ph)(22b)].
Sugar-based modular ligands (Fig. 5) provided low-to-good enantioselectivities (4–88% e.e.) in allylic alkylation of 5, depending on the sugar, linker and protecting group used [13]. β-d-glucose, β-d-galactose, α-d-mannose or a glucosamine derivatives were used as sugar residues, 1,2-ethanodithiol, 1,3 propanodithiol, 1,2 benzyldithiol or 1,2 benzenedithiol as linkers and acetyl, benzyl, benzoyl and pivaloyl as protecting groups R. The (S)-alkylated product 6 (Scheme 2a) was obtained with high e.e.’s (86% at 293 K, 90% at 273 K) when the bis-thioglycoside contained β-d-glucose, 1,2-ethanodithiol and pivaloyl groups. In general, the best results were achieved using 1,2-ethanodithiol as a linker and β-d-glucose or β-d-galactose as the sugar base. The observed enantiomeric discrimination in the catalytic reaction seems to be related to the steric hindrance of protecting groups in sugars and not to the high stereocontrol of sulphur inversion by exo-anomeric effect [14].

Bis-thioglycoside ligands 24. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
2.2 (S,S)-heterodonor
2.2.1 Thioether–thiophosphines
C1-symmetric binaphthyl (S,S)-heterodonor ligands 25 were synthesised and tested in allylic alkylation of 5, giving moderate-to-good enantiomeric excesses of alkylated product 6 (31% 25a; 74% 25b) at 273 K. The coordination to Pd(II) was investigated by NMR and the preliminary results suggest that both sulphur atoms are coordinated to the metal centre [15].
New chiral ferrocenyl thioether–thiophosphine ligands with a stereogenic plane were prepared and applied in allylic asymmetric reactions. These (S,S)-ligands proved to be efficient asymmetric inductors, giving good yields and excellent enantioselectivities (88–99% of e.e.). The level of enantioselectivity achieved with 26a–f is amongst the best in allylic substitution reactions using (S,S)-heterodonor ligands [16] (Fig. 6).

Chiral thioether–thiophosphines.
3 (S,X)-donor bidentate ligands
Several types of heterodonor chiral ligands containing a thioether group have been developed for application in asymmetric allylic substitutions. Mixed (S,N)- and to a lesser extent (S,P)-donor ligands have played a dominant role.
3.1 (S,N)-donor ligands
3.1.1 Thioether–oxazolines
Some of the most successful mixed (S,N)-donor ligands have been the thioglucose-derived ligands containing a chiral oxazoline moiety (27a–d) (Fig. 7). These ligands afforded e.e.’s between 90 and 97% in the palladium allylic alkylation of dimethyl malonate to 5. The effects of the thiosugar substituents on enantioselectivity were mild. However, if a thiocyclohexyl group replaces the thiosugar (27e), the enantiomeric excess falls to 75% [17]. Ligand 27f, related to 27e, but generating a smaller chelate complex after coordination to Pd, produced higher enantioselectivity (81%) [18].

Thioether–oxazoline ligands 27. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
Uemura and co-workers also reported the synthesis of ligands 28 and 29 (Fig. 8). Their use as chiral auxiliaries in the Pd-catalysed asymmetric allylic alkylation reactions provided e.e.’s up to 90%. The best enantiomeric excess was obtained with ligand 28 for substrate 3-acetoxy-1,3-diphenyl-1-propene. These ligands were also applied in allylic alkylation of 3-acetoxy-1,3-dimethyl-1-propene and 3-acetoxy-1-cyclohexene with low enantioselectivities (e.e.’s up to 49%) [18].
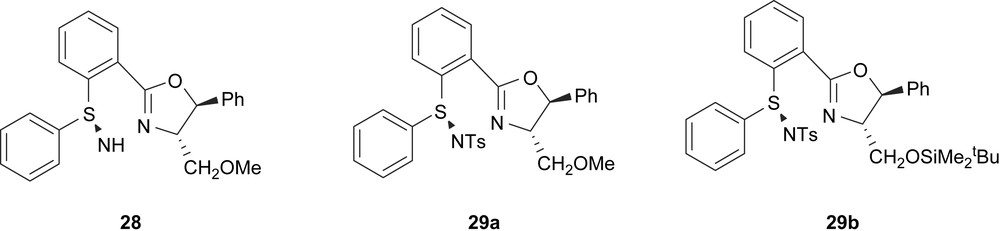
Ligands developed by Uemura and co-workers.
Several groups have developed chiral ferrocenyl ligands for application in asymmetric catalysis. In particular, three different kinds of thioether derivatives of ferrocenyloxazolines have been tested in asymmetric allylic alkylations.
The first kind of ferrocenyloxazoline ligands contains the thioether group tethered to the oxazoline moiety (ligands 30, Fig. 9). These ligands were applied in the model Pd-catalysed asymmetric allylic alkylation of 5 and the best asymmetric induction was obtained using 30b [19]. This is probably due to the increase in tether length between the donor atoms, which coordinate to palladium and bring the asymmetric environment closer to the allylic ligand in the catalytic species.
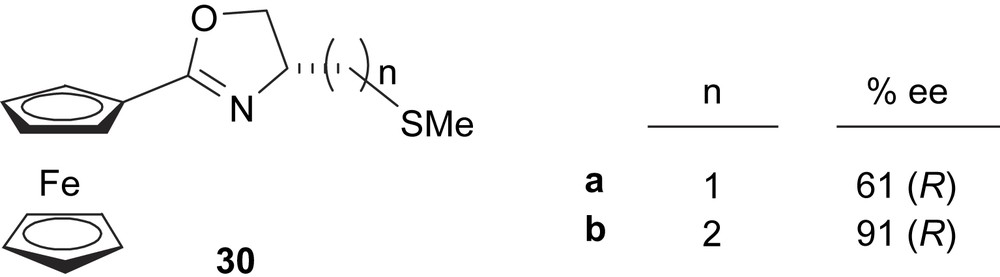
Thioether–ferrocenyloxazoline ligands 30. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
The second kind of ferrocenyloxazolines applied to this process contains the thioether group attached to the cyclopentadienyl rings producing 1,2- or 1,1′-disubstituted ferrocene derivatives (ligands 31–33, Fig. 10). These ligands were applied in the Pd-catalysed asymmetric allylic alkylation of 5 (Scheme 2a). The results indicated that the chiral stereocentre in the oxazoline moiety is necessary to obtain high enantioselectivities. Therefore, ligands 31a–e, 32 and 33 containing one chiral stereocentre on the oxazoline ring produced better enantioselectivities (e.e.’s up to 98%) than ligands 31f–h bearing achiral oxazoline substituents (e.e.’s up to 72%) [20].

Thioether–ferrocenyloxazoline ligands 31–33.
The NMR study of the 1,3-diphenylallyl palladium complexes 34 and 35, containing the thioether–ferrocenyloxazoline ligands 31b and 33b, respectively, shows a mixture of two isomers. These studies reveal that the major structure is the M-type (Scheme 4), which is the same conformation as the X-ray solid-state structure for the palladium complex 34. The structural data are in agreement with the absolute configuration observed for the substitution product. Comparing diastereomeric excesses of the Pd intermediates and enantiomeric excesses of 6, it has been observed that e.e.’s are higher than d.e.’s, thus M-34 and W-35 must react faster than W-34 and M-35, respectively.
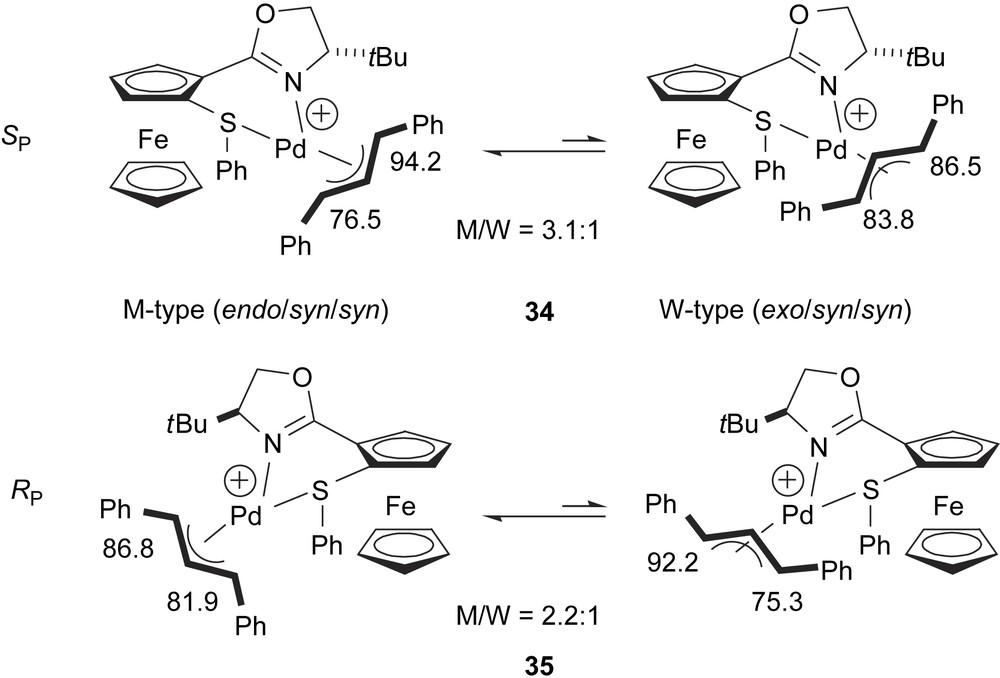
1,3-Diphenylallyl palladium isomers for cations 34 and 35. Figures indicate 13C chemical shifts (ppm) of the terminal allyl carbon atoms.
The third kind of ferrocenyloxazoline ligands developed for the Pd-catalysed asymmetric allylic substitution reactions are compounds 36 (Fig. 11). These thioether–ferrocenyloxazolines, without a stereogenic plane, afforded low-to-moderate enantioselectivities and poor yields [21]. These catalytic results indicate that the matching effect between stereocentre and stereogenic plane in ferrocene-based S,N-donor ligands may provide high levels of asymmetric induction in palladium-catalysed allylic substitutions.
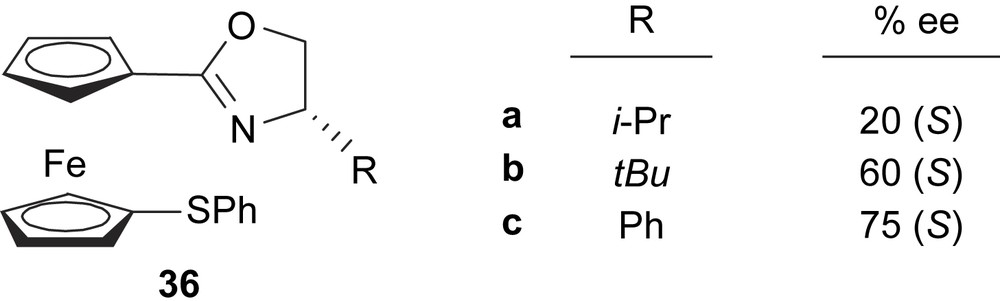
Thioether–ferrocenyloxazoline ligands 36. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
Another group of ligands containing a stereogenic plane are ligands 37–39 (Fig. 12). These ligands are based on the [2,2]paracyclophane backbone containing both a stereogenic centre and plane. The best enantioselectivity was obtained with ligand 39 that contains the thioether groups attached to the alkyl chain backbone. It is interesting to note that the planar chirality controls the sense of enantioselectivity. Therefore, 37 with Sp planar chirality afforded the alkylated product in (R)-configuration, whereas 38 with Rp planar chirality provided the (S)-enantiomer [22].

[2,2]Paracyclophane-based thioether–oxazoline ligands 37–39. In brackets, the enantiomeric excesses of alkylated product 6 are shown.
Chiral oxazoline thioether ligands with an axis-fixed (40 and 41) and -unfixed (42) biphenyl backbone have been prepared and applied in alkylation of 5 catalysed by palladium complexes (Fig. 13). In general, the activity of the catalytic systems with 40 and 41 was low; however, with (R)-axial ligands, the activity improved. The best enantioselectivity (e.e.’s up to 81%) was obtained using ligand 42a, which has a free rotation biphenyl axis [23].

Biphenyl-based thioether–oxazoline ligands 40–42.
Dithioether–bis(oxazoline) ligand (43) with an unfixed biphenyl backbone has also been tested in the Pd-catalysed allylic alkylation reaction (Fig. 14). In contrast to other analogous biphenyl derivatives, such as type-42 ligands, the selectivity was lower (45% e.e. vs. 81% e.e.) [4d].
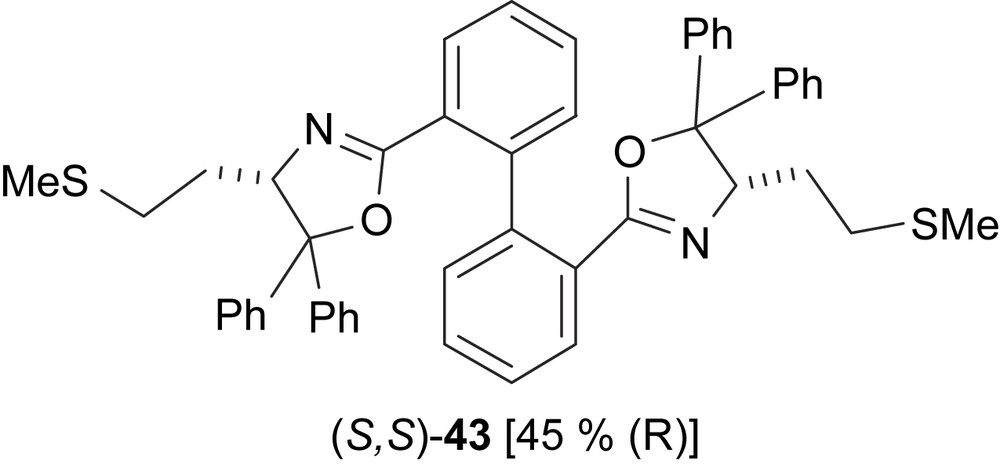
Dithioether–bis(oxazoline) ligand 43. In brackets, the enantiomeric excesses of alkylated product 6 are shown.
Gladiali and co-workers developed the thioether–oxazoline ligands 44 with chiral binaphthyl backbones as chiral auxiliaries for Pd-catalysed asymmetric allylic alkylation of 1,3-diphenylallyl esters (Fig. 15). In this reaction, the methyl-substituted ligand 44a led to complete conversion in a few hours with e.e. up to 66%, whereas the i-propyl analogue 44b produced a catalyst of poor activity (50% conversion within 22 h), which gave rise to a racemic product [24].
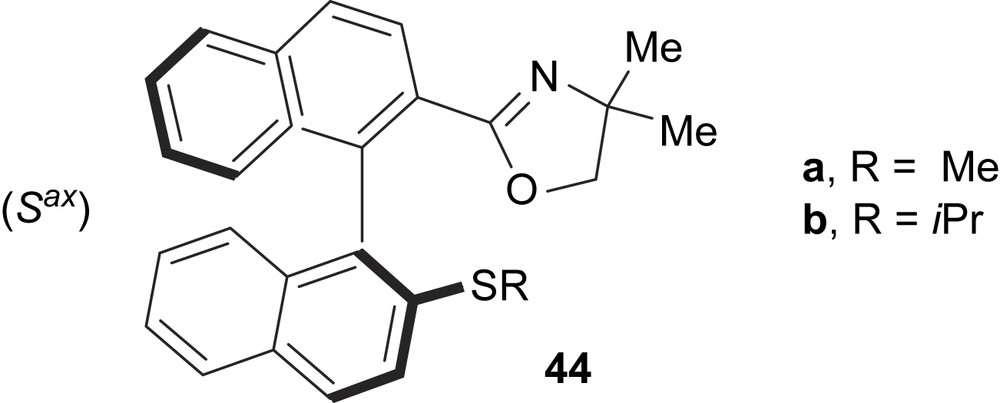
Thioether–oxazoline ligands 44a and 44b.
Recently, a new series of oxazoline-1,3-dithianes 45 have been developed and applied in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5 (Fig. 16). Enantiomeric excess depends strongly on the steric properties of the substituent in the oxazoline moiety, while the substituents on the dithianyl ring have no influence on the reaction outcome. Therefore, the best enantioselectivities were obtained with ligand 45g (e.e.’s up to 90%) [25].
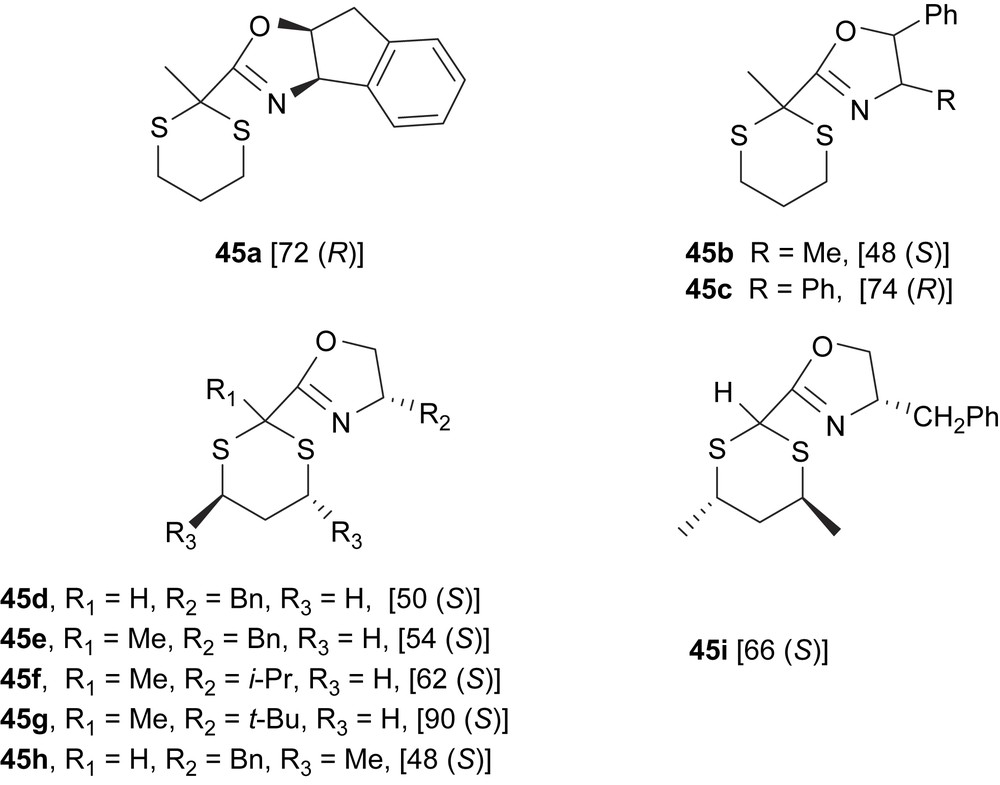
Oxazoline-1,3-dithiane ligands 45. In brackets, the enantiomeric excesses of alkylated product 6 are shown.
3.1.2 Thioether–pyridines
Chelucci and co-workers developed a series of epimeric phenylthiotetrahydroquinolines 46a and 46b as chiral auxiliaries for Pd-catalysed asymmetric allylic alkylation of 5 with e.e.’s up to 83% [26] (Fig. 17).
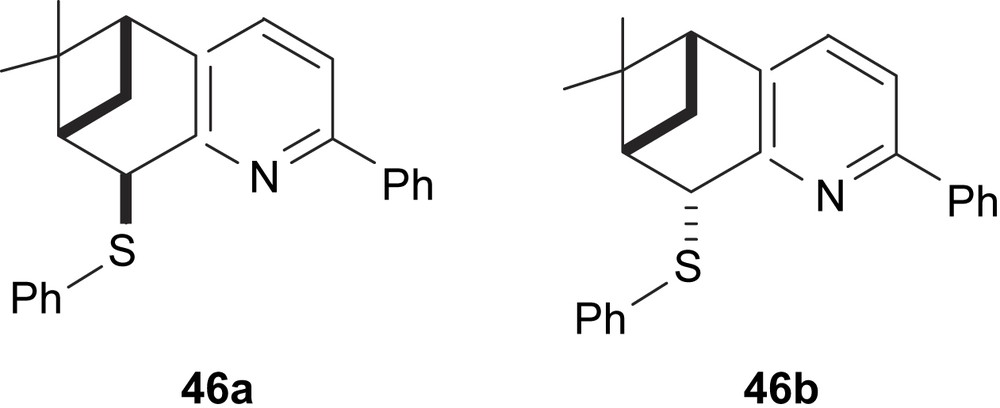
Thioether–pyridine ligands 46.
Chiral thioalkyl-pyridine ligands containing a (R)-(−)-fenchone-derived moiety (47) have proven to be effective asymmetric inductors in the palladium-catalysed allylic alkylation (Fig. 18). The results indicated an effect of the thioether substituent on enantioselectivity. Therefore, the best enantioselectivities were obtained with ligands that contain an ethyl or benzyl thioether substituent. Enantioselectivities were further increased using the (−)- or (+)-benzyl derivatives (up to 98%) by lowering the reaction temperature to 0 °C [27].
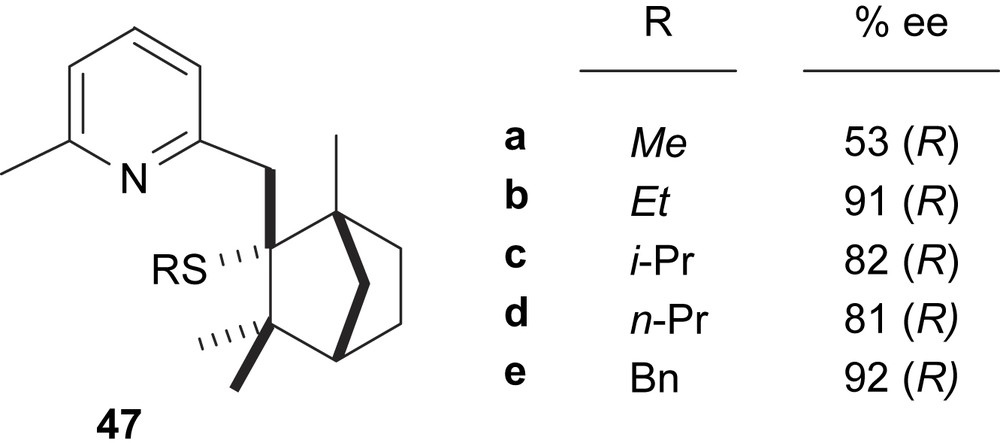
Thioether–pyridine ligands 47. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
Palladium catalytic systems with thiophenyl-pyridine ligands 48–50 bearing a (+)-camphor-derived moiety were applied in the asymmetric allylic alkylation of 5 with dimethyl malonate (Fig. 19). The best enantioselectivity (e.e.’s up to 77%) was obtained with ligand 48 with a phenyl group at the 6-position of the pyridine ring [28].
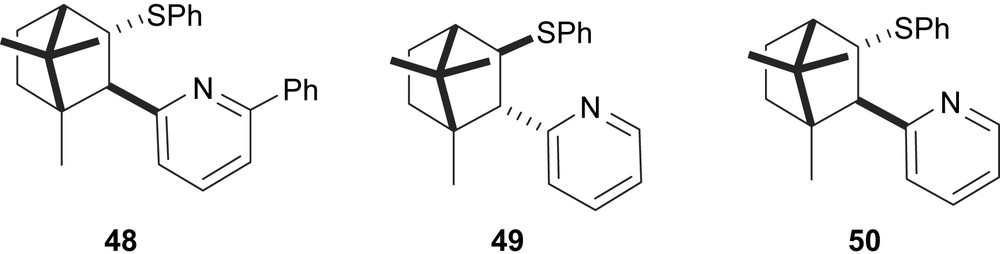
Thioether–pyridine ligands 48–50.
The chiral ligand 1-(2-thiomethyl-1-naphthyl)isoquinoline 51 (Fig. 20) has been synthesised and tested in allylic alkylation of substrate 5 with poor success (total conversion in two weeks with 2% e.e.) [29].

Thioether–pyridine ligand 51.
3.1.3 Thioether–imines
A series of (S)-valinol (52), (R)-phenylglycinol (53) and (S)-tert-leucinol (54) derived imine ligands containing thioether group have been developed for the palladium-catalysed asymmetric allylic alkylation (Fig. 21). Excellent enantioselectivities were obtained (e.e.’s up to 96%) and the results indicated that enantioselectivity is not sensitive to electronic effects caused by substituents on the imine group, whereas the steric bulk in ortho-position of the aryl group attached to the imine has a subtle effect. The bulkiness of the aryl substituent at sulphur (ligand 52k) provided the highest enantioselectivity (96% e.e.). However, the replacement by a methyl group (ligand 52j) only reduced the enantiomeric excess to 90%. The increase in steric hindrance of the backbone (ligand 54) caused a decrease in both e.e.’s and yields [30].
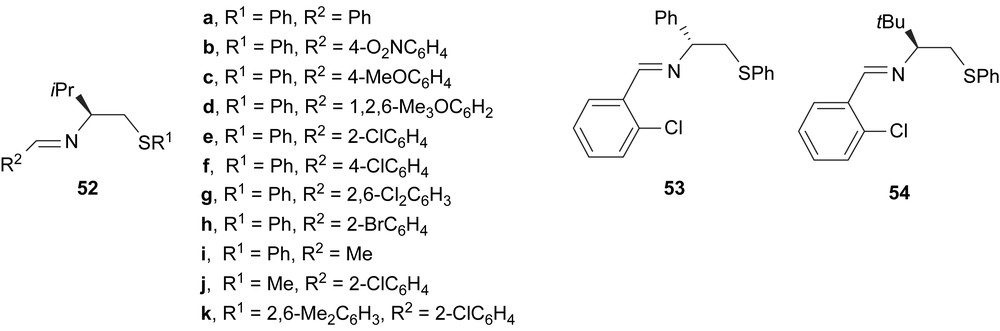
Thioether–imine ligands 52–54.
In contrast to other chiral (S,N)-donor ligands, the authors have proved that for these systems, the nucleophilic attack on the allyl group is not trans to the sulphur atom, but trans to the imine donor, and so the enantioselectivity is controlled by the steric environment of the chelate ligand more than by electronic effects. The X-ray structure of complex [Pd(η3-1,3-PhC3H3Ph)(52e)]ClO4 shows that the allyl group adopted a W conformation and the sulphur phenyl substituent is trans to the isopropyl group, avoiding an unfavourable interaction. In addition, one structure is present in solution, showing a syn/syn allyl arrangement with a W conformation.
In 2003, Dai and co-workers developed the ferrocenyl–imino ligands 55 and 56 containing a stereogenic plane (Fig. 22) to be used in allylic alkylation of dimethyl malonate to 5 and 3-acetoxycyclohexene, affording 86 and 82% of enantioselectivity, respectively [31].
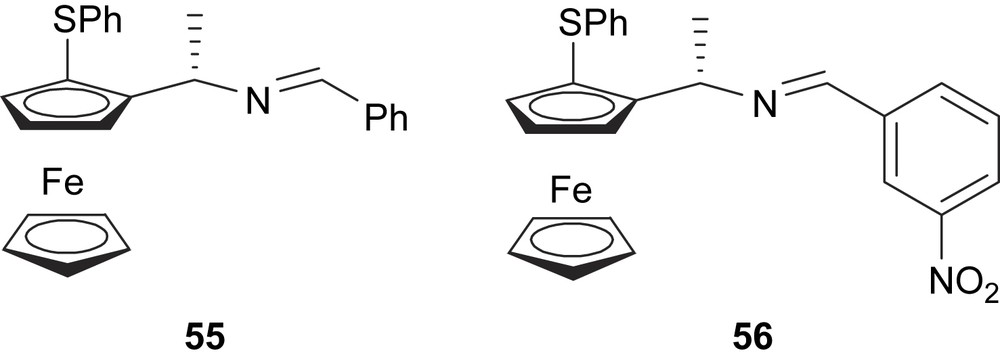
Ferrocenyl thioether-imine ligands 55 and 56.
Recently, 2-iminoalkyl ferrocenyl derivatives were prepared and tested in asymmetric allylic alkylation, obtaining low-to-modest e.e.’s (15–72%) [32]. Better results (61–95% of e.e.) were obtained by Tokuda, employing sterically congested 2-iminothioethers to alkylate 5 with dimethyl malonate. The authors suggest that the M-type π-allyl palladium intermediate is favoured, since it minimizes the steric hindrance between substrate and ligand aromatic moieties; so the nucleophile preferentially attacks trans to sulphur, which explains the high enantiomeric excesses and the configuration of the alkylated product 6 [33].
3.1.4 Thioether–amines
A series of norephedrine-based thioether–amine ligands 57 (Fig. 23) were applied in the model allylic alkylation (Scheme 2a) with low-to-good enantioselectivities (9–72%). The best enantioselectivity was obtained with ligand 57f with tert-butyl group in the thioether moiety [34].
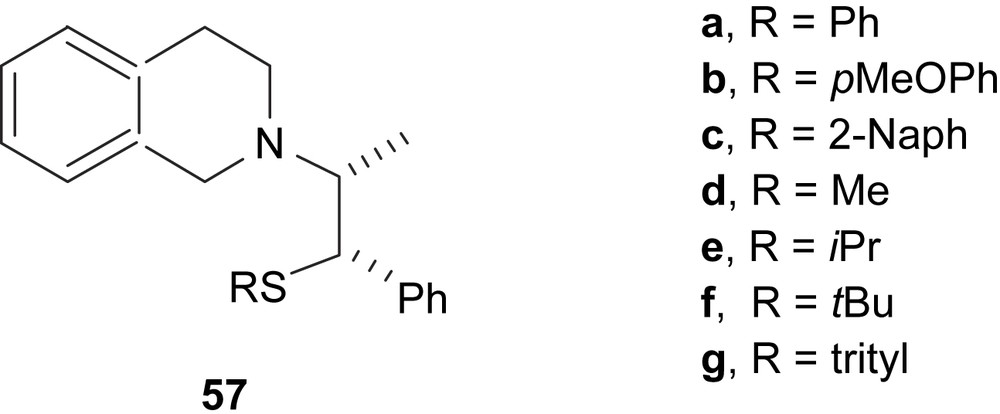
Thioether–amine ligands 57 derived from ephedrine.
In 2004, Braga and co-workers developed a series of modular thioether–oxazolidine ligands 58 and 59 for allylic alkylation of 5 (Fig. 24). By carefully selecting the ligand components, they obtained an enantioselectivity of 81% (S) with ligand 58d. This ligand is an analogue of ligand 58a, but has more steric crowding in the oxazolidine ring. Ligand 59, which generates a larger bite angle after coordination to palladium, gave the lowest enantioselectivity (e.e.’s up to 6% (S)) [35].

Thioether–oxazolidine ligands 58 and 59. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
The same authors also reported the synthesis of thioether–aziridine ligands for application in the Pd-catalysed asymmetric allylic alkylation (Fig. 25). The catalytic results indicated that the influence of the benzyl group attached to nitrogen is important in obtaining high asymmetric induction and the substituent at the sulphur atom has a small effect in the conversion. Therefore, the best enantioselectivity (up to 99% (S)) was obtained with ligand 61c, which contains an electron-withdrawing benzyl group attached to nitrogen [36].

Thioether–aziridine ligands 60 and 61.
Recently, Anderson and co-workers reported the synthesis of a series of ferrocenyl thioether–amine ligands 62 for the allylic alkylation of rac-3-acetoxy-1,3-diphenyl-1-propene (Fig. 26) with low-to-moderate enantioselectivities. The best e.e.’s (up to 59%) were attained with methyl substituents in the thioether and amine groups (ligand 62c) [37].
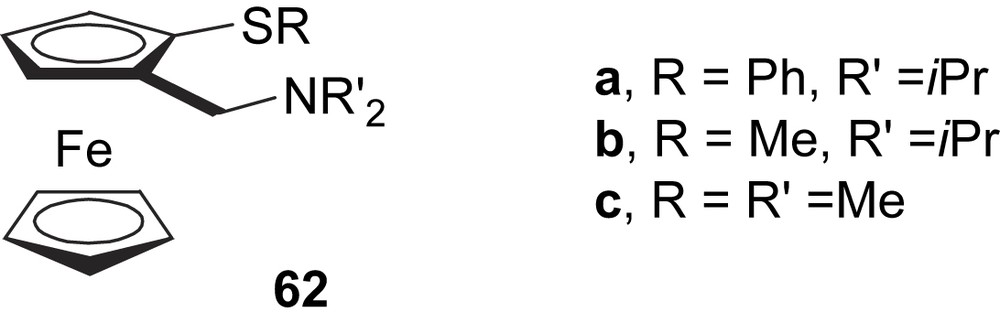
Ferrocenyl thioether–amine ligands 62.
3.1.5 Thioether–imidazolines
Two thioether–imidazoline ligands, 63 and 64, have been developed and applied in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 3-pivaloxy-1,3-diphenylpropene (Fig. 27). The best enantioselectivity was obtained with ligand 63 (up to 96%) [38].
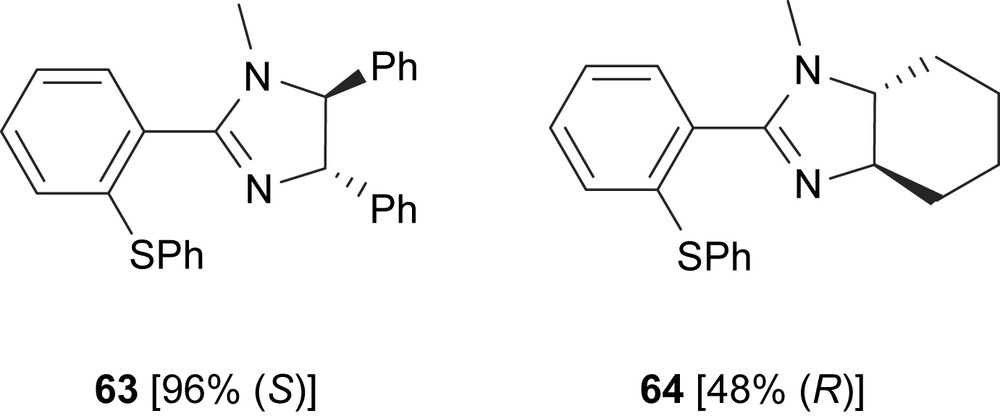
Thioether–imidazoline ligands 63 and 64. In brackets, the enantiomeric excesses of alkylated product 6 are shown.
3.2 S,P-donor ligands
3.2.1 Thioether–phosphines
The catalytic results using ferrocenylphosphine–thioether ligands 65–69 (Fig. 28) indicated that enantioselectivity is better when the phosphine group is attached to the Cp ring (ligands 65–67) rather than when it is attached to the thioether unit (ligands 68 and 69). Furthermore, by comparing ligands 65–67, it can be seen that enantioselectivities are not affected by the presence of an additional stereogenic unit or by the length of the thioether chain. The structural studies of a 1,3-diphenylallyl palladium complex containing ligand 65a, [Pd(η3-1,3-PhC3H3Ph)(65a)]PF6, indicate that the small substituents on thioether groups favour the nucleophilic attack in the cis position to the S-donor moiety [31a,39].

Ferrocenyl thioether–phosphine ligands 65–69. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
Carretero and co-workers developed a new series of successful ferrocenyl thioether–phosphine ligands 70–71 (Fig. 29) for Pd-catalysed asymmetric allylic substitution of 5 (e.e.’s up to 97%). Enantiomeric excess depends strongly on the steric properties of the substituent in the thioether moiety and the electronic properties of the phosphine group. Therefore, for high enantioselectivities, a bulky group on the thioether moiety and electron-withdrawing substituents on the phosphorus are needed [40].
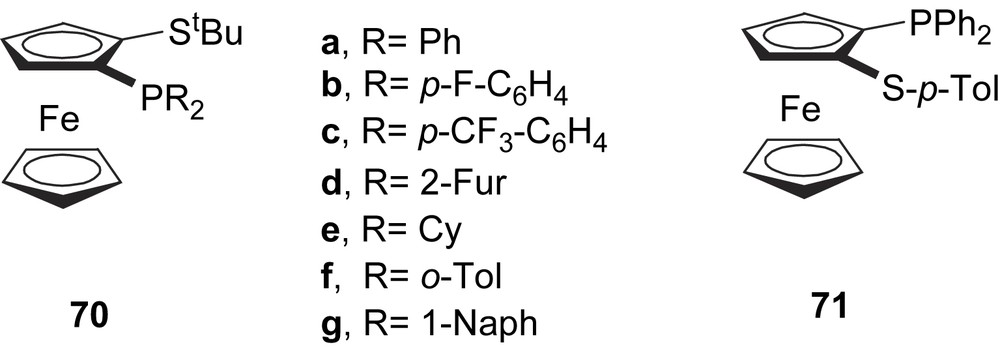
Ferrocenyl thioether–phosphine ligands 70–71.
The ferrocenylphosphine–thiosugar ligand 72 (Fig. 30), with several chiral sources, afforded an e.e. of 88% in the palladium allylic alkylation. However, when the thiosugar moiety is the only source of chirality on (S,P)-ligand (73a,b, Fig. 30), only asymmetric inductions of 64% (R) and 53% (R), respectively, were reached. Good activities but low enantiomeric excesses were obtained with a similar thioether–phosphine ligand using a chiral norborneol fragment (74). The authors attributed their performance to the effects of chain size between the sulphur donor and the kind of stereogenic unit [41].
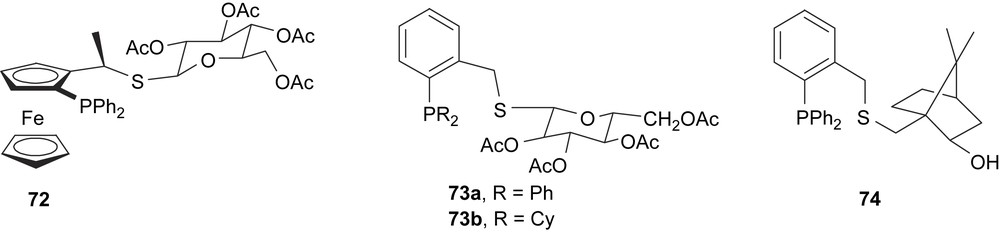
Chiral thioether–phosphine ligands 72–74.
The 1,3-diphenylallyl palladium complexes [Pd(η3-1,3-PhC3H3Ph)(74)]BF4, [Pd(η3-1,3-PhC3H3Ph)(73a)]BF4 and [Pd(η3-1,3-PhC3H3Ph)(73b)]BF4 were obtained as a mixture of isomers. Dynamic studies indicate that the η3–η1–η3 isomerisation occurs by dissociation of the carbon trans to sulphur and cis to phosphorus, in contrast to the non-substituted allyl complex [Pd(η3-1,3-C3H5)(74)]BF4, due to the steric hindrance between the PPh2 group and the phenyl allyl substituent. From these results, the authors suggest that this isomerisation process is sterically, and not electronically, controlled. For ligand 72, the study of corresponding 1,3-diphenylallyl palladium complex indicates that the nucleophilic attack takes place trans to the phosphine moiety.
The (P,S)-hybrid ferrocenyl ligand 75 was developed by Kang and co-workers and used as chiral auxiliary in the model Pd-catalysed asymmetric allylic alkylation (Scheme 2a), with moderate enantioselectivity (38% e.e.) (Fig. 31) [42].
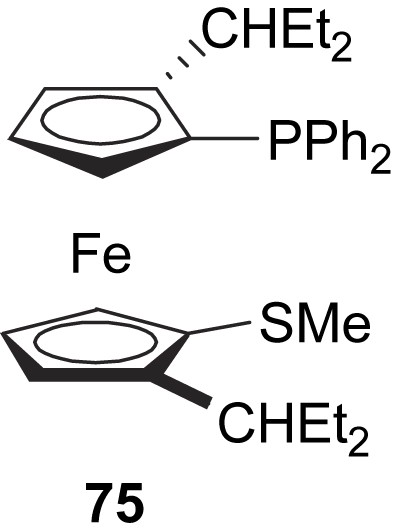
Chiral thioether–phosphine ligand 75.
The thioether–phosphine ligand 76a (Fig. 32) with a binaphthyl backbone was applied in asymmetric allylic alkylation of dimethyl malonate to 5 with enantioselectivity of 91% [43a]. Later, Gladiali and co-workers proved the ligand 76b in the same reaction obtaining 60% of e.e [43b].
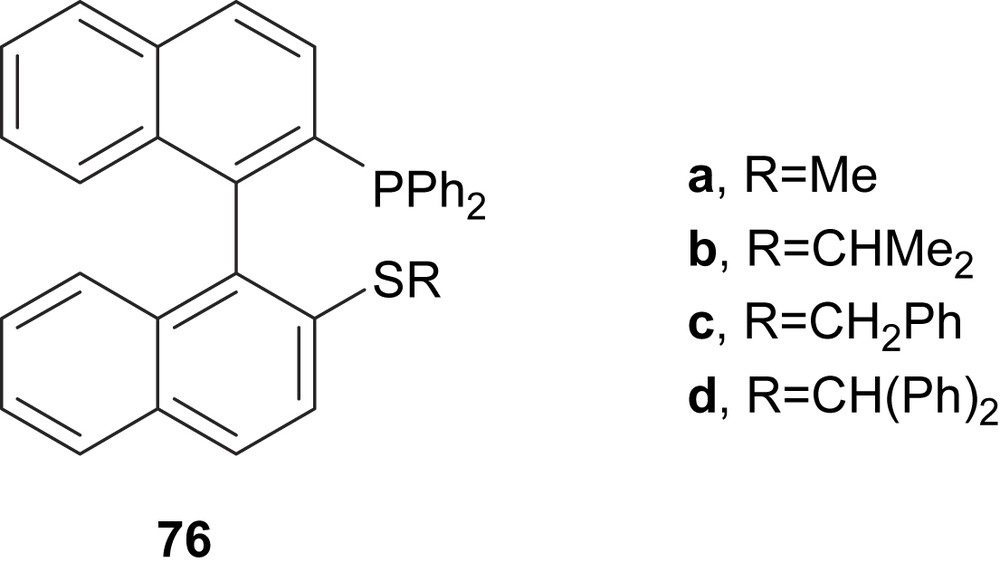
Thioether–phosphine ligands 76.
In 2004, Zhang and Shi tested the ligands 76a–d in asymmetric allylic alkylation of 5 (Scheme 2a) achieving enantiomeric excesses up to 96% (76a). The authors observed an interesting reversal of the absolute configuration in the alkylated product 6 by changing of steric bulkiness of ligand 76 [43c].
A series of (S)-proline-derived phosphine ligands bearing thioether functionalities (77–79) were prepared and proved in palladium-catalysed asymmetric allylic alkylation, affording enantioselectivities from 31 to 88%, depending on the steric bulkiness of the sulphur atom substituents (Fig. 33) [44].
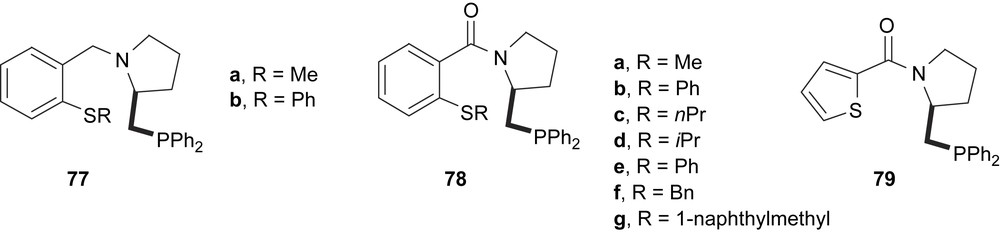
Chiral thioether–phosphine ligands 77–79.
Chiral phosphinooxathiane ligands 80–82 have been successfully used in alkylation and amination reactions of substituted allyl acetates (Fig. 34). The chiral norbornane-based phosphinooxathianes provided moderate-to-excellent levels of enantioselectivity (52–94%) in allylic alkylation of 5 with dimethyl malonate and enantioselectivities of 13–85% with low regioselectivities to the branched product in the allylic alkylation of substrate 13 (Fig. 2). The model allylic amination reaction (Scheme 2b) was also explored with these chiral phosphinooxathianes, and asymmetric inductions of 46–86% e.e. were achieved [45].

Chiral thioether–phosphine ligands 80–82.
Chiral phosphinooxathiolanes, also norbornane derived, were tested in the same catalytic reactions and conditions. Enantiomeric excesses up to 82 and 66% for alkylation and amination reactions, respectively, were attained using these new ligands [46].
Additionally, Nakano and co-workers developed a new xylofuranose-based phosphinooxathiane ligand, 83, that provided high enantioselectivities (e.e's up to 91%) in the Pd-catalysed asymmetric allylic substitutions of 5 (Fig. 35) [47].
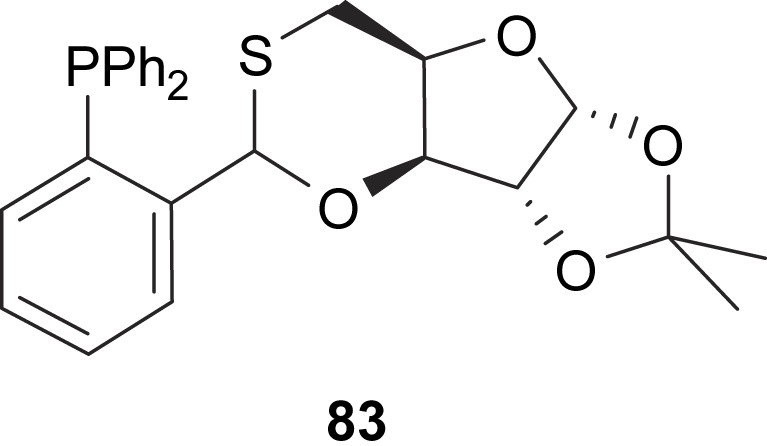
Chiral thioether–phosphine ligand 83.
Recently, Molander and co-workers developed a series of cyclopropane-based phosphine–thioether ligands (84–86) for the palladium-catalysed allylic alkylation of 5 with dimethyl malonate (Fig. 36). Varying the ligand substituents on the phosphorus, sulphur and carbon chain revealed that 86c provided the optimal configuration for this reaction, producing high yield and enantioselectivity [48].
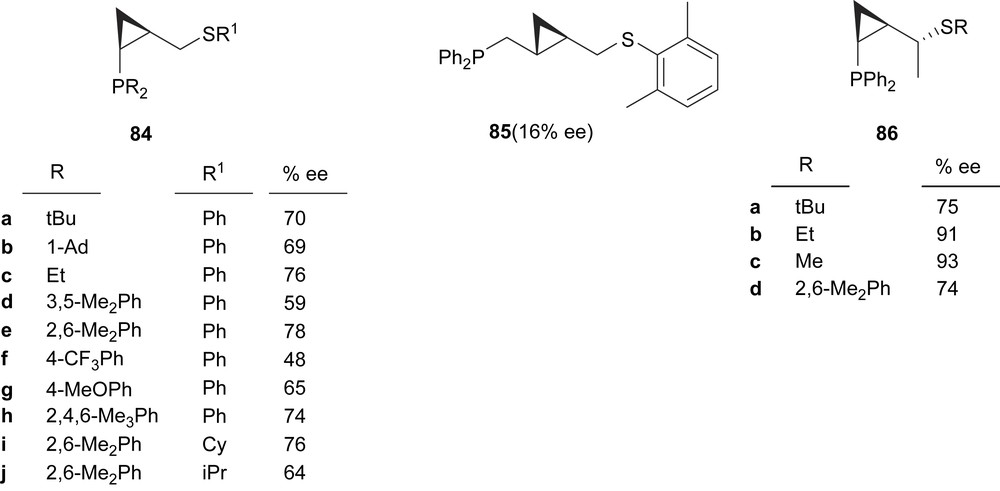
Thioether–phosphine ligands 84–86. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
3.2.2 Thioether–phosphites
To date, two families of thioether–phosphite ligands have been applied to this process.
Thioether–phosphite ligands with furanoside backbone 87 have been applied in the palladium-catalysed allylic alkylation and amination substitution reactions providing up to 58 and 67% e.e., respectively (Fig. 37). The catalytic results reveal that the thioether moiety hardly affects the enantioselectivity. It is thus assumed that the nucleophilic attack takes place trans to the thioether group [49].
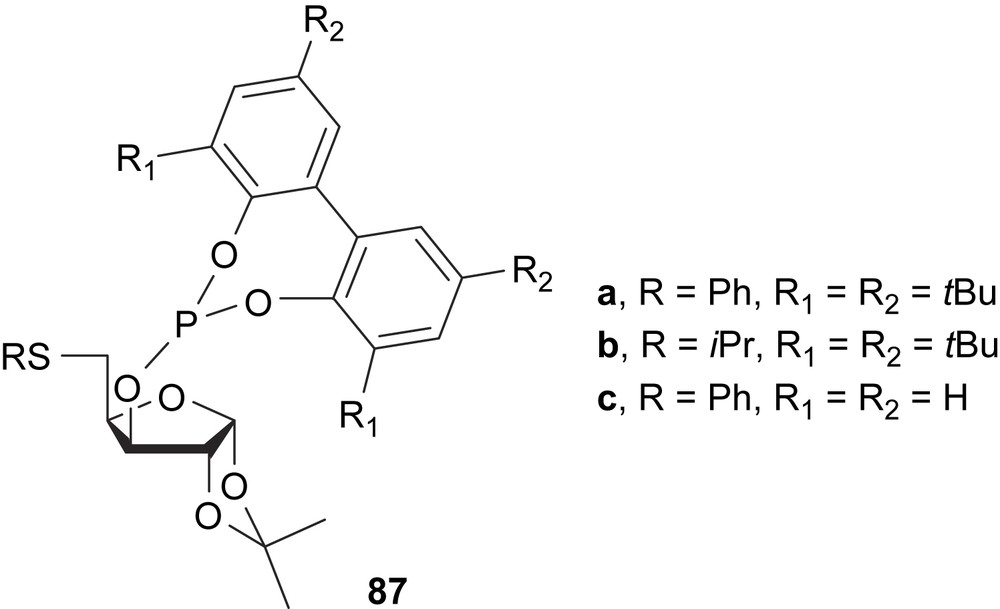
Furanoside thioether–phosphite ligands 87.
The allylic alkylation reaction of unsymmetrical substrates catalysed by palladium-containing binaphthylphosphite–thioether ligand 88 as chiral inductor was investigated. Yields up to 70% were reached, but in all cases, both the regio- and enantioselectivities were moderate [50] (Fig. 38).
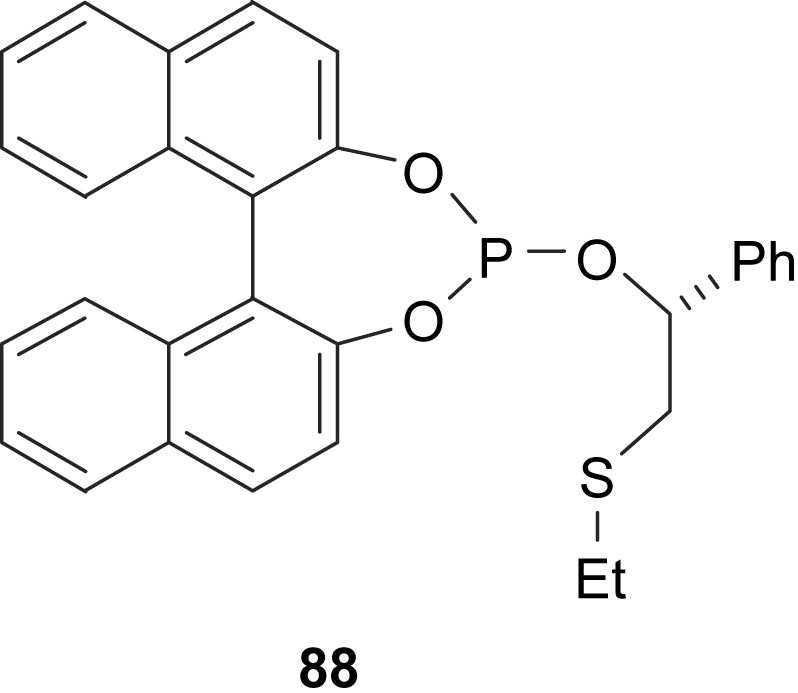
Thioether–phosphite ligand 88.
3.2.3 Thioether–phosphinites
The first family of mixed thioether–phosphinite ligands (89–95) was developed by Evans and co-workers, and was successfully applied in enantioselective palladium-catalysed allylic substitutions of several acyclic and cyclic substrates (Fig. 39) [51]. Electronic and steric effects of the ligands on the enantioselectivity of the allylic alkylation reaction (Scheme 2a) were studied by varying sulphur, backbone and phosphorous substituents. The best results were obtained with 89a and 90a, containing bulky alkyl substituents in both backbone and thioether group. In allylic alkylation of 5 with dimethyl malonate, 89a and 90a afforded enantiomeric excesses of 90 and 98%, respectively. In contrast, 89a was superior in the model allylic amination reaction, giving 99% e.e. A variety of other heteroatom nucleophiles in allylic substitution of 5 were investigated, but low yields and enantioselectivities were attained.

Thioether–phosphinite ligands 89–95.
The regioselective allylic alkylation of trisubstituted propenyl acetates was also explored with 89a and 90a, affording high yields and asymmetric inductions up to 94% of e.e. In addition, this family of ligands was tested in the palladium-catalysed allylic substitution of cyclic substrates. Ligand 91a afforded enantiomeric excesses of 91–97% in quantitative yields in the alkylation and amination of several cycloalkenyl acetates [51].
Related to these ligands, two allylic palladium intermediates have been characterised, in both the solution and solid state, 96 and 97 containing ligands 90a and 89a and the 1,3-diphenylallyl and cyclohexenyl groups, respectively (Fig. 40). The X-ray diffraction data indicate a stronger trans influence of the phosphinite group [52].
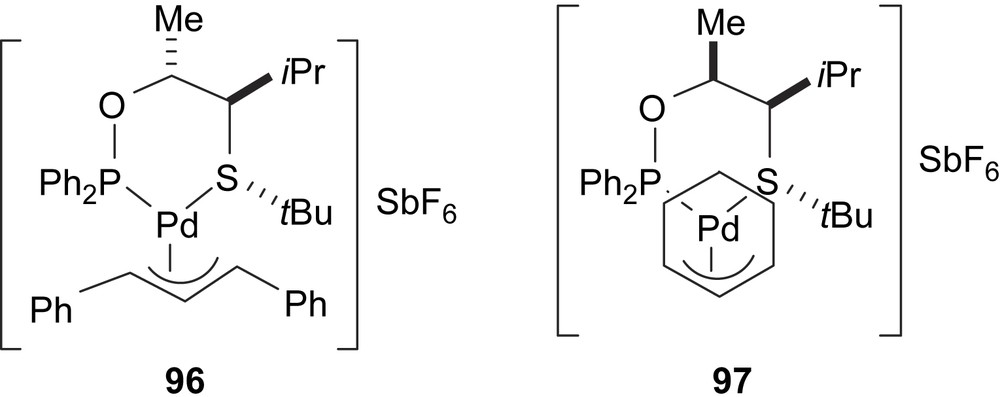
Allyl palladium complexes 96 and 97.
In solution, 96 exists as a mixture of two diastereomers, in a 2.3:1 ratio, due to the relative position of the central allylic carbon and the tert-butyl substituent on the sulphur moiety. The major isomer was assigned to an exo arrangement (both groups pointing in the same direction), which is consistent with the structure observed in solid state. In contrast to 96, NMR data of complex 97 reveal the presence of mainly one allyl palladium isomer (ratio > 98:2), consistent with the X-ray structure.
Recently, a series of thioether–phosphinite ligands (98) with a furanoside backbone have been tested in the Pd-catalysed allylic substitution reactions of several acyclic and cyclic substrates (Fig. 41). The results indicated that enantioselectivity depends strongly on the steric and electronic properties of the substituents in the thioether moiety and the substrate structure. By carefully selecting the ligand parameters, good enantioselectivities were obtained for hindered linear substrates (e.e.’s up to 95%) and for unhindered cyclic substrates (e.e.’s up to 91%). The strong trans influence of the phosphinite moiety, together with the effect of the thioether moiety on enantioselectivity, confirms that the nucleophilic attack takes place on the allyl terminus located trans to the phosphinite moiety. This contrasts with related thioether–phosphite ligands 87, where the nucleophilic attack was trans to the thioether group [53].
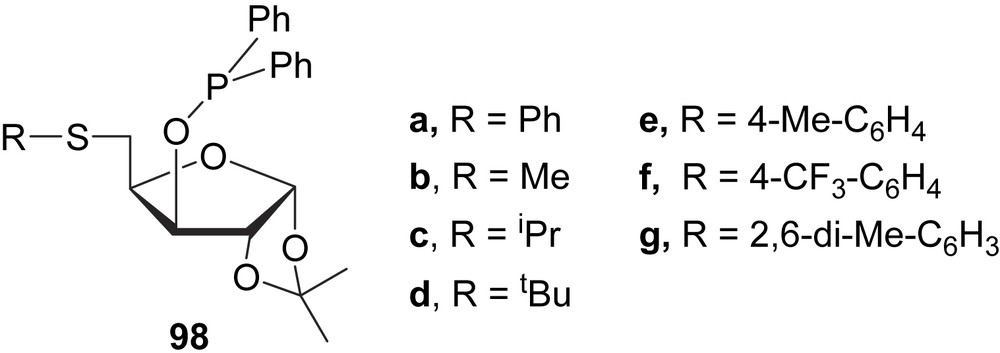
Sugar-based thioether–phosphinite ligands 98.
Phosphinite β-thioglycosides have been prepared and evaluated in allylic alkylation of 5 under Trost conditions. The alkylated product 6 was obtained in high yield in all cases, but with variable e.e.’s (60–92%), depending on the substituent at the sulphur atom. The bulky t-butylthioglycoside derivative was also an efficient inductor in the amination reaction (Scheme 2b) affording 94% of e.e. [54].
3.2.4 Thioether–phosphinamidites
Recently, Chan and co-workers developed a series of ferrocene-based thioether–phosphinamidite (Fig. 42), and successfully applied them in the asymmetric allylic substitution of 5 (e.e.’s up to 93%). A mild effect of the thioether substituent on enantioselectivity was observed; however, activities are better when a small ethyl substituent in the thioether group is present [55].
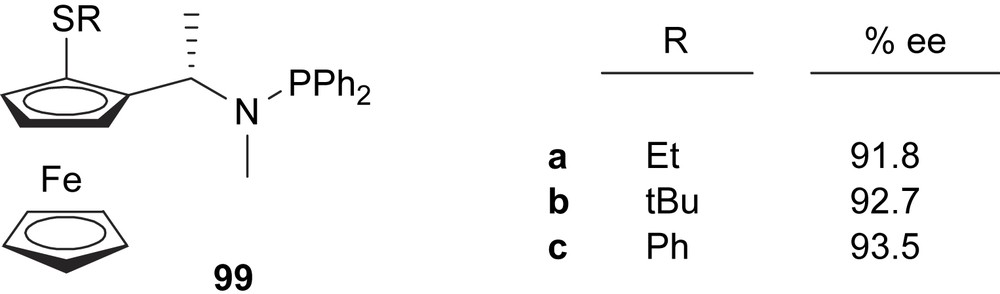
Thioether–phosphineamidite ligands 99. This figure also shows the enantioselectivities obtained in the Pd-catalysed asymmetric allylic alkylation of dimethyl malonate to 5.
3.3 Other mixed thioether ligands
Other mixed thioether ligands such as (S,Se)-donor [39,56] and (S,X)-polydentate donor ligands [27,28] were applied to this process, but only the tridentate chiral pyridine–dithioether ligands 100a–e (Fig. 43) provided good enantiomeric excess (78–85% e.e.) [27].

Tridentate pyridine–dithioether ligands 100.
4 Final remarks
Chiral thioethers are an alternative coordinative group to be successfully applied in asymmetric allylic substitution. Particularly, the use of the chiral heterodonor ligands containing sulphur functionalities is a highly efficient strategy to control the enantioselectivity through the different electronic effects of the donor atoms. Furthermore, the electronic and steric properties of these kinds of ligands can be easily modified by the groups attached at the sulphur atom, modulating in this way, the strength of the M–S bond and changing the thioether trans influence in metal complexes. In fact, different catalytic performances have been obtained due to the variety of electronic combinations of (S,X)-ligands.
An additional feature is the creation of a new chiral sulphur centre upon thioether complexation, which represents an advantage, since it generates a chiral pocket nearer to the metal centre. Nevertheless, the difficulty in controlling the sulphur inversion and therefore, the number of diastereomeric complexes in solution, is the reason for the enantioselectivity erosion in asymmetric reactions using metal–thioether catalysts. However, this problem can be solved by appropriately tuning the ligand, as has been shown in this review.
Acknowledgements
We thank DGAPA-UNAM (Project PAPIIT-IN202902), the Spanish Ministerio de Educación, Cultura y Deporte (CTQ2004-04412/BQU) and the Generalitat de Catalunya (Distinction to M.D.). We also gratefully recognise the valuable help provided by Prof A.M. Masdeu-Bultó from URV (Spain).