

Since Crabtree and co-workers discovered that [Ir(pyridine)(PCy3)(COD)]PF6 catalyses the hydrogenation of unfunctionalised olefins with high turnover frequencies in non-coordinating solvents [1], subsequent studies have focused on investigations into the mechanism of iridium-catalysed hydrogenation [2], applications in stereoselective organic synthesis [3] and, most recently, enantioselective hydrogenations using chiral P, N ligands [4].
Our interest in developing chiral analogues of Crabtree's catalyst stemmed from the fact that whilst asymmetric hydrogenation is clearly an attractive synthetic method [5], the Monsanto -l-DOPA process being a well-known example from industry [6], the range of olefins that can be hydrogenated with high enantiomeric excess is limited. Rhodium and ruthenium catalysts require the presence of a polar functional group next to the CC bond which can coordinate to the metal centre and help in achieving high levels of activity and stereocontrol [5,7].
Some years ago, we introduced cationic iridium complexes with chiral P, N ligands as catalysts that overcome these limitations. The original ligand was a phosphino-oxazoline (PHOX) [8] and the optimal counter-anion was found to be the large, weakly coordinating tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF). These catalysts allowed the hydrogenation of stilbenes to be carried out with high levels of enantioselectivity [9]. The nature of the anion proved to be crucial in avoiding catalyst deactivation and reducing the moisture-sensitivity of the reactions. Under optimised conditions turnover numbers of >5000 could be achieved (Scheme 1) [10].

Highly enantioselective hydrogenation of a stilbene.
Subsequent studies in these laboratories have extended the range of substrates which can be hydrogenated with high levels of enantioselectivity by systematic variation of the structure of the P, N ligands [11]. The threonine-derived phosphinite P, N ligands (ThrePHOX) have proven to be especially versatile for highly efficient and enantioselective hydrogenations of a wide range of unfunctionalised and certain functionalised olefins (Scheme 2) [12].
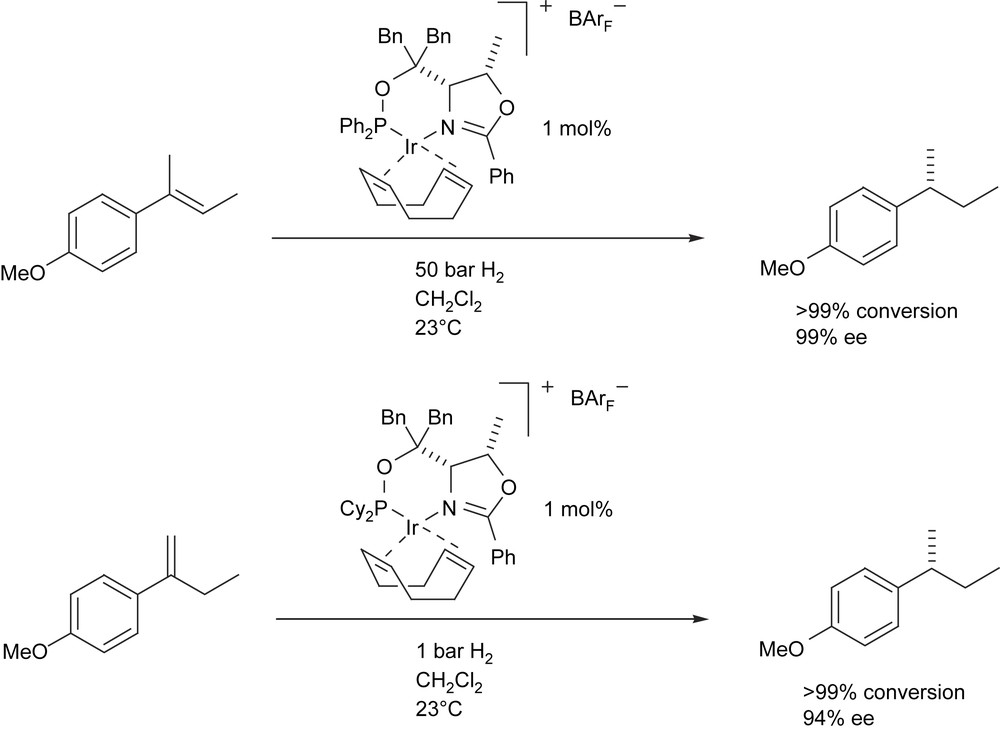
Highly enantioselective hydrogenations using IrThrePHOX catalysts.
Other research groups have also become interested in asymmetric hydrogenation of unfunctionalised olefins using chiral analogoues of Crabtree's catalyst and, during the last few years, have reported a number of efficient ligands [13].
Until recently, highly enantioselective hydrogenation of purely alkyl-substituted olefins has not been possible. From a synthetic point of view, a catalyst which is able to hydrogenate the CC double bond of a substrate without the need for any specific functional group or aryl substituent would allow the reaction to be applied to a much wider range of substrates. In view of the encouraging results obtained with pyridine–phosphinites of type 1 [14], we prepared a series of bicyclic analogues 2, since we expected the more rigid geometry imposed by the additional ring to raise enantioselectivity (Fig. 1). Iridium complexes incorporating the five- and six-membered ring derivatives 2 (n = 1,2) proved to be efficient catalysts, inducing enantioselectivities generally higher than those of the analogous ligands 1 and we have found that bicyclic pyridine–phosphinite complexes of this type are highly selective catalysts for hydrogenation of simple, purely alkyl-substituted olefins [15].
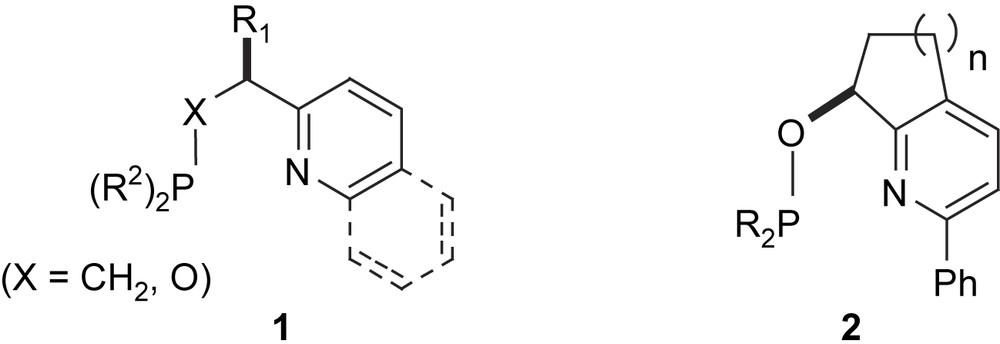
Pyridine–phosphinite ligands 1 and their bicyclic analogues 2.
Initially, we used the (E)- and (Z)-olefins 3 as test substrates, the methoxyphenyl group having been introduced to facilitate product analysis by GC or HPLC on chiral columns. Most ligands that had given high enantioselectivities with alkenes containing an aryl substituent at the CC bond performed poorly with (E)- and (Z)-3. Pyridine-based ligands 4, 5 and 6 were exceptions, giving full conversion and ee values between 83% and 98%. Among a series of oxazoline- and imidazoline-derived ligands, two derivatives 7 and 8 induced reduction with almost 90% ee. However, these two ligands performed well only with one of the two substrate isomers. The best enantioselectivities were obtained with the bicyclic pyridine–phosphinite ligand 4, giving 93% and 98% ee for the (E)- and (Z)-isomers, respectively. Consistent with previous studies, the (E)- and (Z)-isomers were converted to the products 9 having opposite configurations (Fig. 2).
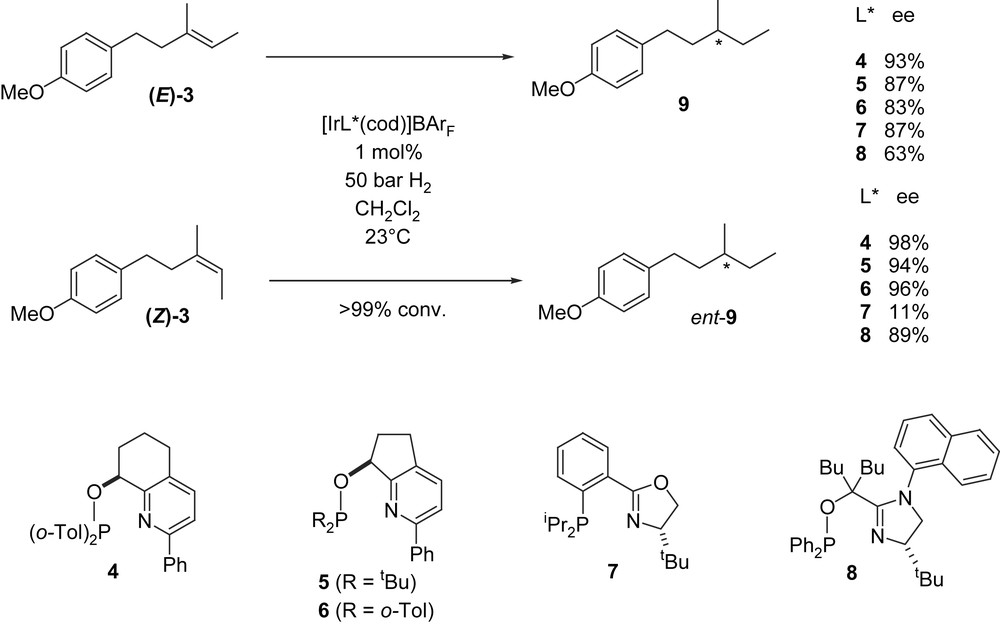
Highly enantioselective hydrogenation of olefins bearing a remote aryl moiety.
These results encouraged us to search for a suitable substrate devoid of any heteroatom or aryl group, but still allowing reliable determination of the enantioselectivity after hydrogenation. Cyclohexylalkene 10 was found to meet these requirements: it was readily prepared in high isomeric purity (E/Z > 99:1) following published procedures and the ee of the hydrogenation product 11 could be determined by GC on a chiral column. This substrate proved to be more demanding than alkenes (E)- and (Z)-3 and most ligands studied gave ee values below 30%. We identified only two ligands, the bicyclic pyridine–phosphinites 4 and 5, which induced enantioselectivities of >80% ee. Nonetheless, the ee value of 92% obtained with ligand 5 shows that highly enantioselective hydrogenation of purely alkyl-substituted olefins is possible (Fig. 3).

Highly enantioselective hydrogenation of a purely alkyl-substituted olefin.
To demonstrate the potential of our catalysts for enantioselective synthesis of biologically relevant molecules, we studied the hydrogenation of γ-tocotrienyl acetate 12. This reaction involves reduction of three CC bonds and creates two new stereocentres in a single step. Because the two pro-chiral double bonds are both (E)-configured, the sense of asymmetric induction at the two reaction sites is expected to be the same, leading either to the (RR)- or (SS)-configuration depending on the absolute configuration of the catalyst. We screened various iridium catalysts derived from different ligand types in the hydrogenation of tocotrienol derivatives. Whereas oxazoline-based ligands showed disappointingly low stereoselectivities, imidazolines and pyridine–phosphinites gave encouraging results. The best stereoselectivity was achieved with the iridium catalyst derived from pyridine–phosphinite ligand 4, which gave almost exclusively the natural (RRR)-isomer of γ-tocotrienol acetate 13 (Scheme 3).
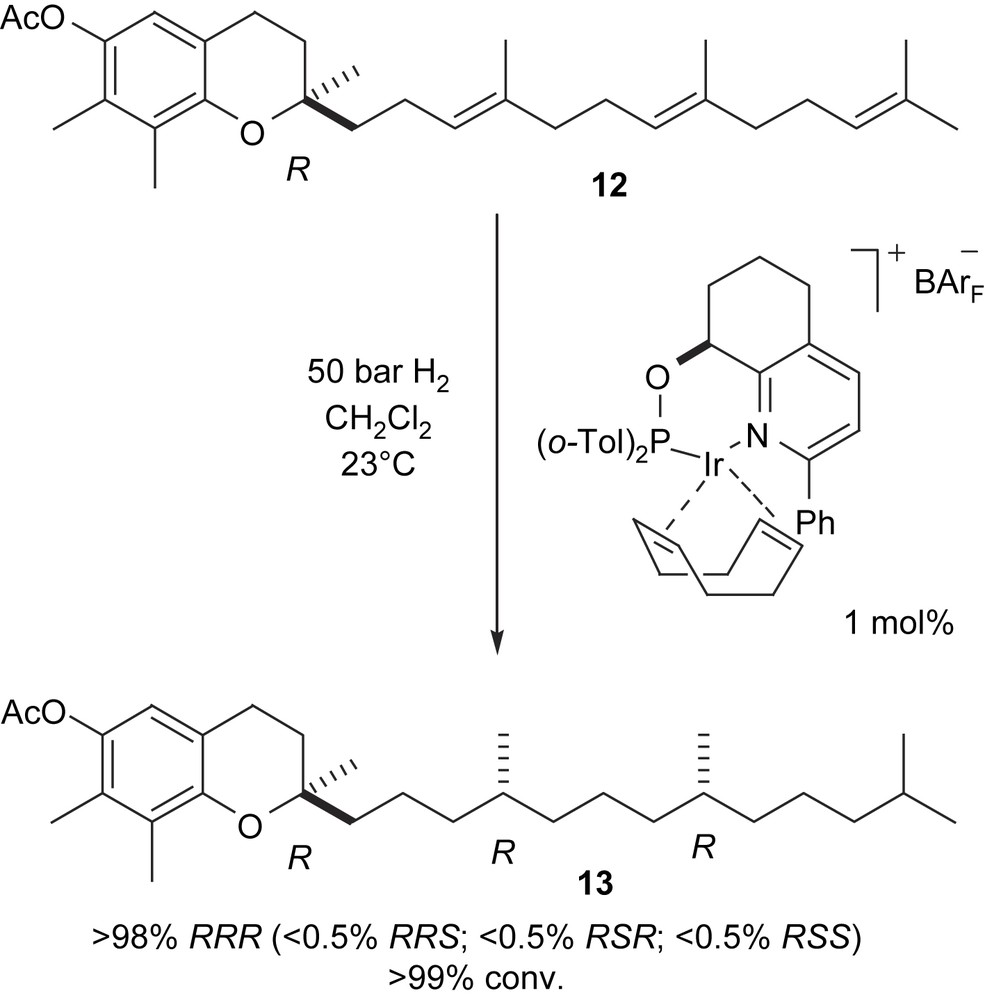
Highly enantioselective hydrogenation of γ-tocotrienyl acetate.
Further systematic studies using this ligand class have revealed that they can be highly selective in asymmetric hydrogenation of furans and benzofurans, for which no catalysts were known before [16]. In previous studies of furyl-substituted alkenes, we had found that with certain Ir complexes derived from oxazoline-dialkyl-phosphinites, both the olefinic CC bond and the furan π system were reduced. The stereoselectivities were, however, moderate. Catalysts incorporating ligands 5 and 14 proved to be more efficient and induced good to excellent enantioselectivities in the hydrogenation of a series of substituted furans and benzofurans. As expected, the benzene ring of substrates 15 and 16 was not reduced (Fig. 4).
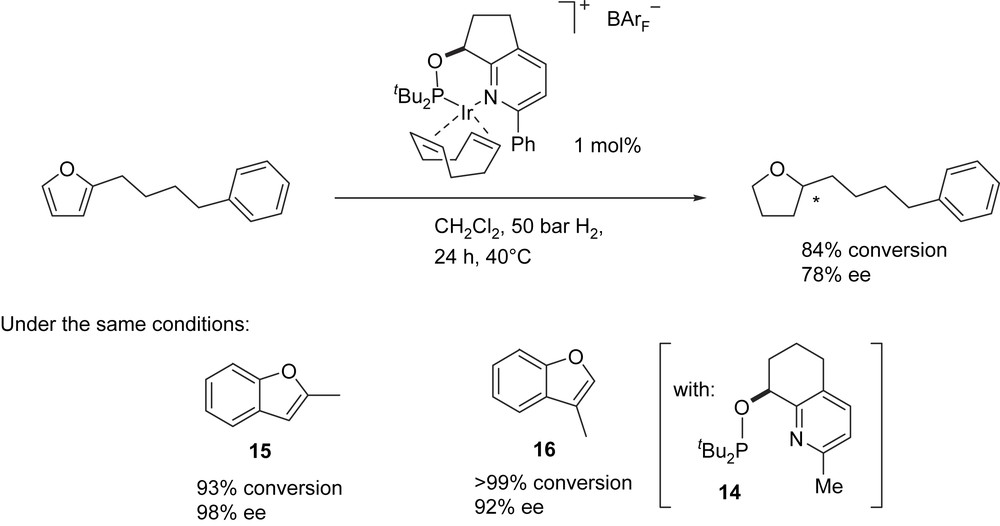
Enantioselective hydrogenation of furans and benzofurans.
Ligands with bulky, electron-rich (tBu)2P groups were found to be best suited for this class of substrate. Ligands with cyclohexyl substituents at the P atom gave lower conversion and ee, whereas catalysts with analogous diphenylphosphinite ligands showed essentially no activity. Because of the low reactivity of the furan and benzofuran π systems, elevated temperatures and relatively long reaction times were necessary. The benzofuran carboxylate ester 17, in particular, reacted only sluggishly, albeit with near perfect enantioselectivity (Fig. 5).

Highly enantioselective hydrogenation of functionalised furans.
Overall, ligands like 5 and 14 open up an attractive enantioselective route to tetrahydrofuran and benzodihydrofuran systems, which are structural motifs found in many natural products and biologically active compounds.
An enantioselective ligand synthesis was not reported as part of these hydrogenation studies. It has subsequently been found that kinetic resolution of the pyridyl alcohol ligand precursors by copper(II) boron-bisoxazoline-catalysed acylation can be highly selective, providing straightforward access to both enantiomers of the ligand precursor. Using one of the anionic, boron-bridged bisoxazoline ligands recently developed in our group [17], an S value (krel) of around 100 was obtained when the pyridyl ring was fused to a six-membered ring and substituted in the α-position with a phenyl group. Analogous bisoxazoline ligands were found to be less selective (Scheme 4) [18].
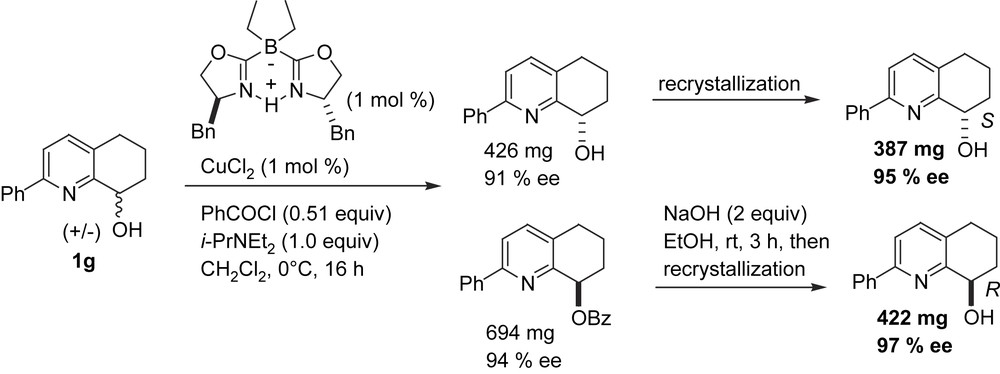
Efficient kinetic resolution of pyridyl alcohol ligand precursors by Cu(II)(borabox) catalysed acylation.
Access to the optically enriched bicyclic pyridyl alcohol ligand precursors is also possible by enantioselective reduction of the corresponding ketones [19] and enzymatic resolution of pyridyl alcohols [20].
The mechanism of olefin hydrogenation using Crabtree's catalyst and chiral analogues has been studied over several years. The earliest mechanistic study in this area was carried out by Crabtree and co-workers, who used NMR spectroscopy to detect olefin dihydride intermediate 18 which was formed during hydrogenation of cyclooctadiene using [Ir(pyridine)(PCy3)COD]PF6 in dichloromethane at 0 °C (Scheme 5) [2].
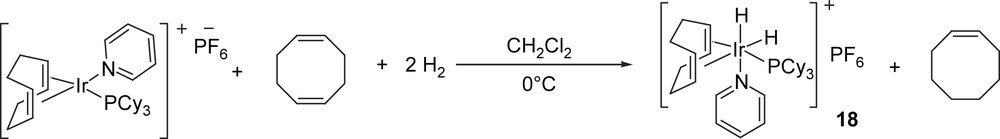
Olefin dihydride intermediates observed during the hydrogenation of cyclooctadiene.
Since oxidative addition of hydrogen to the iridium centre was found to be rapid at low temperature and alkyl hydride intermediates were not observed, the olefin dihydride intermediate 18 was thought to be the resting state during catalysis with migratory insertion being the rate-limiting step.
In a complementary study, we have shown that when [Ir(PHOX)COD]BArF complex 19 was treated with hydrogen at −40 °C for 5 min in [D8]-THF, olefin dihydride intermediates are formed, which were characterised by NMR spectroscopy. Two new signals appeared in the hydride region that were assigned to a single dihydride complex formulated as [Ir(PHOX)(H)2(COD)]BArF. The 2J(H, P) values of 20 Hz clearly showed that both hydrides were disposed cis to the phosphorus atom. Observation of NOE contacts between the hydride in the apical position and the isopropyl substituent, as well as the ortho proton of one of the P-phenyl rings, is consistent with the structure 20c (Fig. 6) [21].

Stereoselective formation of olefin dihydrides and solvate dihydrides using a chiral ligand.
Highly selective formation of isomer 20c results from H2 addition to the more sterically encumbered face of the starting complex. The high preference for this pathway over the alternative pathway leading to complex 20d is likely due to steric factors: dihydrogen addition to the sterically more accessible face, leading to isomer 20d, would build up steric strain between the chelating COD ligand and the isopropyl group in the oxazoline ring and the pseudoaxial P-phenyl group. The predominance of isomer 20c over 20a or 20b is consistent with Crabtree's findings, who convincingly demonstrated that, in the reaction of H2 with [Ir(pyridine)(PR3)COD]PF6, the formation of an Ir–H bond trans to the N ligand is electronically favoured [22]. When the solution containing complex 20c was warmed to 0 °C under hydrogen and kept at that temperature for 30 min, gradual consumption of isomer 20c was observed, accompanied by the appearance of two new hydride complexes 21c and 21d with concomitant formation of cyclooctane.
The hydrogenation of complex 19 was carried out in [D8]-THF, since in dichloromethane a complex mixture of hydrido complexes was observed. This prevented the observation of intermediates formed during olefin hydrogenation, since oxygen donor solvents completely suppress catalysis. Nevertheless, taken together, these results do indicate that oxidative addition to iridium is rapid at low temperature, with migratory insertion being a slower step. That iridium solvate dihydrides 21 can be isolated and characterised in solution following hydrogenolysis of the cyclooctadiene ligand is an indication that the ‘dihydride route’ could play a major role in iridium-catalysed hydrogenation [5a,23].
Recent work in our laboratories has resulted in the characterisation of an iridium dihydride solvate species in dichloromethane which incorporates a chiral phosphinite oxazoline ligand. Thus detection of intermediates formed during catalytic hydrogenation may become possible and work in this area is ongoing.
However, since the structures of reactive intermediates formed during hydrogenation have been difficult to determine experimentally, a computational approach may yield useful information about the potential reaction pathways. Steric effects obviously play a major role in these systems, so we decided to carry out quantum mechanical calculations on the complete catalyst and substrate structures instead of resorting to simpler model compounds. In order to test the method on an experimentally characterised reaction pathway, we calculated the structures shown in Fig. 6 at the B3LYP level of theory, using the LANL2DZ effective core potential for iridium and explicit basis sets of 6-31G(d,p) quality for all other atoms. The fully minimised structures of the four possible cis-dihydride complexes, which can be formed by oxidative addition of H2 to [Ir(PHOX)(COD)]+, were calculated. The most stable calculated structure corresponded to the reaction product that was shown to be formed exclusively in the NMR experiment. Higher energy isomers corresponding to 20a and 20d were also located. The four possible [Ir(PHOX)(H)2(solvent)2]+ complexes resulting from hydrogenation of the cyclooctadiene ligand were also studied and again the two most stable structures corresponded to the isomers observed in the NMR experiments, indicating that the computational approach used leads to experimentally meaningful results [21].
Based on this work, we began a systematic DFT study of the potential reaction pathways leading to enantioselective hydrogenation. When considering which intermediates may be formed during catalysis, one of the first issues which becomes apparent is what ligands are coordinated to iridium during catalysis. The difficulty is that an olefin dihydride iridium complex which incorporates a bidentate P-, N-ligand has a sixth coordination site available for an additional ligand and the nature of this ligand may be important in determining which of the competing pathways is lower in free energy. Although coordination of a second molecule of olefin seems highly unlikely due to steric hindrance, both dihydrogen and dichloromethane may be effective ligands for iridium.
The two situations are indicated in the general mechanisms shown below. The first is analogous to the well-established mechanism for rhodium diphosphine-catalysed hydrogenation of olefins [5a,23]. An IrIII olefin dihydride intermediate 22, in which dichloromethane is coordinated to the iridium, undergoes migratory insertion to give an alkyl hydride 23. Subsequent reductive elimination would give an IrI complex, which is then expected to undergo ligand substitution and rapid oxidative addition of hydrogen, eventually leading to an IrIII intermediate, such as solvate dihydride 24 (Fig. 7).
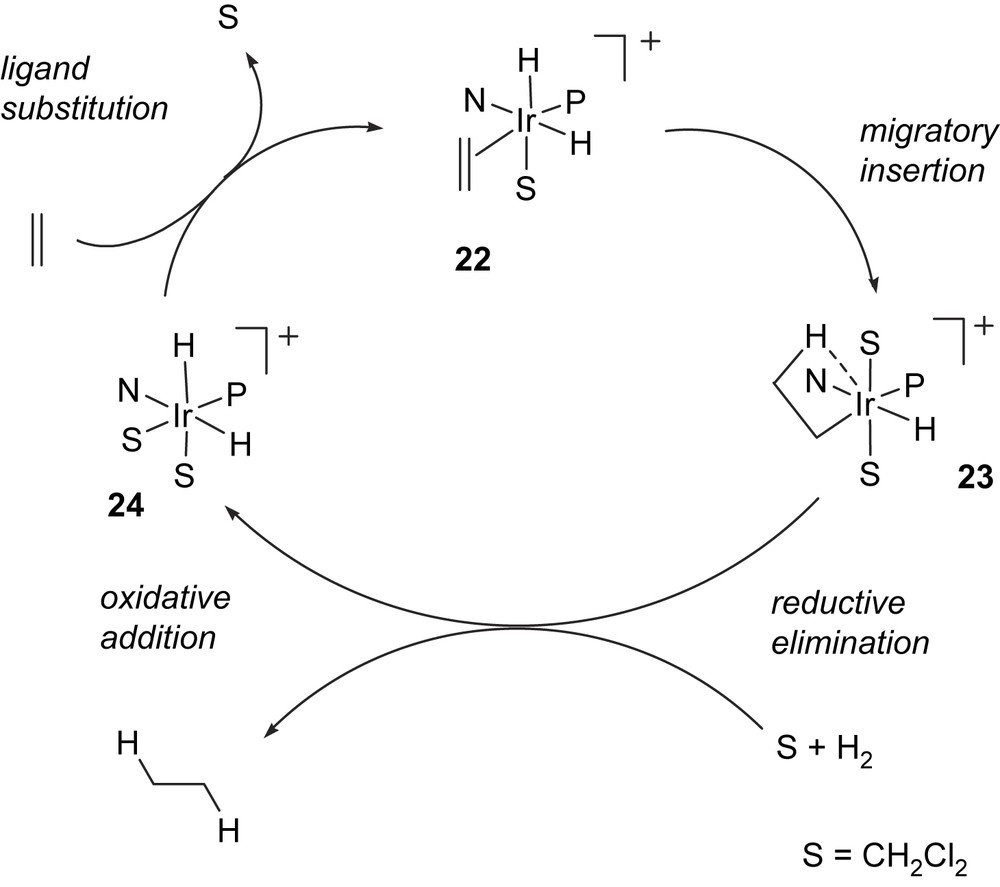
A catalytic cycle for olefin hydrogenation involving IrI–IrIII intermediates.
Alternatively, olefin dihydride intermediate 25 could be formed in which dihydrogen is coordinated to the iridium in a η2 fashion instead of dichloromethane. Migratory insertion may be accompanied by cleavage of the H–H bond leading to an IrV alkyl hydride 26. Reductive elimination produces an IrIII dihydride 27 which undergoes ligand substitution to return the olefin dihydride intermediate 25 (Fig. 8).
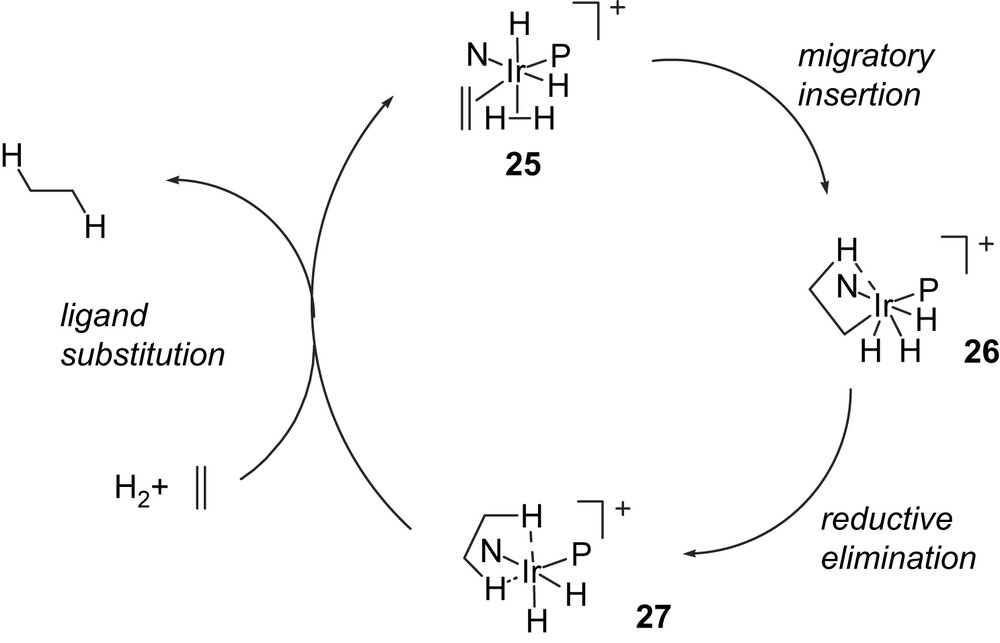
A catalytic cycle for olefin hydrogenation involving IrIII–IrV intermediates.
Theoretical investigations into the reaction mechanism carried out by Brandt et al. originally led to a proposed catalytic cycle involving IrIII–IrV intermediates, in which the dihydrogen co-ligand undergoes oxidative addition to the iridium during migratory insertion [24]. Since an extremely truncated model for the ligand and substrate (ethylene) was used, and as steric factors are obviously important in determining the coordination chemistry of the iridium during catalysis, it seems dangerous to rule out an alternative IrI–IrIII cycle on this basis. However, subsequent studies of the Andersson group, which included DFT calculations of the full catalyst and substrate structures, were exclusively based on the postulated IrIII–IrV cycle [13d,e]. From calculated transition states of the migratory insertion of [Ir(P∧N)(H)2(η2-H2)(η2-olefin)]+ complexes, they derived a simple qualitative model for rationalising the observed enantioselectivities.
Burgess, Hall and co-workers [13b] also reported calculations on the complete ligand and substrate structures. The key finding was that calculations carried out at the PBE level of theory required much less computational time than, and were in reasonable agreement with, those carried out at the B3LYP level. Assuming an Ir(III)–Ir(V) cycle, calculations led to the correct selectivity order for three different substrates. Unfortunately, no calculations based on an IrI–IrIII cycle were reported. Our work is not yet completed, but preliminary findings do indicate that calculated pathways beginning from [Ir(P∧N)(H)2(η2-H2)(η2-olefin)]+ complexes lead to predicted enantioselectivities which are opposite to the experimentally observed values. Whilst competing pathways involving solvated IrI–IrIII intermediates are somewhat higher in energy (by a few kcal/mol), calculated transition states are in reasonable agreement with the experimentally observed enantioselectivity. Taking into account that CH2Cl2 as the solvent is present in much higher concentration than H2, the IrI–IrIII and IrIII–IrV cycles become energetically very similar, making it difficult to distinguish between them based on calculations alone.
Chen and Dietiker have reported an experimental investigation of the hydrogenation of styrene by the [Ir(PHOX)(COD)]BArF by means of electrospray ionization tandem mass spectrometry which suggests that the catalytic cycle proceeds by way of IrI and IrIII intermediates, presumably by a ‘dihydride’ catalytic cycle indicated below (Fig. 9) [25].
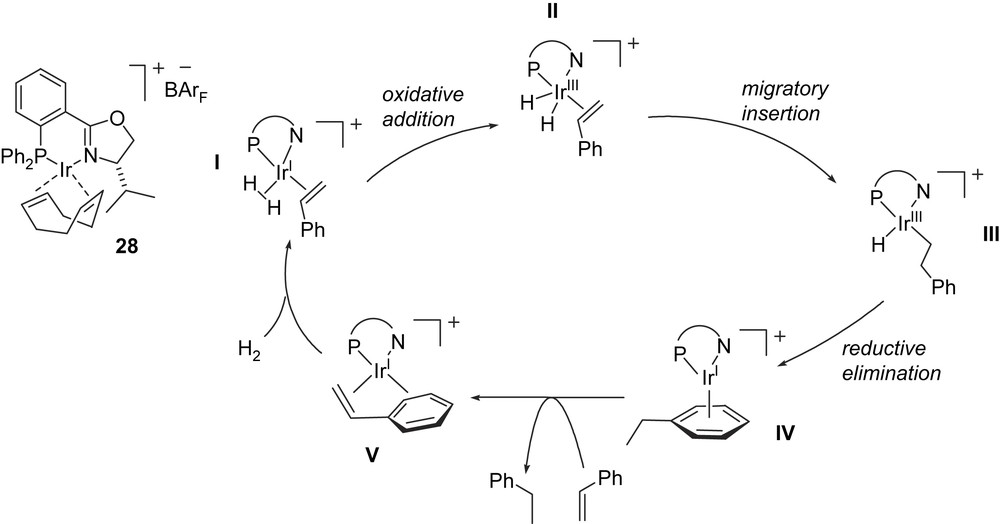
Proposed catalytic cycle via an IrIII dihydride intermediate.
Gas-phase reactions of selectively prepared intermediates were examined and found to be instructive with regard to the catalytic cycle. [Ir(PHOX)(H2)]+ ions, obtained from a solution of [Ir(PHOX)(COD)]BArF 28 and H2, were subjected to multiple collisions with ethylbenzene to produce an ion with the composition [Ir(PHOX)(ethylbenzene)]+. This ion, assumed to be species IV, was isolated in the gas phase according to its m/z ratio and subsequent collision-induced dissociation of IV with argon led to facile loss of dihydrogen, producing the styrene complex V. Thus the iridium styrene complex V has been linked to the iridium ethylbenzene complex IV mechanistically, although under these conditions the catalytic cycle is traversed backwards, forming V after irreversible loss of hydrogen. In a second instructive experiment V is treated under soft conditions (initial collision energy of 6.9 kcal/mol or less) with D2 gas. The sole observable products are V, [D1]V and [D2]V. This result demonstrates that species I–III are intermediates in hydrogenation, since they must be formed for isotope exchange in V to occur. No evidence for the participation of IrV complexes was found, since when the selectively prepared sample of V was treated with D2, no [D3]V was observed. The authors argue that during this experiment a species of the composition [Ir(PHOX)(styrene)(D2)2]+ had the opportunity to form and if such species are intermediates in hydrogenation, IrV alkyl hydride intermediates which incorporate three Ir–D bonds (and an alkyl group with one deuterium atom) would be formed. Given the reversibility of elementary steps in the gas-phase reaction, at least partial incorporation of more than two deuterium atom into the styrene substrate would be expected.
At present it is still too early to draw definitive conclusions regarding the mechanism of Ir-catalysed asymmetric hydrogenation. Experimental data favour an IrI–IrIII cycle, whilst computational studies have shown that pathways via IrIII–IrV intermediates are at least energetically feasible. In some cases, calculations based on an IrIII–IrV pathway were shown to be consistent with the observed enantioselectivities [13]. However, this should not be used as an argument in favour of an IrIII–IrV cycle, because alternative IrI–IrIII cycles were not calculated in these studies.
In conclusion, recent studies in the design of chiral P-, N-ligands based on conformationally rigid, bicyclic pyridyl phosphinites have extended the scope of iridium-catalysed asymmetric hydrogenation to include simple, purely alkyl-substituted olefins and substituted furans. These substrate classes are important from a synthetic point of view and their efficient and highly enantioselective hydrogenation constitutes a significant advance. Challenges remain, however, in developing efficient and selective catalysts for other substrate classes such as tetrasubstituted olefins [26]. Our knowledge of the reaction mechanism is developing more slowly, but a combined computational and experimental approach promises eventually to lead to valuable insights into the reaction mechanism and the factors responsible for enantioselectivity.