

1 Introduction
The synthesis of ligands which combine hard and soft donor functionalities is a subject of considerable current interest, in particular with respect to the development of new reactive, hemilabile metal–ligand systems and more selective homogeneous catalysts [1,2]. In particular, numerous metal complexes containing P,N type ligands are catalytically active in a wide range of reactions of both academic and industrial interest [2,3]. Various unsymmetrical P,N-type bidentate ligands have recently been applied to CO/alkene copolymerization reactions [4].
Using phosphinito- and phosphonito-oxazoline ligands as P,N chelates in Pd(II) complexes, we could recently isolate and fully characterize intermediates in the CO/ethylene stepwise insertion reaction [4a]. Cationic Pd(II) complexes containing the diphenylphosphino-P,N type ligands 1b or 1c have been investigated as catalysts for ethylene oligomerization [5,6] and CO/ethylene copolymerization [4h]. These ligands have allowed us to prepare and fully characterize initial intermediates in the sequential CO/ethylene or CO/methyl acrylate coupling reactions, without the need for using excess methyl acrylate [4b]. Despite the considerable interest in the alternating copolymerization of olefins with CO and with polar monomers, such as methyl acrylate [4b,7], up to date only few metal complexes featuring CO/methyl acrylate coupling have been isolated and characterized [4b,g,8–13]. Therefore, investigating the behaviour of other functional ligands bearing substituents on the phosphorus donor atom with different stereoelectronic properties should be of particular interest for these types of reactions. We have now extended our studies with the diphenylphosphino-P,N ligands 1b,c to the new isopropyl derivative 1a and studied the reactivity of its Pd(II) complexes.
2 Results and discussion
2.1 Ligand synthesis
The ligand (i-Pr)2PCH2(oxazoline) (1a) was obtained in quantitative yield (95%) by the one-pot procedure previously used to synthesize 1b [4h], which consists first of the deprotonation of the corresponding 2-methyl-2-oxazoline in THF at −78 °C, followed by the addition at this temperature of SiClMe3, to form the N-silyl derivative, and the reaction with P(i-Pr)2Cl (Eq. (1)).

In the 1H NMR spectrum of ligand 1a, the PCH2 (δ = 2.35, 2JPH = 1.0 Hz) and NCH2 (δ = 3.75, 3JHH = 9.3 Hz) protons appear as a broad doublet and broad triplet, respectively, indicating a small 5JHH coupling, while the CH protons of the i-Pr substituents on phosphorus appear as a septet of doublets (δ = 1.75, 2JPH = 1.4 Hz, 3JHH = 7.1 Hz). The 31P{1H} NMR spectrum contains a singlet at δ 6.4 and the characteristic νCN band for the oxazoline ring appears in the IR spectrum at 1658 cm−1 (Table 1). In contrast to ligand 1b, which was obtained as a white powder, [4h] ligand 1a is a pale yellow oil that can be exposed to air for short periods of time, but is best kept under an inert atmosphere. Comparative spectroscopic data are presented in Table 1.
Selected IR and NMR data for the ligands and complexes
| IR | NMRc | |||
| νCN | νCO | 1H | 31P | |
| 1a | 1658a (s) | 6.4 | ||
| 1b | 1660a (s) | −15.8 | ||
| 2a | 1639b (s) | 0.51 Pd–CH3 (d, 3JPH = 2.3) | 57.3 | |
| 2b | 1647a (s) | 0.55 Pd–CH3 (d, 3JPH = 2.7) | 33.1 | |
| 3a | 1642b (m) | 0.51 Pd–CH3 (s) | 63.4 | |
| 3b | 1633b (s) | 0.60 Pd–CH3 (s) | 37.4 | |
| 4a | 1642a (m) | 1697a (m) | 54.4 | |
| 4b | 1644a (s) | 1704a (s) | 22.0 | |
| νCN/CO | νC(O)OMe | |||
| 5a | 1635b (s) | 2.45 C(O)CH3 (s) | 62.3 | |
| 5b | 1634b (s) | 2.45 C(O)CH3 (s) | 34.4 | |
| 7a | 1639b (s) | 1679b (s) | 2.55 C(O)CH3 (s) | 59.5 |
| 7b | 1633b (s) | 1683b (s) | 2.52 C(O)CH3 (s) | 32.8 |
a In CH2Cl2.
b In KBr, cm−1.
c In CDCl3, ppm, J in Hz.
2.2 Synthesis of the complex [PdClMe(P,N)] (2a)
Reaction of 1a with 0.8 equiv of [PdClMe(COD)] (COD = cycloocta-1,5-diene) afforded [PdClMe(P,N)] (P,N = 1a) (2a) in quantitative yield (Scheme 1).

Synthesis of complexes 2a,b and 3a,b. All reactions were performed at room temperature in CH2Cl2.
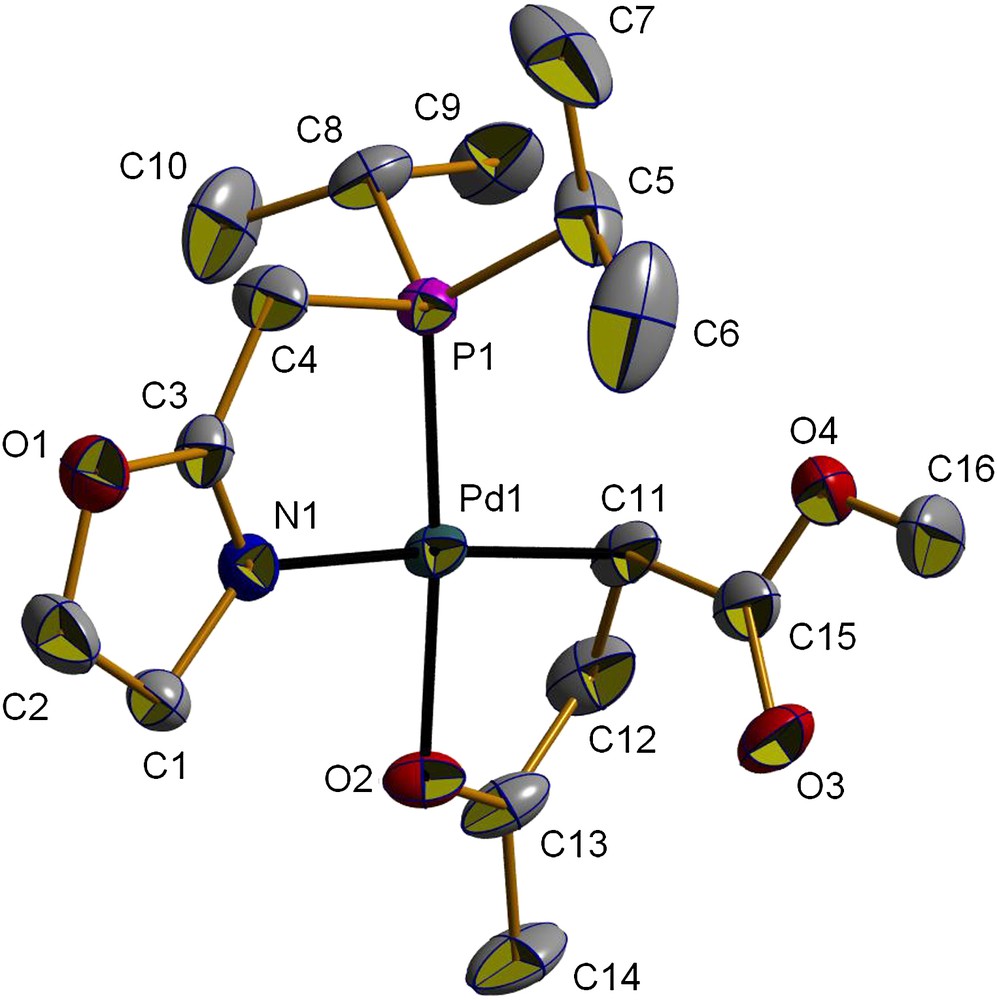
Coordination of the ligand resulted in a shift of the νCN absorption to 1639 cm−1 (Table 1). The presence of a single peak in the 31P{1H} NMR spectrum of 2a suggests the formation of a single isomer. In the 1H NMR spectrum, the Pd–Me resonance of 2a appears as a doublet at δ 0.51, of which the magnitude of the coupling constant (3JPH = 2.3 Hz) indicates a cis relationship between the two groups with the largest trans influence, namely the phosphorus atom and the methyl group. The PCH2 protons appear as a doublet of triplets (δ 2.51, 2JPH = 8.8, 5JHH = 2.0 Hz) owing to a 5JHH coupling between the PCH2 and NCH2 protons. A similar 5JHH coupling has been observed for 2b and related palladium complexes stabilized by the ligand 1b [4b,h]. The CH protons of the i-Pr substituents of the phosphorus appear as a doublet of septets (δ = 2.19, 2JPH = 8.6, 3JHH = 7.1 Hz). Other characterizing data are given in Section 3. Single crystals of 2a suitable for X-ray diffraction were obtained by slow diffusion of hexane into a CH2Cl2 solution of the complex. A view of its molecular structure is shown in Fig. 1 and selected distances and angles are given in Table 2.
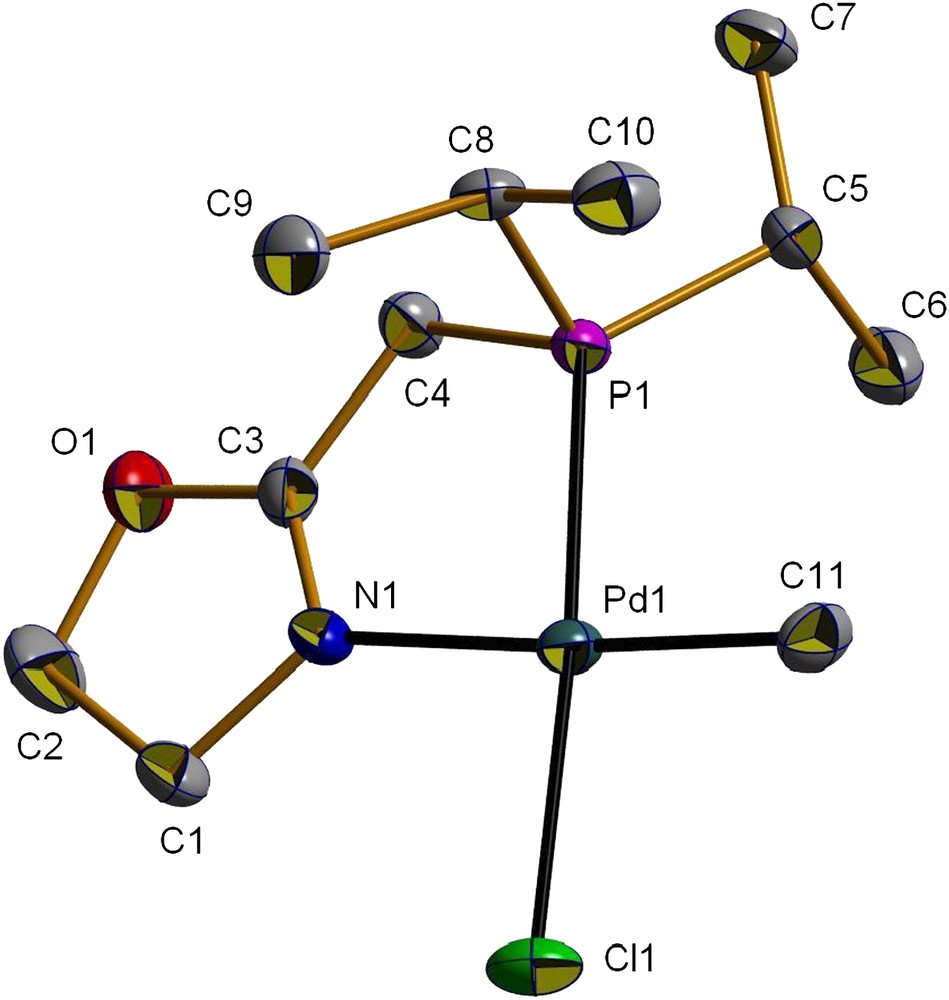
ORTEP view of the structure of complex 2a (H atoms omitted). Displacement ellipsoids are drawn at 50% probability level.
Selected bond lengths [Å] and angles [°] in complexes 2a, 3a, 6 and 7a
| 2a | 3a | 6 | 7a | |
| Pd1–N1 | 2.138(2) | 2.135(2) | 2.008(5) | 2.076(6) |
| Pd1–P1 | 2.2058(7) | 2.1817(7) | 2.2140(14) | 2.222(2) |
| Pd1–C11 | 2.042(3) | 2.031(3) | 2.049(7) | |
| Pd1–O2 | 2.177(2) | 2.112(5) | ||
| Pd1–Cl1 | 2.3724(7) | 2.3220(14) | ||
| Pd1–Cl1iv | 2.4357(13) | |||
| N1–C3 | 1.276(3) | 1.274(4) | 1.284(7) | 1.278(10) |
| C3–C4 | 1.488(4) | 1.488(4) | 1.475(7) | 1.480(12) |
| C4–P1 | 1.852(3) | 1.839(3) | 1.833(5) | 1.845(8) |
| O2–C13 | 1.232(10) | |||
| C13–C12 | 1.506(14) | |||
| C12–C11 | 1.538(13) | |||
| N1–Pd1–P1 | 82.68(6) | 83.63(6) | 83.18(14) | 84.1(2) |
| N1–Pd1–C11 | 175.32(10) | 174.2(1) | 174.7(3) | |
| N1–Pd1–O2 | 96.73(8) | 94.3(2) | ||
| N1–Pd1–Cl1 | 94.19(6) | 176.71(14) | ||
| N1–Pd1–Cl1iv | 94.09(14) | |||
| P1–Pd1–O2 | 177.76(6) | 174.3(2) | ||
| P1–Pd1–C11 | 92.99(8) | 91.55(10) | 100.1(2) | |
| P1-Pd1-Cl1 | 175.80(3) | 93.54(5) | ||
| P1–Pd1–Cl1iv | 176.93(5) | |||
| C11–Pd1–O2 | 88.2(1) | 81.8(3) | ||
| C11–Pd1–Cl1 | 90.22(8) | |||
| Cl1–Pd1–Cl1iv | 89.19(5) | |||
| Pd1–Cl1–Pd1iv | 88.22(5) |
As observed for 2b [4b], the coordination geometry around the palladium centre in 2a is very close to square planar. As expected, the Me group is positioned cis to the phosphorus donor atom, which is consistent with the fact that the groups with the largest trans influence avoid being mutually trans to one another [4b,14,15]. The P,N-chelate bite angle [82.68(6)°] is close to that reported for 2b [82.2(3)°] [4b], and is consistent with that in other five-membered ring complexes of the type [PdClMe(P,N)] [4f,g,15,16].
Although no classical intermolecular hydrogen bonds were detected in 2a, a non-conventional C–H⋯Cl hydrogen interaction is present, which involves the Cl atom bound to palladium and the unique PC–H atom of an isopropyl group of a neighbouring molecule (C8–H⋯Cl1, Table 3). This results in an infinite, one-dimensional wavelike chain structure (Fig. 2). Interactions of this type have been recently reported for related complexes [PdClMe(P,N)] and [PdCl2(P,N)] (P,N = 2-(2,6-dimethylphenyl)-6-(diphenylphosphinomethyl)pyridine) [15].
Hydrogen-bonding parameters (Å, deg) for compounds 2a and 3a
| Complex | D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
| 2a | C8–H⋯Cl1i | 1.00 | 2.77 | 3.610(3) | 142 |
| 3a | C4iii–H⋯O4 | 0.99 | 2.26 | 3.215(4) | 160 |
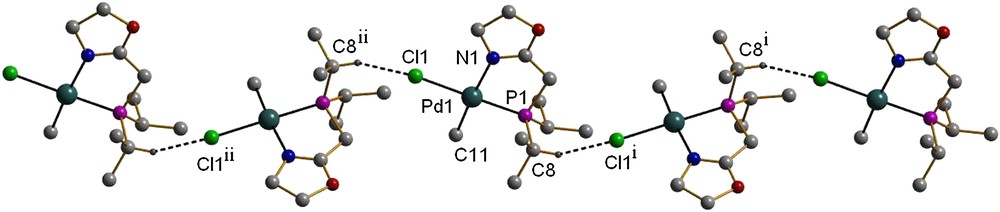
View of the crystal structure of complex 2a showing the PC8–H⋯Cl1 interactions. Symmetry codes: (i) x, 0.5 − y, 0.5 + z; (ii) x, 0.5 − y, −0.5 + z.
2.3 Synthesis of the complex [PdMe(P,N)OTf] (3a)
The complex [PdMe(P,N)OTf] (P,N = 1a, OTf = OSO2CF3) (3a) was prepared similarly to 3b [4h] by treatment of a dichloromethane solution of 2a with AgOTf (Scheme 1). After filtration through Celite to remove the AgCl formed, the solvent was evaporated under reduced pressure and the desired complex was obtained as a solid and washed with diethylether and pentane. Selected IR and NMR data are reported in Table 1. As for 2a, the singlet resonance at δ 0.51 for the Pd–Me protons is characteristic of the coordination geometry in 3a. The absence of 3JPH coupling constant (or too small value to be detected) indicates a cis relationship between the phosphorus atom and the methyl group. There was no spectroscopic indication of a coordinated solvent molecule, consistent with the triflate anion being a better donor than CH2Cl2 and remaining coordinated to the metal in solution. Note, however, that the 19F{1H} NMR signal for 3a in CDCl3 is very similar to that in 5a and 7a. As expected, the formation of cationic Pd(II) complexes is observed in solvents such as MeCN [4h]. When compared to the 31P{1H} NMR data of its less electrophilic but also neutral analogue 2a, the downfield shift observed (Δδ = 6.1 ppm) is larger than when going from 2b to 3b (Δδ = 4.3 ppm) (Table 1).
X-ray quality single crystals of 3a were grown by slow diffusion of hexane into a dichloromethane solution of the complex; its molecular structure is represented in Fig. 3 and selected bond lengths and angles are given in Table 2. It confirmed that the triflate anion is directly bonded to Pd, with a Pd–O2 distance of 2.177(2) Å. The geometry around Pd is square planar, as shown by the values of the P1–Pd1–O2 and N1–Pd1–C11 angles of 177.76(6)° and 174.20(11)°, respectively. The bond lengths and angles involving the P,N chelate are similar to those found in 3b [4b].

ORTEP view of the structure of complex 3a (H atoms omitted). Displacement ellipsoids are drawn at the 50% probability level.
Examination of the crystal packing of complex 3a reveals an intermolecular C4–H⋯O4 hydrogen-bonding interaction involving a triflate oxygen and a PC4–H hydrogen atom of an adjacent molecule [C4⋯O4 = 3.215 (4) Å, Table 3], which leads to a one-dimensional chain arrangement in the solid state (Fig. 4).

View of the crystal structure of complex 3a showing the C4iii–H⋯O4 interaction. Only one of the H atoms at C4 is shown. Symmetry code: (iii) x, 1 + y, z.
2.4 CO/ethylene or CO/methyl acrylate insertion reactions
The reaction of complex 3a with CO in CH2Cl2 at room temperature was monitored by 31P{1H} NMR spectroscopy. CO insertion into its palladium methyl bond produced in less than 1 h the acyl derivative [Pd{C(O)Me}(P,N)(OTf)] (P,N = 1a) (4a) (Scheme 2), which was only observed in solution and characterized by the large high-field shift of its 31P{1H} NMR resonance (Δδ = −9.0, Table 1) and by infrared spectroscopy. The IR absorption band due to the CO stretching appears at 1697 cm−1, which is typical for such acyl complexes [4b,e,g]. Previous efforts to isolate the analogous complex 4b have not been successful because of the instability of the complex, and no satisfactory elemental analyses could be obtained [4h]. Therefore, no attempt was made to isolate 4a. In a previous study with ligands 1b and 1c, we have demonstrated the existence of a temperature-dependent equilibrium between the triflate, acyl complex, [Pd{C(O)Me}(P,N)OTf] (P,N = 1c), and a cationic carbonyl, acyl Pd(II) complex [Pd{C(O)Me}(P,N)(CO)]OTf [4b], and a similar equilibrium is expected to occur in the case of 4a, i.e. at room temperature the triflate anion coordinates to the palladium centre.
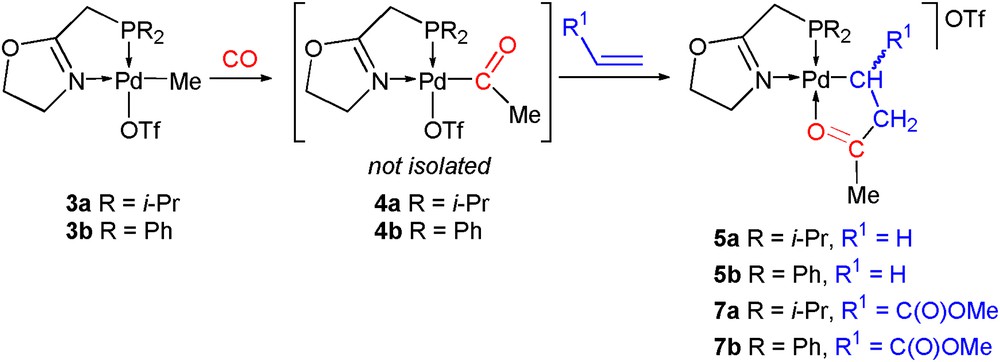
All reactions were performed at room temperature in CH2Cl2.
As previously observed for 4b [4b], ethylene insertion into the palladium–acyl bond of 4a to the five-membered C,O chelate complex (5a) was completed in less than 1 h (31P{1H} monitoring). The IR spectrum shows only one absorption band at 1635 cm−1, corresponding to the CN and CO stretching vibrations (νCN/CO, Table 1), which represents a shift for the CO band of 62 cm−1 to a lower wave number with respect to that for 4a. The 31P{1H} NMR spectrum of 5a contains a singlet at δ 62.3 which is shifted to low field relative to that of 4a (δ 54.4, Table 1). In the 1H NMR spectrum of 5a, the Pd–CH2 protons give rise to a well-resolved triplet of doublets (δ 1.65, 3JHH = 6.2, 3JPH = 1.6 Hz), whereas the CH2CO protons appear as a broad triplet (δ 3.09, 3JHH = 6.2 Hz), indicating a smaller JPH coupling constant.
Attempts to crystallize 5a by layering a CH2Cl2 solution with hexane were unsuccessful. Nevertheless, this procedure afforded a crystalline material suitable for X-ray crystallography, which subsequently revealed to be the dinuclear palladium complex trans-[Pd(μ-Cl){(i-Pr)2PCH2(oxazoline)}]2(OTf)2 (6) containing a di-μ-chloro bridge and two equivalent five-membered P,N-chelate rings. A view of the centrosymmetric dinuclear cationic complex in 6 is shown in Fig. 5, and selected bond distances and angles are given in Table 2. According to the Cambridge Structure Database, 6 is the first dinuclear complex of the type [Pd(μ-Cl)(P,N)]2, and only the second of the type [Pd(μ-X)(P,N)]2 (X = halide) to be characterized by X-ray crystallography [16]. The coordination geometry around the palladium atoms in 6 is close to square planar with the nitrogen donor atoms in transoid relationship with respect to the Pd–Pd axis, and is essentially similar to those of cyclopalladated species containing a five-membered chelate ring [16,17]. The coordination planes (mean planes passing through Cl1, Cl1iv, N1, P1 and through Cl1, Cl1iv, N1iv, P1iv) form an angle of 25.47(1)° and the distance between two palladium atoms is 3.3126(5) Å, which excludes the presence of metal–metal bonding. The length of the Pd–Cl bond trans to P [2.4357(13) Å] is longer than that trans to N [2.3220(14) Å], which parallels the different trans influences exerted by the phosphorus and the nitrogen atoms [14,15,18].

View of the structure of the dicationic complex in 6 (H atoms and triflate anions omitted). Displacement ellipsoids are drawn at 50% probability level. Symmetry code: (iv) 1 − x, y, 3/2 − z.
In order to determine the fate of the growing polymer chain upon formation of 6, the 13CO-labeled isotopomer of 5a was prepared by bubbling 13CO into a solution of [PdMe(P,N)OTf] (3a), which was subsequently treated with ethylene according to the described procedure. In the 1H NMR spectrum of the 13CO-labeled isotopomer of 5a, the methyl group [13C(O)CH3] appears as a doublet due to coupling of the protons with the 13C of the labeled 13CO, with a coupling constant (2JHC = 5.8 Hz) which is in agreement with values reported in a related study with 13CO and propene [19]. The 13C{1H} NMR spectrum displays 1JCC of 40.3 and 40.1 Hz for the [13C(O)CH3] and [CH213C(O)CH3], respectively, in agreement with values reported for a 13CO-labeled isotopomer of a CO/methyl acrylate insertion product [8].
Complex 5a is stable in the solid state for several weeks; however, it decomposes progressively in solution by β-hydrogen elimination. Keeping a CDCl3 solution of the 13CO-labeled isotopomer of 5a in a NMR tube over a period of 10 days at room temperature affords a few crystals of complex 6, which deposit along with some palladium metal. The 31P{1H} NMR spectrum of the remaining solution shows only one singlet at δ 56.4, indicating that the only P-containing species in solution is the dinuclear complex 6. In the 13C{1H} NMR spectrum, the disappearance of the signal for the 13CO-labeled 5a is accompanied by the appearance of a new singlet at δ 199.0 corresponding to methyl vinyl ketone resulting from β-hydrogen elimination from complex 5a (Scheme 3).
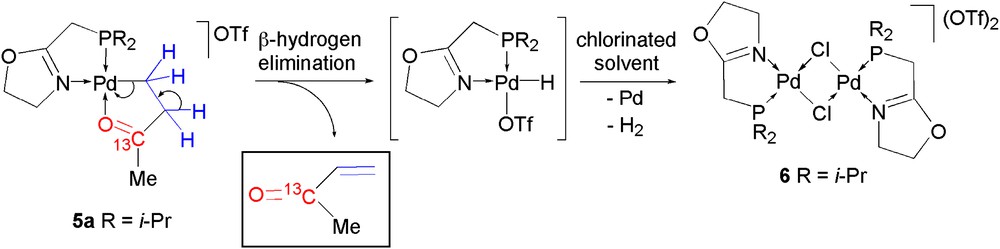
β-hydrogen elimination from complex 5a with formation of methylvinylketone and the dinuclear complex 6.
Further confirmation came from GC–MS analysis of the solution after trap-to-trap distillation. The fragmentation pattern found for Me13C(O)CHCH2 corresponds perfectly, plus one unit, to that for MeC(O)CHCH2.
Palladium hydride complexes resulting from CO–ethylene copolymerization termination reactions usually decompose into dicationic complexes of the type [Pd(L)2] (L = bidentate ligand) [4e,20]. However, Consiglio recently reported the structure of a dinuclear complex of the type [Pd(μ-Cl)(L)]2(OTf)2 (L = diphosphine ligand) obtained by replacement of a CH3 group with a chloride from CHCl3 [21].
The insertion of methyl acrylate into the palladium acyl bond of 4a was regioselective (2,1-insertion) and afforded (7a) within 1 h (31P{1H} monitoring). The low-field shift in the 31P{1H} resonance of 7a relative to that of 4a (Δδ = 5.1, Table 1) is smaller than when considering 7b and 4b (Δδ = 10.8, Table 1) [4b]. The 1H NMR spectrum of 7a contains methyl signals for the inserted methyl acrylate at δ = 2.55 [C(O)Me] and 3.63 [C(O)OMe], the CH and CH2 protons were identified by means of 2D (COSY and HSQC) NMR experiments and resonate at δ = 2.55 and 3.07, respectively (see Section 3). The latter resonance appears, surprisingly, as a slightly broadened singlet. Complex 7a is stable in the solid state for several weeks, and in contrast to 5a, no significant decomposition is observed in a CDCl3 solution over a period of 10 days.
The solid-state structure of 7a was unambiguously established by single-crystal X-ray diffraction (Fig. 6) and it represents, to the best of our knowledge, only the fourth methyl acrylate–acyl coupling product to be structurally characterized [4b,g]. This structural determination confirms the formation of a five-membered (C,O) chelated product, which makes β-hydrogen elimination more difficult [4b,22]. The Pd–C distance in 7a of 2.049(7) Å (Table 2) is close that in 7b [2.046(4) Å] [4b], the most significant difference between these two structures being the value of the Pd1–P1 bond length, which is longer in 7a than in 7b by about 0.02 Å. Although it has not yet been possible to insert a CO molecule into the Pd–C11 bond of 7a or 7b (1 atm, 25 °C), our results provide further insight into the comparative reactivity of ethylene and methylacrylate into a Pd–carbon bond, and the stability of the corresponding insertion products. Although (partial) dissociation in solution of the triflate ligand of 3a and 4a cannot be completely ruled out, which would render the complexes more electrophilic (see above), we assume that it remains coordinated to the Pd(II) centre. This obviously does not prevent insertion reactions to take place because the triflate ligand can be displaced by CO or the olefin. In all cases, ethylene or methylacrylate insertion occurred into the Pd–C(O)Me and not into the Pd–Me bond. The increased basicity of the P(i-Pr)2 group compared to PPh2 did not translate into significant reactivity differences [4a,b].
View of the structure of the cation in 7a (H atoms omitted). Displacement ellipsoids are drawn at 50% probability level.
3 Experimental section
The 1H, 13C{1H}, 31P{1H} and 19F{1H} NMR spectra were recorded at 300.13, 75.48, 121.49 and 282.38 MHz, respectively, on FT Bruker AC300, Avance 300, unless otherwise stated. IR spectra in the range 4000–400 cm−1 were recorded on a Bruker IFS66FT and a Perkin Elmer 1600 Series FTIR. Elemental analyses were preformed by the ‘Service de microanalyse, université Louis-Pasteur (Strasbourg, France)’. All reactions were carried out under purified N2, using Schlenk techniques, and the solvents were freshly distilled under nitrogen prior to use. [PdClMe(COD)] (COD = 1,5-cyclooctadiene, C8H12) was prepared according to literature procedures [23].
3.1 Preparation and spectroscopic data for 1a
To a THF solution (75 mL) of 2-methyl-2-oxazoline (0.5 mL, 5.87 mmol) in a 250-mL flask at −78 °C, was added dropwise a solution of n-butyllithium in hexane (3.67 mL, 1.6 M, 5.87 mmol). The mixture was stirred for 1 h and degassed SiClMe3 (0.75 mL, 5.87 mmol) was added. The mixture was further stirred for 1 h at −78 °C and P(i-Pr)2Cl (0.95 mL, 5.87 mmol) was added. The solution was stirred until it reached room temperature. After evaporation of the solvent under reduced pressure, the oily residue was dissolved in toluene (60 mL) and the solution was filtered through Celite. After evaporation of the toluene under vacuum, 1a was obtained as a pale yellow oil (1.12 g, 95%). IR (CH2Cl2): 1658 (s, νCN) cm−1. 1H NMR (300.13 MHz, CDCl3, room temp.): δ 1.02 [d, 3JHH = 7.1 Hz, 6H, (CH(CH3)(CH3))2], 1.06 [dd, 3JPH = 2.3 Hz, 3JHH = 7.1 Hz, 6H, (CH(CH3)(CH3))2], 1.75 [septd, 2JPH = 1.4 Hz, 3JHH = 7.1 Hz, 2H, (CH(CH3)2)2], 2.35 (br d, 2JPH = 1.0 Hz, 2H, PCH2), 3.75 (br t, 3JHH = 9.3 Hz, 2H, NCH2), 4.18 (t, 3JHH = 9.3 Hz, 2H, OCH2). 13C{1H} (75.48 MHz, CDCl3, room temp.): δ 18.6 [d, 2JPC = 10.1 Hz, (CH(CH3)(CH3))2], 19.6 [d, 2JPC = 15.8 Hz, (CH(CH3)(CH3))2], 21.4 (d, 1JPC = 25.6 Hz, PCH2), 23.6 [d, 1JPC = 13.9 Hz, (CH(CH3)2)2], 54.5 (s, NCH2), 67.5 (s, OCH2), 167.3 (d, 2JPC = 7.5 Hz, CN). 31P{1H} NMR (121.49 MHz, CDCl3, room temp.): δ 6.4 (s).
3.2 Preparation and spectroscopic data for [PdClMe(P,N)] (2a)
To a solution of ligand 1a (0.48 g, 2.38 mmol) in CH2Cl2 (20 mL) was added solid [PdClMe(COD)] (0.50 g, 1.90 mmol, 0.8 equiv) at room temperature and the resulting mixture was stirred overnight. The solvent was then evaporated under reduced pressure. The resulting white residue was washed with diethylether (10 mL) and pentane (2 × 10 mL) and dried under vacuum to give a white powder (0.61 g, 89%). IR (KBr): 1639 (s, νCN) cm−1. 1H NMR (300.13 MHz, CDCl3, room temp.): δ 0.51 (d, 3JPH = 2.3 Hz, 3H, PdCH3), 1.20 [dd, 3JPH = 16.2 Hz, 3JHH = 7.1 Hz, 6H, (CH(CH3)(CH3))2], 1.26 [dd, 3JPH = 19.0 Hz, 3JHH = 7.1 Hz, 6H, (CH(CH3)(CH3))2], 2.19 [dsept, 2JPH = 8.6 Hz, 3JHH = 7.1 Hz, 2H, (CH(CH3)2)2], 2.51 (dt, 2JPH = 8.8 Hz, 5JHH = 2.0 Hz, 2H, PCH2), 3.96 (tt, 3JHH = 9.7 Hz, 5JHH = 2.0 Hz, 2H, NCH2), 4.54 (t, 3JHH = 9.7 Hz, 2H, OCH2). 13C{1H} (75.48 MHz, CDCl3, room temp.): δ −10.9 (d, 2JPC = 2.9 Hz, PdCH3), 17.6 [s, (CH(CH3)(CH3))2], 18.3 [d, 2JPC = 4.6 Hz, (CH(CH3)(CH3))2], 21.5 (d, 1JPC = 23.6 Hz, PCH2), 23.9 [d, 1JPC = 26.0 Hz, (CH(CH3)2)2], 52.0 (s, NCH2), 72.0 (s, OCH2), 171.8 (d, 2JPC = 15.4 Hz, CN). 31P{1H} NMR (121.49 MHz, CDCl3, room temp.): δ 57.3 (s). Anal. Calcd for C11H23ClNOPPd: C, 36.89; H, 6.47; N, 3.91. Found: C, 36.84; H, 6.30; N, 3.72.
3.3 Preparation and spectroscopic data for [PdMe(P,N)OTf] (3a)
To a solution of complex 2a (0.51 g, 1.42 mmol) in CH2Cl2 (20 mL) was added AgOTf (0.44 g, 1.71 mmol, 1.2 equiv). The reaction mixture was protected from room light with an aluminum foil and stirred for 2 h at room temperature. The solution was then filtered through dry Celite and the solvent was evaporated under reduced pressure. The residue was washed with diethylether (10 mL), pentane (2 × 10 mL) and dried under vacuum overnight. Complex 3a was obtained as a light beige powder (0.65 g, 97%). IR (KBr): 1642 (m, νCN) cm−1. 1H NMR (300.13 MHz, CDCl3, room temp.): δ 0.51 (s, 3H, PdCH3), 1.23 [dd, 3JPH = 16.2 Hz, 3JHH = 7.0 Hz, 6H, (CH(CH3)(CH3))2], 1.29 [dd, 3JPH = 18.6 Hz, 3JHH = 7.0 Hz, 6H, (CH(CH3)(CH3))2], 2.20 [dsept, 2JPH = 8.8 Hz, 3JHH = 7.0 Hz, 4H, (CH(CH3)2)2], 2.58 (dt, 2JPH = 9.1 Hz, 5JHH = 2.0 Hz, 2H, PCH2), 4.00 (tt, 3JHH = 9.7 Hz, 5JHH = 2.0 Hz, 2H, NCH2), 4.57 (t, 3JHH = 9.7 Hz, 2H, OCH2). 13C{1H} (75.48 MHz, CDCl3, room temp.): δ −7.4 (d, 2JPC = 3.0 Hz, PdCH3), 17.6 [s, (CH(CH3)(CH3))2], 18.3 [d, 2JPC = 3.4 Hz, (CH(CH3)(CH3))2], 21.8 (d, 1JPC = 27.0 Hz, PCH2), 24.4 [d, 1JPC = 29.3 Hz, (CH(CH3)2)2], 52.3 (s, NCH2), 72.1 (s, OCH2), 171.5 (d, 2JPC = 12.8 Hz, CN). 31P{1H} NMR (121.49 MHz, CDCl3, room temp.): δ 63.4 (s). 19F{1H} NMR (282.4 MHz, CDCl3): δ −78.1 (s). Anal. Calcd for C12H23F3NO4PPdS: C, 30.55; H, 4.91; N, 2.97. Found: C, 30.30; H, 4.67; N, 2.74.
3.4 Preparation and spectroscopic data for (5a)
A solution of 3a (0.17 g, 0.36 mmol) in CH2Cl2 (25 mL) was stirred under 1 atm CO at room temperature for 1 h, the CO was then replaced by 1 atm ethylene and the solution was further stirred for 1 h. The workup was as described for 2a, and afforded 5a as an orange powder (0.13 g, 68% yield). IR (KBr): 1635 (m, νCN and νCO) cm−1. 1H NMR (300.13 MHz, CDCl3, room temp.): δ 1.25 [dd, 3JPH = 16.6 Hz, 3JHH = 7.0 Hz, 6H, (CH(CH3)(CH3))2], 1.27 [dd, 3JPH = 18.9 Hz, 3JHH = 7.0 Hz, 6H, (CH(CH3)(CH3))2], 1.65 (td, 3JHH = 6.2, 3JPH = 1.6 Hz, 2H, PdCH2), 2.23 [dsept, 2JPH = 8.8 Hz, 3JHH = 7.0 Hz, 2H, (CH(CH3)2)2], 2.45 [s, 3H, C(O)CH3], 2.85 (dt, 2JPH = 9.4 Hz, 5JHH = 1.9 Hz, 2H, PCH2), 3.09 (br t, 3JHH = 6.2 Hz, 2H, PdCH2CH2), 3.98 (tt, 3JHH = 9.7 Hz, 5JHH = 1.9 Hz, 2H, NCH2), 4.72 (t, 3JHH = 9.7 Hz, 2H, OCH2). 13C{1H} (75.48 MHz, CDCl3, room temp.): δ 11.9 (s, PdCH2), 17.7 [s, (CH(CH3)(CH3))2], 18.3 [d, 2JPC = 3.1 Hz, (CH(CH3)(CH3))2], 21.3 (d, 1JPC = 28.3 Hz, PCH2), 24.5 [d, 1JPC = 28.7 Hz, (CH(CH3)2)2], 27.9 [d, 4+5JPC = 1.4 Hz, CH2C(O)CH3], 50.7 (s, PdCH2CH2), 52.3 (s, NCH2), 72.2 (s, OCH2), 173.9 (d, 2JPC = 13.1 Hz, CN), 233.7 [d, 3+4JPC = 0.7 Hz, CH2C(O)CH3]. 31P{1H} NMR (121.49 MHz, CDCl3, room temp.): δ 62.3 (s). 19F{1H} NMR (282.4 MHz, CDCl3): δ −78.6 (s). Anal. Calcd for C15H27F3NO5PPdS: C, 34.13; H, 5.16; N, 2.65. Found: C, 34.04; H, 4.93; N, 2.41.
3.5 Preparation and spectroscopic data for (7a)
A solution of 3b (0.15 g, 0.32 mmol) in CH2Cl2 (20 mL) was stirred under 1 atm CO at room temperature for 1 h, after this period the CO atmosphere was replaced with nitrogen and methyl acrylate (29 μL, 1 equiv) was added and the solution was further stirred for 1 h. The workup was as described for 2a, and afforded 7a as a beige powder (0.16 g, 85% yield). IR (KBr): 1639 (m, νCN and νCO), 1679 (m, νCO) cm−1. 1H NMR (400.13 MHz, CDCl3, room temp.): 2D (COSY and HSQC) and 31P decoupled NMR experiments were used to determine chemical shifts and coupling constants; δ 1.18–1.47 [complex m, 12H, (CH(CH3)2)2], 2.36 [dsept, 2JPH = 1.8 Hz, 3JHH = 7.0 Hz, (CH(CH3)2)(CH(CH3)2)], 2.55 [s, 3H, C(O)CH3], 2.57 [sept, 3JHH = 7.1 Hz, 1H, (CH(CH3)2)(CH(CH3)2), overlapping with the C(O)CH3 signal], ABMNX spin system (A = B = M = N = H, X = P, δA 2.88, δB 3.07, 2JAB = 18.8, 2JAX = 9.5, 2JBX = 9.4, 5JAH = 1.6, 5JBH = 1.9 Hz, 2H, PCH2), 3.07 [appearance of br s, 2H, CH2C(O)CH3, overlapping with PCH2 signal], 3.63 [s, 3H, C(O)OCH3], 3.92 (m, 1H, NCHH), 4.05 (m, 1H, NCHH), ABMN spin system (A = B = M = N = H, δA 4.70, δB 4.81, 2JAB = 10.7, 3JAH = 8.4, 3JBH = 8.7 Hz, 2H, OCH2), although it was not possible to clearly observe the signal of the PdCH proton, COSY and HSQC experiments indicate that it resonates at 2.55 ppm, thus overlapping with the signals for the C(O)CH3 and (CH(CH3)2)(CH(CH3)2) protons. 13C{1H} (75.48 MHz, CDCl3, room temp.): δ 16.2 [d, 2JPC = 5.6 Hz, CH(CH3)], 17.7 [d, 2JPC = 1.0 Hz, CH(CH3)], 18.8 [d, 2JPC = 2.3 Hz, CH(CH3)], 18.9 [s, CH(CH3)], 21.2 (d, 1JPC = 29.9 Hz, PCH2), 23.1 [d, 1JPC = 26.4 Hz, (CH(CH3)2)(CH(CH3)2)], 25.5 [d, 1JPC = 29.2 Hz, (CH(CH3)2)(CH(CH3)2)], 27.0 (d, 2JPC = 2.3 Hz, PdCH), 28.2 [d, 4+5JPC = 2.4 Hz, CH2C(O)CH3], 50.7 [s, CH2C(O)CH3], 51.6 [s, C(O)OCH3], 51.9 (s, NCH2), 72.6 (s, OCH2), 174.8 (d, 2JPC = 13.8 Hz, CN), 177.7 [s, C(O)OCH3], 233.2 [d, 3+4JPC = 2.2 Hz, CH2C(O)CH3]. 31P{1H} NMR (121.49 MHz, CDCl3, room temp.): δ 59.5 (s). 19F{1H} NMR (282.4 MHz, CDCl3): δ −78.6 (s). Anal. Calcd for C17H29F3NO7PPdS: C, 34.85; H, 4.99; N, 2.39. Found: C, 34.59; H, 4.83; N, 2.14.
3.6 Crystal structure determinations
Crystals of 2a, 3a and 7a suitable for an X-ray diffraction study were obtained by slow diffusion of hexane into a CH2Cl2 solution of the respective complex at 5 °C. Crystals of 6 were obtained by slow diffusion of hexane into a CH2Cl2 solution of complex 5a at 5 °C or, alternatively, by slow evaporation of a CDCl3 solution at room temperature. Diffraction data were collected on a Kappa CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) (Table 4). Data were collected using phi-scans and the structures were solved by direct methods using the SHELXL 97 software [24,25], and the refinement was performed by full-matrix least squares on F2. No absorption correction was used. All non-hydrogen atoms were refined anisotropically with H atoms introduced as fixed contributors (dC–H = 0.95 Å, U11 = 0.04).
X-ray diffraction data for the structural determination of complexes 2a, 3a, 6 and 7a
| 2a | 3a | 6 | 7a | |
| Formula | C11H23ClNOPPd | C12H23NOPPd·CF3SO3 | C20H40Cl2N2O2P2Pd2·2(CF3SO3) | C16H29NO4PPd·CF3SO3 |
| Mr | 358.12 | 471.74 | 984.32 | 585.84 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P1¯ | C2/c | P21/a |
| a [Å] | 8.3170(2) | 8.3800(2) | 24.6530(7) | 9.4210(2) |
| b [Å] | 10.6290(3) | 10.2020(3) | 8.6270(3) | 25.2850(4) |
| c [Å] | 16.6060(4) | 11.3650(4) | 18.9330(7) | 9.9140(2) |
| α [°] | 86.1340(8) | |||
| β [°] | 95.509(2) | 80.6550(8) | 116.4510(14) | 96.68(5) |
| γ [°] | 73.1650(17) | |||
| V [Å3] | 1461.21 | 917.43(5) | 3605.2(2) | 2345.6(3) |
| Z | 4 | 2 | 4 | 4 |
| Dcalc [kg m−3] | 1.628 | 1.708 | 1.814 | 1.659 |
| μ [mm−1] | 1.544 | 1.255 | 1.425 | 1.009 |
| T [K] | 173(2) | 173(2) | 173(2) | 173(2) |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| θmax [°] | 30.03 | 30.06 | 29.14 | 30.01 |
| Data set [h, k, l] | −11/11, −14/14, −23/23 | −11/11, −11/14, −14/16 | −33/33, −10/11, −25/25 | −11/13, −35/32, −13/13 |
| Tot., unique data, R(int) | 7268, 4255, 0.0305 | 7018, 5346, 0.0218 | 7943, 4829, 0.0374 | 19315, 6815, 0.0672 |
| Observed data [I > 2σ(I)] | 3151 | 4298 | 3192 | 5607 |
| No. reflns, No. params | 4255, 145 | 5346, 208 | 4829, 202 | 6815, 250 |
| R1, wR2, GOF | 0.0373, 0.0921, 1.058 | 0.0382, 0.0918, 1.076 | 0.0609, 0.1671, 1.068 | 0.0992, 0.2525, 1.128 |
The crystallographic material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material CCDC 630553-630556 and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number). See http://www.ccdc.cam.ac.uk/deposit for crystallographic files in .cif format.
Acknowledgements
We are grateful to Prof. R. Welter and Dr. A. DeCian (ULP Strasbourg) for the crystal structure determinations. This work was supported by the European Commission (Palladium Network HPRN-CT-2002-00196, PhD grant to MA), the ‘Centre national de la recherche scientifique’ (France) and the French ‘Ministère de la Recherche’.