

1 Introduction
Recent challenges in highly acidic nuclear waste partitioning involve the separation of minor actinides such as americium and curium from long lived fission products.[1–4] Various solutions have been proposed [4–9], but current protocols still lack the ability to effectively separate trivalent actinides from chemically and physically similar trivalent lanthanides in concentrated nitric acid solution.
Under certain conditions, ligands containing soft sulfur or nitrogen donors will exhibit selectivity for Am(III) over Ln(III) [4,10–14]. For instance, a synergistic mixture of di-2-ethylhexyl dithiophosphoric acid (HDEHDTP) (Fig. 1) and tributylphosphate (TBP) has a separation factor (SF = DAm/DEu) of 60 for the partition of Am(III) over Eu(III) [10]. Work done by Zhu et al. have demonstrated that purified Cyanex 301 [bis(2,4,4-trimethylpentyl)dithiophosphinic acid, HBTMPDTP] was able to separate Am(III) from Eu(III) even more efficiently than the HDEHDTP/TBP mixture, with the separation factor of 5900 [15,16]. Unfortunately these acidic organophosphorus reagents will only differentiate between Am(III) and lanthanides in solutions less acidic than pH of 3 (pKa of Cyanex 301 is 2.6) [16]. Since the acidity of waste solutions in nuclear preprocessing is typically in the range of 1–6 M HNO3, there must be a significant adjustment in the acidity of the waste solution for these sulfur reagents to be effectively utilized, and this step significantly complicates the partition process.
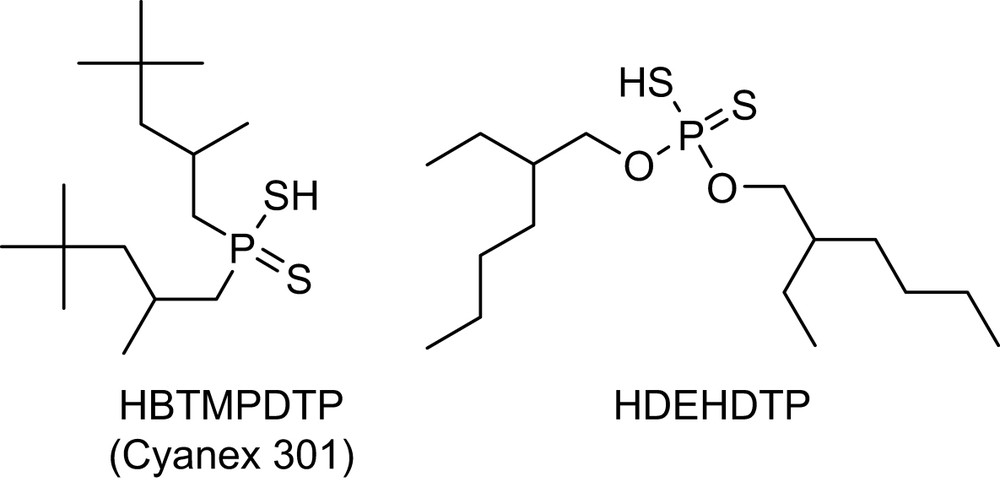
Examples of sulfur-based extractants.
To date, no effective extraction system has been developed to separate trivalent lanthanide and actinides under highly acidic conditions, and current protocols suffer from major shortcomings. Most methods are limited by either low extent of ligand dissociation in highly acidic medium, low efficiency in terms of values of distribution coefficients, and/or poor hydrolytic and radiological stability [17–19]. These problems significantly limit the application of presented protocols such as Cyanex 301, HDEHDTP/TBP or aromatic dithiophosphinic acids [8,20] in large scale nuclear waste clean-up and reprocessing operations.
With our recent success in selectively binding actinides with C3-symmetric ligands containing three carbamoylmethylphosphine oxide (CMPO) arms [21], the ligand syntheses were modified to incorporate softer phosphine sulfide donors in place of the phosphine oxide groups to form carbamoylmethylphosphine sulfide (CMPS) binding groups. In our previous work with triphenoxymethane ligand systems, this base has been shown to favor a conformation with the three phenolic oxygen atoms orientating themselves in an “all up” conformation relative to the central methine hydrogen both in solid state and in solution.[22] Tethering three CMPS moieties to this platform via these phenol oxygens satisfies the requirement for proximate metal binding of the CMPS groups. With three CMPS donor arms, these ligands will present the metal center with a much softer set of ligand donors (three carbonyl oxygens and three phosphine sulfides) in comparison to the tris-CMPO ligand, and unlike the dithiophosphinic acid groups described above, the ligand should be stable at high acid concentrations. Herein, we describe the synthesis, characterization and f-element binding properties of a new class of tris-CMPS ligands.
2 Results and discussion
2.1 Synthesis and extraction data
The synthetic methodology to obtain tris-CMPS compounds follows procedures described for synthesis of tris-carbamoylmethylphosphine oxide (tris-CMPO) derivatives (Fig. 2) [21–25], with p-nitrophenyl (diphenylphosphoryl) acetate replaced by (diphenyl-phosphinothioyl)-acetic acid (5) as presented in Fig. 3.
Tris-carbamoylmethylphosphine oxide (tris-CMPO).
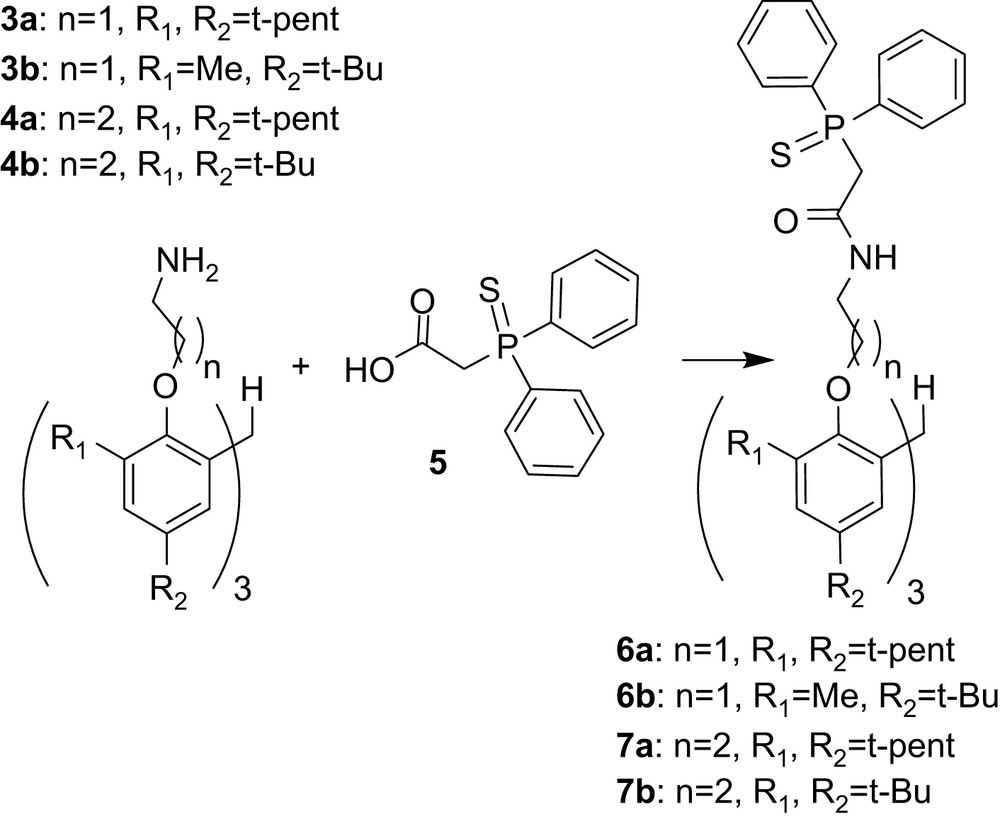
Synthetic protocol for the synthesis of tris-CMPS extractants.
The tris-CMPO compounds showed very good selectivity for tetravalent actinides and lacked the ability to efficiently bind Ln(III) and An(III) [21,26]. It was anticipated that the much softer character of the basic phosphine sulfide groups in the new extractant (tris-CMPS) would induce slightly stronger attraction of the ligand for trivalent actinides in comparison to lanthanides; thus possibly affording some discrimination between these two groups of elements in liquid–liquid extraction experiments. To compare the performance of both types of ligands, preliminary extraction experiments were performed on a series of trivalent lanthanides and tetravalent thorium.
In the first experiment, the binding properties of CMPO, 1, and CMPS, 6a, derivatives with a shorter (two carbon) spacer between the triphenoxymethane base and binding units were compared (Fig. 4). The results revealed very low extraction efficiency of 6a for all studied cations. In contrast to the harder phosphine oxide, the phosphine sulfide compound was not able to take advantage of the difference in the oxidation states of f-element cations. The soft, neutral phosphine sulfide groups are incompatible with hard acids such as tetravalent thorium. In order to test the influence of the flexibility of the ligating CMPS arm on the extraction pattern, derivative 7a, was prepared. It contained an additional carbon atom in each of the arms linking the CMPS donors to the triphenoxymethane platform. The increase in the length of the arm in 7a had no apparent influence on the extraction event.
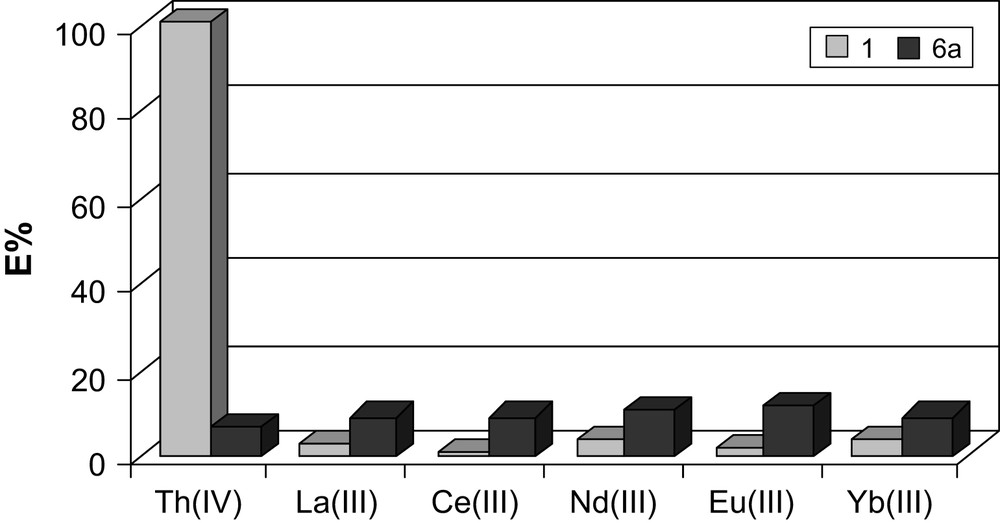
Comparison of metal binding by ligands 1 and 6a expressed by extraction percentage (%E). Aqueous phase: 10−4 M metal nitrate in 1 M HNO3, organic phase: 10−3 M of ligand in methylene chloride.
Compounds 2 and 7a differ only in the identity of the atom attached to the phosphine group (Fig. 5). The cavity size in both extractants should be quite similar and upon expansion of the length of the arms, much like 2, 7a was expected to be able to slightly better adopt the geometry required by the metal ion, and show some binding improvement. Also, the extraction experiments performed on 241Am(III) and 152Eu(III) presented the tris-CMPS as ineffective extractants, and the expected selectivity for americium over europium was not observed [25]. For 7a, the sulfur atoms do not appear to participate in binding to the hard f-element and 7a exhibited very low affinity for all test trivalent lanthanide ions (Table 1).
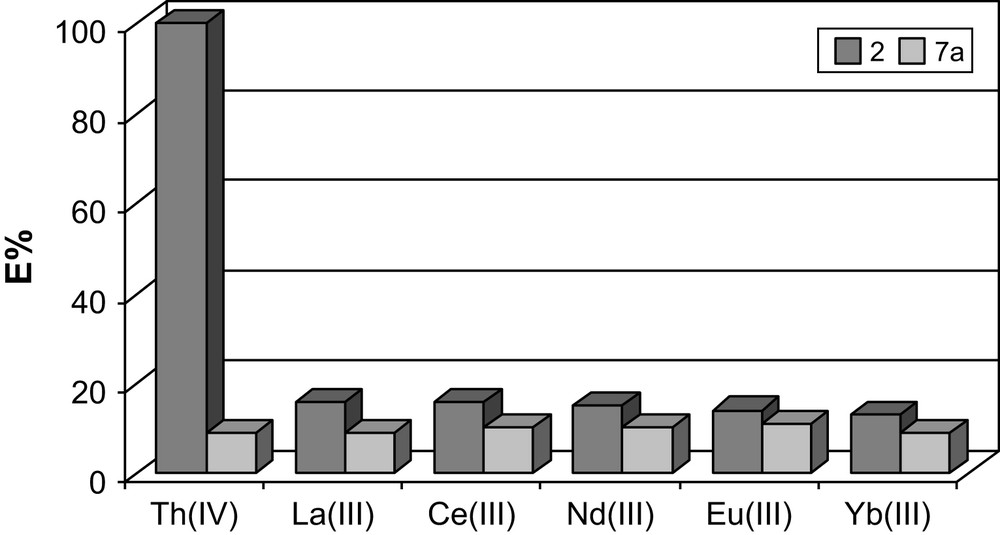
Comparison of metal binding by ligands 2 and 7a.
Extraction percentage (%E) for ligands: 1, 2, 6a and 7a
| Ligand | 1 | 2 | 6a | 7a |
| Th (IV) | 100 | 100 | 7 | 9 |
| La (III) | 3 | 16 | 9 | 9 |
| Ce (III) | 1 | 16 | 9 | 10 |
| Nd (III) | 5 | 15 | 10 | 10 |
| Eu (III) | 2 | 14 | 11 | 11 |
| Yb (III) | 4 | 13 | 9 | 9 |
2.2 Crystal structure analysis
Single crystals of ligand 7a·CH2Cl2·Et2O were grown by slow diffusion of ether into the concentrated solution of ligand in dichloromethane. In the crystal structure of 7a presented in Fig. 6, the average length of the carbonyl bonds is similar to the length of carbonyl bonds in the CMPO equivalent 2 [1.225(7) and 1.232(4) Å, respectively]. The average distances between the sulfur and phosphorus in phosphine sulfide moieties (1.952(2) Å) are in the range of typical PS bonds with phenyl substituents on the phosphorus (Ph3PS, PS: 1.951(2)–1.954(4) Å) [27–29], but almost 0.5 Å longer than the distance between phosphorus and oxygen in the phosphine oxide 2 (1.477(3) Å). The P–C(Ph) mean bond length 1.810(6) Å is also similar to the distances found in Ph3PS (1.817(7) Å), as well as to those found in the tris-CMPO compound (1.798(4) Å).
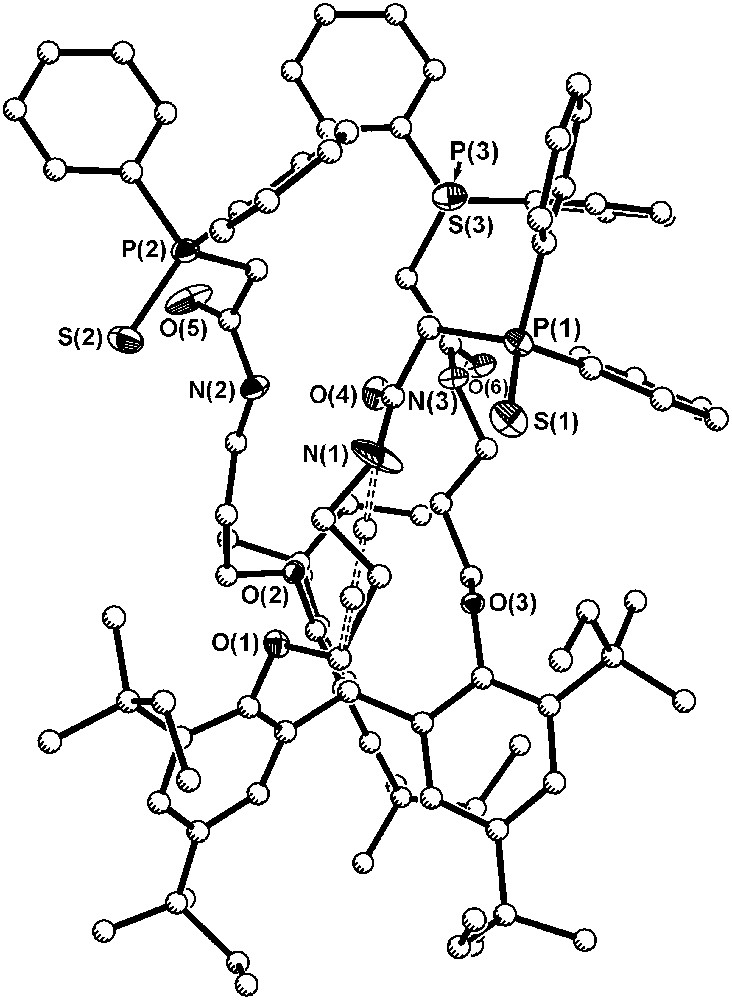
Diagram of the solid-state structure of 7a·CH2Cl2·Et2O (30% probability ellipsoids for N, O, S and P atoms; carbon atoms drawn with arbitrary radii). For clarity, all hydrogen atoms and solvate molecules have been omitted. Dashed line indicates second orientation of a disordered ligand arm.
In order to gain some understanding of the binding attributes of the ligand, a complex of Tb(NO3)3 with ligand 7a was synthesized (Fig. 7). In the solid state, the complex crystallized with two symmetry independent molecules of [7a·Tb(NO3)3] in the asymmetric unit along with four acetonitriles, and the metric parameters of the two terbium centers are essentially indistinguishable. If both the sulfide and carbonyl groups on the CMPS groups were to coordinate to the metal center, the arms would present the terbium metal with a six-membered chelate ring, and in the case of CMPO, this type of interaction forms a very stable structure. With 7a, only the carbonyl groups from the CMPS moiety coordinate to the metal center, and the phosphine sulfide groups are orientated away from the metal. The terbium–oxygen interactions are similar in strength (2.320(3) Å) to the analogous bonds in the terbium complex with tris-CMPO (2.308(5) Å). The carbonyl groups from the CMPS arms represent only three of the nine donors in the coordination sphere of the metal, and the metal accommodates three additional nitrate ions each in a bidentate fashion, fully neutralizing the charge and filling the remaining binding sites on the metal. In the tris-CMPS species, the sulfur atoms are too soft and they cannot displace the nitrate ions from the metal. In the acidic solution, water also may impede binding of the CMPS. Often with sulfur donor ligands, the preparation of lanthanide complexes requires great care to eliminate water from the reaction media, particularly in the case of the acidic organophosphorus ligands with sulfur [30].
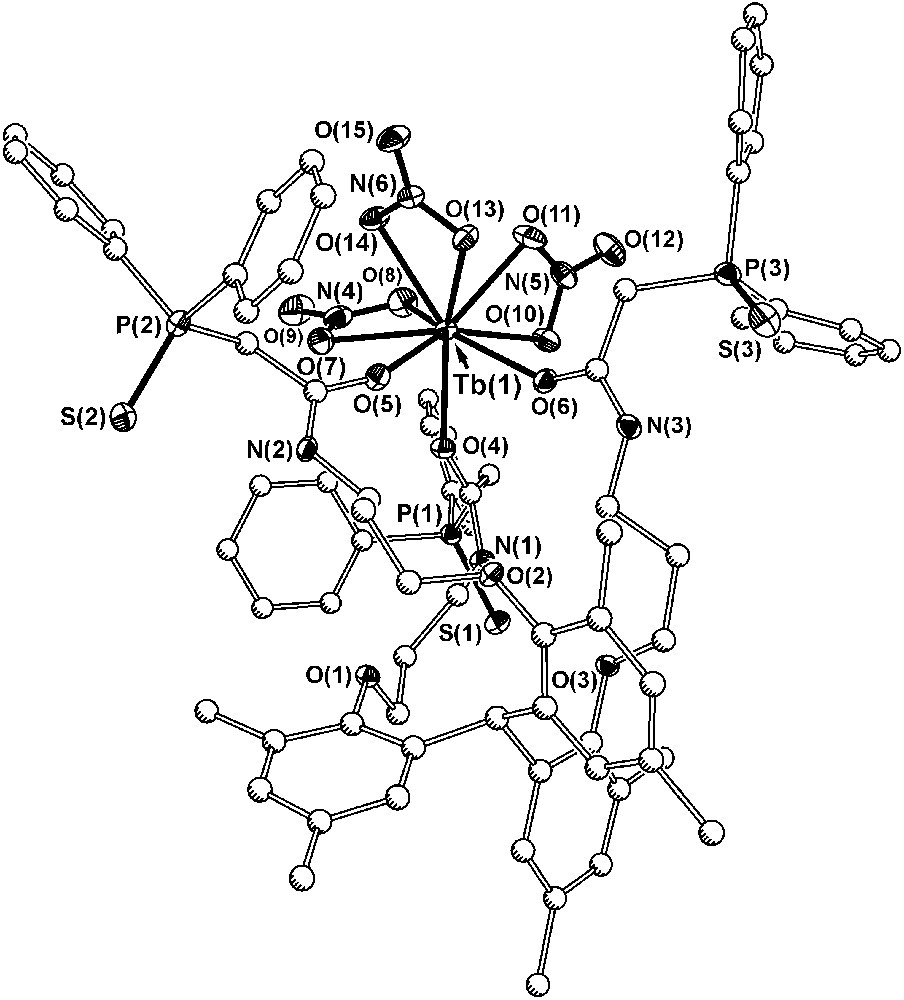
Diagram of one of the two symmetry independent terbium complexes in the structure of [7a·Tb(NO3)3] (30% probability ellipsoids for Tb, N, O, S and P atoms; carbon atoms drawn with arbitrary radii). Hydrogen atoms, the tert-pentyl arms, and solvate molecules have been omitted for clarity and all bonds to carbon have been drawn with open lines.
3 Conclusions
Tripodal phosphine sulfide (CMPS) compounds were synthesized as softer derivatives of the tris-CMPO chelate. Their ability to differentiate between trivalent lanthanides and actinides was tested using 41Am and 152Eu isotopes. Extraction experiments using tris-CMPS compounds revealed that the ligands are not able to bind preferentially trivalent Am over Eu. The very soft nature of the phosphine sulfide donor group was found to be incompatible with the hard tetravalent thorium and the trivalent f-elments, and in contrast to the tris-CMPO, the tris-CMPS analog showed no selectivity for these metal ions.
The solid state structure of tris-CMPS with terbium nitrate demonstrated that the phosphine sulfide portion of the ligand does not participate in metal binding despite the ability of the CMPS arms to form a six-membered chelate ring at the metal center. In sharp contrast to CMPO, the phosphine sulfide group is too soft to displace nitrate ions from the metal, and although no solution studies on the structure of tris-CMPS extractant have been performed, the inability of the ligands to extract metals ions from the highly acidic solutions suggests that soft sulfur atoms do not participate in the coordination of these hard metals in an aqueous, acidic environment.
4 Experimental section
4.1 Synthesis
Amines 3a, b and 4a, b were synthesized as previously described [21,25]. The (diphenyl-phosphinothioyl)-acetic acid, 5, was prepared according to the literature procedure [31].
4.1.1 Compound 6a
A mixture of (diphenyl-phosphinothioyl)-acetic acid (5) (5.40 g, 19.55 mmol), 2-mercaptothiazoline (2.51 g, 21.06 mmol) and 4-dimethylaminopyridine (0.60 g, 4.91 mmol) was stirred in dichloromethane (200 mL) at room temperature for 30 min. N,N′-dicyclohexylcarbodiimide (4.36 g, 21.13 mmol) was then added followed by an additional 15 mL of dichloromethane. After 6 h, solid 3a (4.44 g, 5.27 mmol) was added and the mixture was stirred for an additional 24 h at room temperature. The slurry was filtered and the solvent was removed in vacuo. The product was separated from byproducts by the dissolution in diethyl ether. Addition of methanol to the concentrated solution of the compound resulted in precipitation of solid product that was subsequently filtered and washed with cold methanol. Yield 5.57 g (65%). 1H NMR (CDCl3): δ = 0.46 (t, J = 7.3, 18H; CH2CH3), 1.07 (s, 18H; CCH3), 1.23 (s, 18H; CCH3), 1.41 (q, J = 7.3, 6H; CH2CH3), 1.59 (br, 6H; CH2CH3), 3.37 (br, 12H; OCH2CH2NH), 3.64 (d, J = 14.1, 6H; C(O)CH2P(S)), 6.23 (s, 1H; CH), 6.92 (d, J = 2.1, 3H; Ar), 6.96 (d, J = 2.1, 3H; Ar), 7.38 (m, 18H; Ar), 7.64 (br, 3H; NH), 7.87 (m, 12H; Ar). 13C NMR (CDCl3): δ (CO) = 165.13, 165.07; (aromatic) = 153.1, 142.9, 140.0, 137.8, 131.90, 131.87, 131.7, 131.5, 128.9, 128.7, 127.7, 125.0; (aliphatic) = 70.3 (OCH2), 42.6 (CH2NH2), 42.0, 40.2, 39.3, 37.8, 37.0, 35.5, 29.8, 28.7, 9.8, 9.3. 31P NMR (CDCl3): δ = 38.9. Anal. Calcd for C97H124N3O6P3S3: C, 72.04; H, 7.73; N, 2.60. Found: C, 72.19; H, 7.85; N, 2.57.
4.1.2 Compound 6b
Method I: A mixture of 5 (0.30 g, 1.12 mmol), 4-dimethylaminopyridine (0.13 g, 1.12 mmol) and EEDQ (1.11 g, 4.48 mmol) were dissolved in pyridine (10 mL). After stirring for 1 h 3b (0.18 g, 0.28 mmol) was added, and the reaction mixture was heated to 50 °C for 18 h. After cooling to room temperature, the solvent was removed in vacuo, and the residue was extracted with 9:1 CHCl3/MeOH solution followed by washing with 1 N HCl. Organic phases were collected, dried over MgSO4, and solvent was removed. The solid residue was dissolved in diethyl ether and upon addition of pentane, the product precipitated out of the solution. The compound was purified by column chromatography (SiO2, hexane/ether) to give 0.07 g of ligand 6b in the form of a white solid (18% yield). Method II: 5 (1.90 g, 6.90 mmol), 4-dimethylaminopyridine (0.31 g 2.53 mmol), mercaptothiazoline (0.85 g, 7.13 mmol) and N,N′-dicyclohexylcarbodiimide (1.47 g, 7.13 mmol) were dissolved in dry dichloromethane (50 mL). After few minutes, the solution turned bright yellow and a white solid separated out. The mixture was stirred for an additional 4 h and 3b (1.16 g, 1.84 mmol) was added. The resulting slurry was allowed to stir at room temperature for 48 h. The white solid of byproducts formed in the reaction was filtered, and the solvent was removed in vacuo. Addition of ether to the condensed reaction mixture dissolved the product leaving an amorphous mass of byproducts. The organic solution was decanted, and within several days upon slow evaporation of solvent pure product precipitated from the solution. Yield 0.8 g (31%). 1H NMR (CDCl3): δ = 1.15 (s, 27 H; Ar–C(CH3)3), 2.13 (s, 9 H; Ar–CH3), 3.25 (t, 6 H; Ar–O–CH2CH2), 3.33 (t, 6 H; Ar–O–CH2CH2), 3.65 (d, J(H,P) = 14.1 Hz, 6 H CH2–PSAr2), 6.67 (s, 1 H, C–H), 6.87 (b, 3 H; Ar–H),), 6.95 (b, 3 H; Ar–H), 7.42 (m, 18 H; P–Ar–H), 7.60 (t, 3 H; N–H) 7.89 (m, 12 H; P–Ar–H)). 31P NMR (CDCl3): δ = 38.8. MS [M + H]+ = 1406.5532 (theoretical [M + H]+ = 1406.5596).
4.1.3 Compound 7a
A mixture of 5 (4.73 g, 17.12 mmol), 2-mercaptothiazoline (2.14 g, 17.95 mmol) and 4-dimethylaminopyridine (0.67 g, 5.48 mmol) was stirred in dichloromethane (160 mL) for 30 min. N,N′-dicyclohexylcarbodiimide (5.34 g, 25.88 mmol) was added followed by an additional portion of dichloromethane (20 mL). After 6 h, solid 4a (4.10 g, 4.64 mmol) was added and the mixture was stirred for 24 h. The slurry was filtered and the solvent was removed. The product was separated from the reaction byproducts by dissolution in ether. The crude product was recrystallized from methanol to give 6.70 g (87%) of pure compound. 1H NMR (CDCl3): δ = 0.45 (m, 18H; CH2CH3), 1.10 (s, 18H; CCH3), 1.22 (s, 18H; CCH3), 1.42 (q, J = 7.4, 6H; CH2CH3), 1.57 (q, J = 7.1, 6H; CH2CH3), 1.82 (br, 6H; CH2CH2CH2), 3.30 (br, 12H; OCH2CH2CH2), 3.54 (br, d, J = 13.6, 6H; C(O)CH2P(S)), 6.18 (s, 1H; CH), 6.95 (d, J = 2.1, 3H; Ar), 7.04 (d, J = 2.1, 3H; Ar), 7.40 (m, 18H; Ar), 7.63 (t, J = 5.5, 3H; NH), 7.88 (m, 12H; Ar). 13C NMR (CDCl3): δ (CO) = 164.8, 164.7; (aromatic) = 153.3, 142.4, 139.8, 137.9, 132.7, 131.94, 131.90, 131.6, 131.4, 128.9, 128.7, 127.9, 124.7; (aliphatic) = 69.7 (OCH2), 42.7 (CH2NH2), 42.0, 39.3, 37.9, 37.7, 37.0, 35.3, 30.5, 29.7, 28.7, 9.7, 9.2. 31P NMR (CDCl3): δ = 38.9. Anal. Calcd for C100H130N3O6P3S3: C, 72.39; H, 7.90; N, 2.53. Found: C, 72.00; H, 8.10; N, 2.54.
4.1.4 Compound 7b
A mixture of 5 (2.13 g, 7.71 mmol), 2-mercaptothiazoline (0.97 g, 8.14 mmol) and 4-dimethylaminopyridine (0.29 g, 2.37 mmol) was stirred in dichloromethane (80 mL) for 30 min. The solid N,N′-dicyclohexylcarbodiimide (2.45 g, 11.87 mmol) was then added, followed by an additional 20 mL of dichloromethane. After 6 h, solid 4b (1.69 g, 2.23 mmol) was added and the mixture was stirred for an additional 24 h. The slurry was filtered and the solvent was removed in vacuo. The solid was dissolved in diethyl ether and quickly filtered. Within several days upon slow evaporation of solvent pure product precipitated from the ether solution. Yield 1.44 g (42%). 1H NMR (CDCl3): δ = 1.16 (s, 27H; CCH3), 1.23 (s, 27H; CCH3), 1.85 (br, 6H; CH2CH2CH2), 3.35 (br, m, 12H; OCH2CH2CH2NH), 3.54 (d, J = 14.1, 6H; C(O)CH2P(S)), 6.24 (s, 1H; CH), 7.09 (d, J = 2.3, 3H; Ar), 7.20 (d, J = 2.6, 3H; Ar), 7.40 (m, 18H; Ar), 7.61 (t, J = 5.4, 3H; NH) 7.88 (m, 12H; Ar). 13C NMR (CDCl3): δ (CO) = 164.82, 164.77; (aromatic) = 153.6, 144.5, 142.0, 137.7, 132.8, 132.01, 131.97, 131.7, 131.5, 128.9, 128.8, 127.2, 122.4; (aliphatic) = 70.3 (OCH2), 37.9, 35.7, 34.7, 31.7, 30.6, 25.6. 31P NMR (CDCl3): δ = 38.9. Anal. Calcd for C94H118N3O6P3S3: C, 71.68; H, 7.55; N, 2.67. Found: C, 71.22; H, 7.88; N, 2.53.
4.2 Extraction experiment
The metal extraction experiments followed previously reported procedure [21,32,33]. The extractability of each cation was calculated as %E = 100/(A1 − A)/(A1 − A0), where A is the absorbance of the extracted aqueous phase with the Arsenazo(III) indicator, A1 is the absorbance of the aqueous phase before extraction with the indicator, and A0 is the absorbance of metal-free 1 M nitric acid and the indicator (λLn(III) = 655, λTh(IV) = 665 nm). The errors, based on the precision of the spectrophotometer and the standard deviation from the mean of at least three measurements, were in most cases no higher than 2%.
4.3 Crystallography
Unit cell dimensions and intensity data for all the structures were obtained on a Siemens CCD SMART diffractometer at 173 K. The data collections nominally covered over a hemisphere of reciprocal space, by a combination of three sets of exposures; each set had a different ϕ angle for the crystal and each exposure covered 0.3° in ω. The crystal to detector distance was 5.0 cm. The data sets were corrected empirically for absorption using SADABS. The structures were solved using the Bruker SHELXTL software package for the PC, using either the direct methods or Patterson functions in SHELXS. The space groups of the compounds were determined from an examination of the systematic absences in the data, and the successful solution and refinement of the structures confirmed these assignments. All hydrogen atoms were assigned idealized locations and were given a thermal parameter equivalent to 1.2 or 1.5 times the thermal parameter of the atom to which it was attached. For the methyl groups, where the location of the hydrogen atoms is uncertain, the AFIX 137 card was used to allow the hydrogen atoms to rotate to the maximum area of residual density, while fixing their geometry. In cases of extreme disorder or other problems, the non-hydrogen atoms were refined only isotropically, and hydrogen atoms were not included in the model. Severely disordered solvents were removed from the data using the SQUEEZE function in the Platon for Windows software and the details are reported in the supporting information in the CIF file for each structure. Structural and refinement data and selected bond lengths for the compounds are presented in Tables 2 and 3.
X-ray dataa for the crystal structures of 7a and [7a·Tb(NO3)3] complex
| 7a·CH2Cl2·C4H10O | [7a·Tb(NO3)3]·2CH3CN | |
| Total reflections | 35 243 | 73 828 |
| Unique reflections | 12 730 | 48 890 |
| Θmax(°) | 23.43 | 28.03 |
| Empirical formula | C105H142Cl2N3O7P3S3 | C104H136N8O15P3S3Tb |
| Mr | 1818.21 | 2086.22 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/n | P-1 |
| a (Å) | 14.8551(10) | 16.8425(7) |
| b (Å) | 13.6147(10) | 25.0080(10) |
| c (Å) | 49.329(3) | 27.0255(11) |
| α (°) | 90 | 78.3410(10) |
| β (°) | 96.5800(10) | 74.6450(10) |
| γ (°) | 90 | 87.4780(10) |
| Vc (Å3) | 9910.9(12) | 10749.7(8) |
| Dc (g cm−3) | 1.219 | 1.289 |
| T (K) | 173(2) | 173(2) |
| Z | 4 | 4 |
| μ (Mo Kα) (mm−1) | 0.233 | 0.824 |
| R1 [I ≥ 2σ(I) data]b | 0.0858 [7722] | 0.0510 [22 377] |
| wR2 (all data)c | 0.2182 | 0.1420 |
| GOF | 1.033 | 1.061 |
a Obtained with monochromatic Mo Kα radiation (λ = 0.71073 Å).
b R1 = ∑‖F0| − |Fc‖/∑|F0|.
c wR2 = {∑[w(F02 − Fc2)2/∑[w(F02)2]}1/2.
Selected bond lengths (Å) for compounds: 7a and [7a·Tb(NO3)3]
| 7a | [7a·Tb(NO3)3]Tb(1) | [7a·Tb(NO3)3]Tb(2) | |
| P(1)–S(1) | 1.956(2) | 1.9583(18) | 1.943(2) |
| P(2)–S(2) | 1.943(2) | 1.9539(19) | 1.953(2) |
| P(3)–S(3) | 1.958(2) | 1.9476(19) | 1.952(2) |
| C(53)–O(4) | 1.221(7) | 1.240(6) | 1.233(6) |
| C(70)–O(5) | 1.223(7) | 1.244(6) | 1.231(6) |
| C(87)–O(6) | 1.232(7) | 1.249(6) | 1.247(6) |
| P(1)–CPh(55) | 1.802(6) | 1.808(5) | 1.844(8) |
| P(1)–CPh(61) | 1.812(6) | 1.813(5) | 1.806(6) |
| P(2)–CPh(72) | 1.806(6) | 1.814(5) | 1.817(7) |
| P(2)–CPh(78) | 1.821(6) | 1.809(5) | 1.763(8) |
| P(3)–CPh(89) | 1.802(6) | 1.803(6) | 1.815(6) |
| P(3)–CPh(95) | 1.814(6) | 1.805(6) | 1.814(5) |
| M–O(4) | – | 2.317(3) | 2.301(4) |
| M–O(5) | – | 2.320(3) | 2.343(3) |
| M–O(6) | – | 2.329(3) | 2.319(4) |
| M–O(7) nitrate | – | 2.492(4) | 2.467(4) |
| M–O(8) nitrate | – | 2.434(4) | 2.422(5) |
| M–O(10) nitrate | – | 2.520(4) | 2.425(4) |
| M–O(11) nitrate | – | 2.429(4) | 2.500(4) |
| M–O(13) nitrate | – | 2.492(3) | 2.484(4) |
| M–O(14) nitrate | – | 2.453(3) | 2.462(4) |
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK [CCDC #621057 (7a·CH2Cl2·C4H10O), CCDC #621058 ([7a·Tb(NO3)3]·2CH3CN).
Acknowledgements
The authors gratefully acknowledge financial support from the Nuclear Energy Research Initiative (Grant 02-98) of the Department of Energy (DE-FG07-02SF22614). We would also like to thank Artem Gelis and Monica C. Regalbuto at Argonne National Laboratory for helpful discussions.
Vous devez vous connecter pour continuer.
S'authentifier