

1 Introduction
1.1 Background
Microorganisms have adapted to live in virtually every terrestrial or aquatic environment encountered on Earth, including radioactively contaminated environments. Microbial metabolic processes can influence their local environment, for example, through changing pH and Eh conditions, and can affect radionuclide speciation via a number of mechanisms. In particular, microbial redox reactions in subsurface environments, and their impact on biogeochemical cycles, have received much attention recently. Several important, long-lived radioactive elements have complex redox chemistries which can interact with subsurface biogeochemical cycles leading to changes in radionuclide mobility. Here, we discuss the chemical, microbiological and geochemical influences on these processes.
1.2 Chemical aspects
Chemical speciation (oxidation state, complex form) is a fundamental control on the behaviour of any solute, defining properties such as solubility or reactivity with respect to surfaces. The importance of chemical speciation can be seen clearly in, for example, the differential solubilities of U and Th in seawater. The atom ratio U:Th on the Earth's crust is about 1:4, yet that in seawater is 64,000:1 [1,2] and this difference arises entirely from the different speciation of the two elements. At their most fundamental, both oxidation state stability and complex formation are driven by equilibrium thermodynamics, specifically the Gibbs Free Energy change which allows rationalisation of a diverse range of observations (see e.g. Ref. [3]).
The simplest equilibrium process is homogeneous complex formation, as in the well known example of the UO22+/CO32−/OH− system (Clark et al. [4] and references therein) where, as pH (and hence free CO32−) increases, the system is dominated successively by the complex ions [UO2(CO3)], [UO2(CO3)2]2− and [UO2(CO3)3]4− (Fig. 1). The formation of each complex can be quantified using a straightforward equilibrium constant.
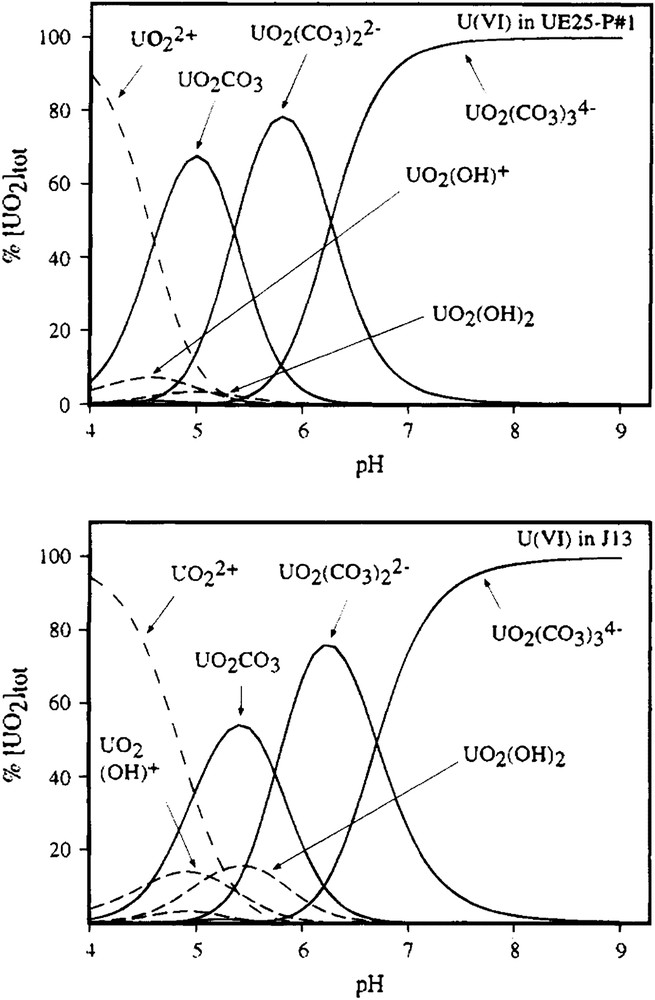
Calculated uranyl species distributions in carbonate solutions used as models for Yucca Mountain groundwaters (from Ref. [4]).
The situation is perhaps more complex in heterogeneous reactions, such as hydrolysis, where reaction may lead to a change in solubility. Pu(IV) provides an excellent, if complicated, example [5]. As the pH increases (and hence the Pu:OH− ratio decreases), the system becomes dominated by [Pu(OH)]4 and these species react to form a high molecular weight polymer, eventually leading to precipitation [6,7]. For the thermodynamic description of a reaction to be valid, the transformation has to be reversible but, in the case of Pu(IV), the initial polymerisation reaction is followed by a series of aggregation and dehydration steps so that it is not possible to describe Pu(IV) hydrolysis by a single equilibrium constant, as it was for the UO22+/CO32− complexes. The thermodynamics of Pu(IV) hydrolysis led to considerable controversy in the early literature although more recent work has clarified the issue considerably. Moreover, because the solid phase transformations can be slow, it can be very difficult to know if the system has reached equilibrium. In these situations, thermodynamic analysis, with its requirement for equilibrium, can only be an approximation and time-dependent effects can be observed; for example, aged Pu polymer is much more difficult to redissolve than freshly precipitated material [8–10].
An understanding of redox transformations is also important. Some transformations are straightforward and involve just electron transfer, for example, U(IV)/U(III) in strong acid solution, where the reaction is simply:
| U4+(aq) + e− → U3+(aq) | (1) |
Others are more complicated, with the making or breaking of chemical bonds being intimately associated with electron transfer. The reduction of Np(V) to Np(IV) in acid medium is an example, and this process can be described:
| NpO2+(aq) + 4H+(aq) + e− → Np4+(aq) + 2H2O | (2) |
Redox reactions are described using standard potentials (E0), which are measured in standard state conditions, generally strong acid. Obviously, a reaction such as reduction of Np(V), in which protons are involved will display a strong dependency on pH (the equilibrium constant will include [H+]4) so that the potential at which Np(IV) becomes thermodynamically favoured over Np(V) will change with pH. This relationship is described using the Nernst equation:
| U(OH)4(aq) + e− → U(OH)3(aq) + OH−. | (3) |
The redox potential for this process will be different from that for reaction (1) (−2.14 and −0.596 V, respectively [11]), not least because we have a different transformation with a different equilibrium constant. Similar effects are observed with ligands other than hydroxide; for example, the U(VI)/U(V) potential shifts from −0.169 V in the presence of CO32− to −0.820 V in its absence [12]. Thus, from the perspective of possible biotransformations, there are two different ways to drive the redox transformation of a metal ion. It is possible either to control electron activity or to complex the ion (through production of a complexant or presenting a specific binding site) in such a way that the redox potential will shift and permit the required redox reaction to occur at the prevailing electron activity.
The aqueous speciation of the mid-actinide elements (U, Np, Pu) is amongst the most complex known (see e.g. Silva and Nitsche [13] and references therein). Oxidation states +3 to +6 are known for U, and +3 to +7 for Np and Pu (+8 is also suggested but not definitively identified for Pu). In environmentally attainable redox conditions, which is a window effectively defined by the chemical stability of water, the oxidation states U(IV,V,VI), Np(IV,V) and Pu(III,IV,V) might realistically be attainable. Oxidation states +3 and +4 form simple ions in solution and, due to their high charge and relatively small size, they are readily hydrolysed. Oxidation states +5 and +6 form the stable, linear dioxo ions AnO2+/2+ (actinyl ions), where An denotes an U, Np or Pu center. Some actinide species, notably U(V), Pu(IV) and Pu(V), are susceptible to disproportionation because the redox potential for self-oxidation is similar to that for self-reduction. Thus, the reaction
| 2Pu4+(aq) + 2H2O → Pu3+(aq) + PuO2+(aq) + 2H+(aq) | (4) |
All the actinide cations which might realistically be encountered in environmental conditions react readily with complexing ligands, particularly those containing “hard” donor atoms such as oxygen. Thus, relatively simple species such as CO32− and CH3CO2− are effective complexants, while more complex molecules such as citrate or siderophores, which can be produced biologically, also bind actinide cations strongly. The general trend in complex stability is:
| An4+ > An3+ ≈ AnO22+ > AnO2+ |
Because of the stability of the linear actinyl ions, complex formation with these species is essentially confined to the equatorial plane whereas the simple An3+ or An4+ ions do not impose comparable stereochemical restrictions. Polydentate ligands such as citrate can, therefore, wrap around the lower oxidation state ions in a way which is not possible for actinyl species. In the case of Pu, relevant potential transformations are discussed in Neu et al. [14].
1.3 Radioactively contaminated land
Nuclear fuel cycle activities have created large volumes of contaminated land in many places. Reprocessing activities, particularly those associated with military Pu production, perhaps have the highest profile (e.g. Mayak in the Former Soviet Union, Hanford in the USA, Sellafield in the UK) and present some difficult challenges because of the presence of complex mixtures of contaminants (e.g. U, transuranic elements, fission and activation products, perhaps with stable metallic and organic co-contaminants). However, by volume, the dominant contaminated land problem is presented by uranium, particularly uranium mining and ore processing sites. These include localities in central Europe, the USA, Australia, South Africa, Namibia, Canada and the Former Soviet Union.
The scale of uranium mining wastes is very large and can be illustrated by examining the legacy of the mining operations in the former East Germany. This is contiguous with ore deposits in the Czech Republic, which have also been extensively exploited and an excellent account is given by Diehl [15]. This region is the third largest uranium mining province on Earth and, between 1946 and 1990, produced around 320,000 tonnes of uranium, often from ores with a grade of only about 0.1%. Ores were extracted and processed conventionally, leaving waste piles which are up to 350 ha in area and contain tens of millions of cubic metres of material. Heap leaching of lower grade ores also occurred, with individual heaps containing up to 7 million tonnes of ore. Finally, in situ leaching was also used. This involves pumping a suitable extractant (often H2SO4 in this case) through porous, uranium-bearing rock and extracting uranium from the leachate. The legacy of these operations includes large volumes of both surface and subsurface contamination. While the details of operations may vary from site to site, and the former East German example may be more extreme than many, it illustrates the scale and complexity of the work required for uranium mine remediation.
1.4 Microbiological aspects
1.4.1 Microorganisms in radioactive environments
A wide range of bacteria, both bacteria and fungi, have been found growing in environments contaminated with radionuclides (e.g. Zhdanova et al. and Nazina [16,17]). These habitats vary from uranium mining and milling sites [18–20], with relatively low levels of radiation, to spent fuel basins [21] and inside the reactor installations at the Chernobyl Nuclear Power Plant [16,22], where microorganisms will be exposed to much higher levels of radiation. For example, Chicote et al. [23] characterized microorganisms attached to the walls of a pool storing nuclear materials at a Spanish nuclear power plant. Six different bacteria (affiliated to β-proteobacteria, Actinomycetales and the Bacillus/Staphylococcus groups) and a fungus (Aspergillus sp.) were identified from the isolates using molecular analysis of the 16S and 18S ribosomal RNA genes, respectively. Zhdanova et al. [16,22,24,25] have extensively investigated the fungal communities growing in and around the Chernobyl Nuclear Power Plant. They found extensive fungal growth in the inner parts of the protective shelter erected over the damaged reactor unit [16]. In total, ∼2000 strains of fungi, representing 200 species in 98 different genera, have been found in and around Chernobyl. Depending on location, the isolates have been exposed to a wide range of ionizing radiation, with doses up to 700 Gy h−1, and this exposure has resulted in the altered genetic composition of some organisms [25,26]. Both proposed and “in-use” repositories for nuclear waste have also been investigated [17,27,28], as the microbial community present in such environments could affect the long-term stability and mobility of the wastes. The Severnyi repository in Eastern Siberia is used for the storage of low-level liquid waste. Liquid radioactive waste has been stored at the site since 1967 and is injected into subsurface horizons. A range of anaerobic prokaryotes including denitrifiers, fermenters, sulphate-reducers and methanogens were cultured from water samples collected from the repository [17]. The numbers of cultivable cells were higher for all the different groups of bacteria, for samples taken from the zone of radioactive waste dispersion, compared to samples taken from outside the zone of dispersion. Organisms isolated were capable of utilizing a wide range of organic and inorganic substrates, including components of the waste such as acetate, sulphate and nitrate. This ability to use some of the waste is perhaps reflected in the higher cell numbers obtained in the zone of dispersion.
As can be seen from these examples, microorganisms are ubiquitous in radioactive environments and there are a range of microbial metabolic processes that can affect the chemical speciation of radionuclides. For example, ligands secreted by microorganisms, either for a specific purpose (as in the case of siderophores produced to obtain iron) or as a by-product of metabolism, can chelate actinide ions and so directly alter the chemical speciation. This can impact solubility, leading to precipitation of the radionuclide or increased solubility, and can affect the redox potential. Here, the different microbial mechanisms for transforming radionuclides are described.
1.4.2 Biosorption
Biosorption is defined as the microbial uptake of metal species by physicochemical mechanisms, for example, adsorption [29]. It is a metabolic-independent process, and so both living and dead microbial biomass can biosorb metals, however, metabolic activity may affect the process, e.g. by causing localized changes in Eh or pH, or by secreting metal-complexing ligands. Biosorption of metals, including radionuclides, has been studied for more than two decades, and reviewed extensively during this period [30–32]. More recent studies have confirmed that a wide range of microorganisms, including bacteria, fungi, lichens and algae, are capable of biosorbing radionuclides [33–37]. Most studies have investigated sorption of U(VI), although sorption of Pu, Am, Np, Th and Tc species have also been studied to a limited extent recently [34,38–43]. Songkasiri et al. [38] investigated sorption of NpO2+ by the bacterium Pseudomonas aeruginosa. With initial Np solution concentrations of ∼3.67 μM and biomass concentrations equivalent to 106 mg/l dry weight biomass, 50–65% of Np was removed from solution over the pH range 6–8. The only study on biosorption of TcO4− investigated cyanobacteria [44]. TcO4− sorption was concentration-dependent and found to increase as pH decreased. All sorbed Tc(VII) was easily desorbed.
Sorption can be either to the cell wall or to extracellular components, for example polysaccharides, glycoproteins or lipopolysaccharides (LPS), associated with the cell wall. A range of functional groups are found in these cell walls and exopolymers, including carboxylate, phosphate, amino and hydroxyl groups. The cell wall of Gram-positive bacteria is composed primarily of peptidoglycan with lesser amounts of other polymers such as teichoic acids; the most likely metal binding sites are the carboxyl groups in peptidoglycan and the phosphoryl groups in the teichoic acids [45]. In Gram-negative bacteria, the cell wall architecture is more complex, as the cells have an additional outer membrane (as well as the cytoplasmic cellular membrane found in all bacteria) with lipopolysaccharide (LPS) groups outside a thin peptidoglycan cell wall. Phosphoryl groups in the LPS are the most probable sites of metal complexation in these organisms [45,46]. A number of studies have investigated the mechanism of U(VI) sorption to bacterial cells and found evidence for coordination to both carboxyl and phosphoryl groups [47–51]. Merroun et al. [49] studied the sorption of U(VI) to Bacillus sphaericus JG-A12, a Gram-positive bacterium isolated from a uranium mining waste pile. The XAS results supported bidentate coordination of U(VI) to carboxyl groups and monodentate coordination to phosphoryl groups. Panak and Nitsche. [39] investigated sorption of Pu(VI) to another B. sphaericus strain, using XAS. Coordination was primarily to phosphate groups and there was no evidence for coordination to carboxyl groups. In fungi, chitin is an important structural component of the cell wall and has been found to be an effective sorbant for radionuclides [29]. Binding of U(VI) is thought to be to amine groups in chitin [52,53] but there are other functional groups present in fungal polymers that would effectively bind radionuclides including carboxyl, phenolic and carbonyl [54].
1.4.3 Intraceullular uptake
Most studies on the intracellular accumulation of radionuclides by microorganisms have focussed on the lighter radioelements such as Cs, Sr and Pb [55–57]. There have been relatively few investigations of intracellular accumulation of actinides and almost all of these have concentrated on U. Bacteria, fungi and algae have all been found to accumulate U intracellularly [33,58–61]. However, accumulation was often found to be independent of metabolism. For example, Volesky and May-Phillips [59] found U(VI) was accumulated both extra- and intracellularly by the yeast Saccharomyces cerevisiae. Uranium was deposited in fine, needle-like crystals and uptake was independent of metabolism. It has been suggested that this intracellular metabolism-independent uptake results from increased cell membrane permeability caused, for example, by the toxic effects of uranium [37]. In a more recent study, Suzuki and Banfield [62] investigated resistance to, and accumulation of uranium by bacteria isolated from acidic, uranium-contaminated land. In one isolate, closely related to Arthrobacter ilicis, uranium was precipitated intracellularly in close association with polyphosphate granules. The cells of this isolate remained viable after accumulation of uranium. They proposed that the intracellular precipitation of uranium acts as a detoxification mechanism. In one study, John et al. [63] reported the intracellular uptake of Pu(IV) in Microbacterium flavescens JG-9. This was an active, metabolism-dependent transport process, with Pu(IV) taken up as a Pu(IV)-desferrioxamine B complex via the siderophore-mediated Fe(III) transport system.
1.4.4 Bioleaching
Microbial leaching of solid metal compounds occurs either through redox transformations, reduction of pH or production of metal-chelating ligands. Leaching can result from both autotrophic and heterotrophic metabolism. Most autotrophic leaching is caused by chemolithotrophic, acidophilic bacteria. These organisms, such as Acidithiobacillus thiooxidans, Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans, use CO2 as their carbon source and obtain energy from the oxidation of reduced sulfur compounds or Fe(II) [29], generating Fe(III) and/or H2SO4.
Metal solubilization can then occur indirectly through oxidation of the metal sulfide (Eq. (5)) or the metal itself by the microbially generated Fe(III).
| MS2(s) + 14Fe3+(aq) + 8H2O → M2+(aq) + 14Fe2+(aq) + 2SO42−(aq) + 16H+ | (5) |
The most well studied radionuclide with regard to microbial leaching is uranium, and in situ bioleaching of low-grade uranium ores is now an important method for extracting uranium [37,64]. Acidithiobacillus and Leptospirillum species are the principal organisms used for extracting U. The mechanisms are either direct oxidation of pitchblende (UO2) or indirect oxidation by Fe(III) (Eq. (6)):
| UIVO2(s) + 2Fe3+(aq) → UVIO22+(aq) + 2Fe2+(aq) | (6) |
Leaching can also result from heterotrophic metabolism and this is most important in the case of fungi [29]. Heterotrophic leaching is mainly caused by production of organic acids, but other processes, such as proton efflux and production of siderophores, can also cause it. Secreted organic acids serve as a source of both protons and metal-chelating ligands and have been found to leach radionuclides from solid forms. For example, citric acid is commonly produced by fungi [65,66]. Citrate forms very stable complexes with uranyl [67] and can leach Pu(IV) and Am(III) from soil particles [68]. The fungi Aspergillus ochraceous and Penicillium funiculosum were able to leach uranium from a range of U-bearing rocks and the mechanism of solubilization was attributed to production of citric and glutamic acids [69,70].
1.4.5 Biomineralization
Radionuclides can be precipitated through the microbial generation of ligands, for example, phosphate, sulphide, carbonate and oxalate. Microbial ligand production causes a high localized ligand concentration around the cell, and the cell surface provides a nucleation site for precipitation, resulting in efficient removal of the radionuclide from solution. One of the most extensively studied systems is phosphate precipitation of radionuclides using the bacterium Serratia (formerly Citrobacter [71]). Inorganic phosphate is generated from the hydrolytic cleavage, by a phosphatase enzyme, of an organic phosphate e.g. glycerol-2-phosphate [72]. In studies using Serratia to generate phosphate, U(VI), Am(III) and Th(IV) were found to be effectively removed from solution [73–75]. In the case of Th(IV), removal from solution was initially poor, as complexation of Th(IV) by citrate (present as a buffer) kept the metal ion in solution [75]. However, the addition of NH4+ was found to significantly enhance Th(IV) removal [74,75]. Up to 90% of Th(IV) was removed from a 300 μM Th(IV) solution, with the metal precipitating as thorium ammonium phosphate. In contrast, Serratia was far less efficient at removing Np(V) and Pu(IV) by phosphate precipitation [76]. However, Np(V) could be removed from solution by reducing Np(V) to Np(IV) [77] (see Section 2.1). Np(IV) then precipitates from solution as the phosphate. Np(V) and Pu(IV) could also be removed by using microbially synthesized LaPO4 (see below) [76].
In addition to direct precipitation by microbially generated ligands, actinide ions can also be removed from solution by biogenic minerals. Microbially enhanced chemisorption of heavy metals (MECHM) is the process whereby microbial cells first precipitate a metal biomineral (“priming deposit”), which then acts as a nucleation focus, or “host crystal” for the subsequent deposition of the metal of interest [73,78]. Examples of priming deposits include FeS, Fe(III) oxides and a range of biogenic metal phosphates (see above). FeS is formed when sulphate-reducing bacteria reduce sulphate to generate H2S, which then reacts with iron. The biogenic FeS can then sorb heavy metals and radionuclides [79]. Fe(III) oxides are made, for example, by aerobic and denitrifying Fe(II)-oxidising bacteria and can also sorb radionuclides [78,80]. One of the best studied example of MECHM involves the generation of phosphate priming minerals, such as hydrogen uranyl phosphate (HUP) or lanthanum phosphate by Serratia. Macaskie and Basnakova [76] used immobilized cells of Serratia to make cell-bound LaPO4, which then effectively sorbed Np and Pu. The HUP crystal structure consists of sheets of uranyl phosphate ions separated by water molecules and accumulation of radionuclides can occur either through intercalation into the interlamellar space between the uranyl phosphate sheets, or, in the case of other hexavalent actinide ions, by displacing uranyl ions in the lattice backbone [78].
Although there is a range of mechanisms by which microbes can affect the chemical speciation of actinides, perhaps the most extensively studied process is microbially mediated redox transformations. These processes can have a profound impact on radionuclide solubilities and have been the focus of recent in situ remediation studies. The rest of this review concentrates on these redox transformations.
2 Redox transformations
In the context of subsurface biogeochemistry, microbial metabolism drives a wide range of redox transformations. The starting point is the well established sequence of terminal electron acceptors (TEAs; e.g. [81]), which are exploited in the order of decreasing free energy yield (O2 > NO3− > Mn(IV) > Fe(III) > SO42− > CO2). This is a useful, though simplified, analysis since any natural system will be heterogeneous – resource quality will vary from place to place; porosity and permeability will be variable; and the microbial community will change both spatially and temporally. The net result is to create a mosaic of microenvironments within the system so that, at a fairly coarse scale, the TEA sequence can provide insight into biogeochemical changes but, at a finer scale, the concept becomes increasingly difficult to use.
Microbial activity can lead to redox changes through either direct or indirect mechanisms. In direct transformations, the trace species of interest, such as UO22+ or TcO4−, can actually be exploited in metabolism because the electron transport chain is sufficiently adaptable to use it as a TEA. Fe(III) is sometimes viewed as a model for U(VI) in biological systems [37] and a wide range of Fe(III)-reducing bacteria have been shown to reduce U(VI) as well. The first biochemical studies on U(VI) reduction focused on the sulphate-reducing bacterium Desulfovibrio vulgaris, and identified a periplasmic cytochrome c3 as the terminal reductase for the radionuclide [82]. Similar mechanisms may be important in Geobacter species [83], but the terminal reductase for U(VI) in this organism remains to be identified unequivocally. A genetic approach has been used to characterise the electron transfer chain in Shewanella putrefaciens [84], and here the authors suggested that U(VI) was reduced via a nitrite reductase. By contrast, the reduction of TcO4− to Tc(IV) by Escherichia coli, Desulfovibrio desulfuricans and Geobacter sulfurreducens is mediated by a hydrogenase enzyme. In the case of E. coli [85] and Desulfovibrio fructosovorans [86], detailed studies of the mechanism have been carried out.
Indirect transformations occur through biological production of redox active species. The reduced members of the Mn, Fe and SO42− TEAs are of most interest because the products of NO3− (except NO2−) or CO2 reduction are not good chemical reductants for kinetic reasons. However, Mn(II), Fe(II) and species such as S2O32− or S2− are chemically reactive and can themselves function as reductants, for example causing transformations such as reduction of UO22+. However, the presence of a potential reductant does not necessarily mean that reduction will occur. For example, Moyes et al. [87] showed that uranium was initially adsorbed by mackinawite (FeS) as a UO22+ surface complex to oxide binding sites on the surface. Only at higher UO22+ concentrations was uranium reduction observed. By contrast, NpO2+ was readily reduced by mackinawite at all concentrations [88]. Similar considerations apply to, for example, Fe(II) in magnetite (Fe3O4) or siderite (FeCO3).
2.1 Selectivity
Although the expected mobile forms of the actinides are the higher oxidation state actinyl ions (UO22+, NpO2+, PuO2+) and these share many chemical similarities, they can differ considerably in redox properties and it is not clear that an organism or process which will cause one to be reduced will necessarily reduce another even though the redox potentials might suggest the reaction should occur. For example, Renshaw et al. [89] showed that G. sulfurreducens could not reduce NpO2+ even though it easily reduced UO22+. The first product of bioreduction of uranium was identified as UO2+, which is unstable with respect to disproportionation, so that the mechanism is as shown in Fig. 2.
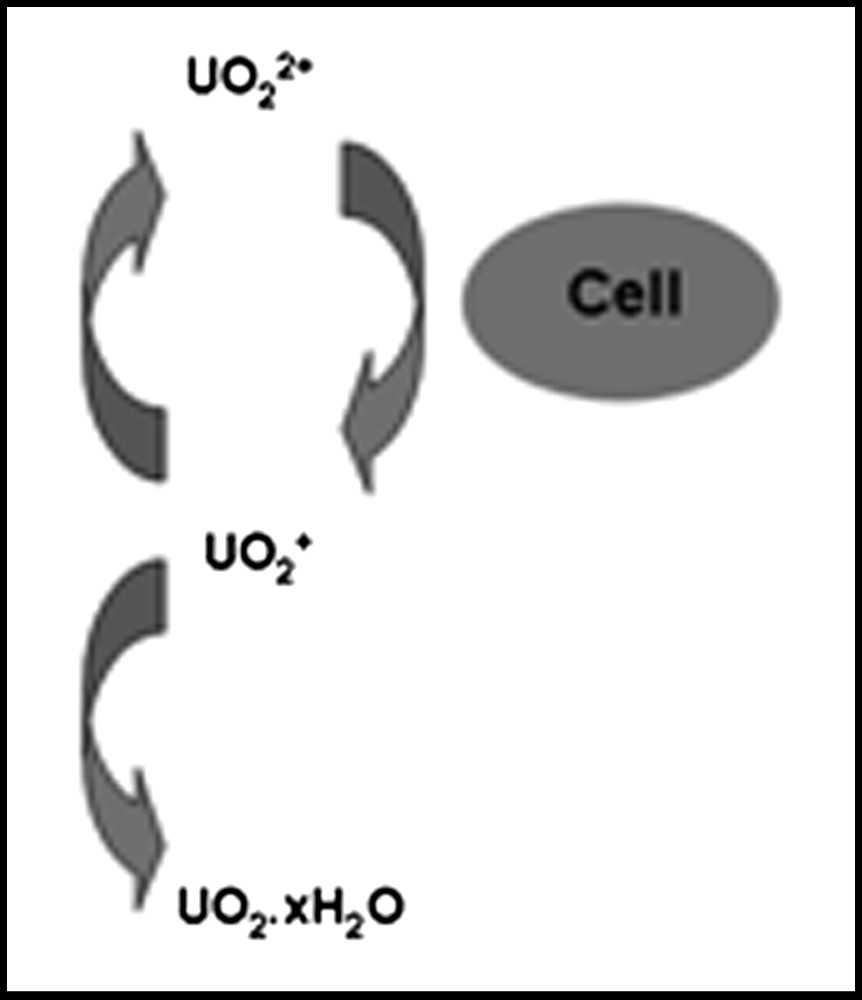
Mechanism of UO22+ reduction by G. sulfurreducens (after Ref. [89]).
This suggests that the enzyme system responsible for uranium reduction is capable of transferring one electron to an actinyl ion. The instability of the resulting U(V) then generates U(IV), whose insolubility drives the reaction. Reduction of Np(V) therefore fails because the enzyme cannot transfer an electron to bound NpO2+. For this reason, it might be expected that PuO2+ should also not be reduced enzymatically by G. sulfurreducens. The potential for bioreduction of Pu(IV) to the more mobile Pu(III) is less clear, particularly as complexation can significantly alter the redox properties. The implications are therefore that a Geobacter-dominated community would be effective at immobilising U and could potentially mobilise Pu from mixed U, Np, Pu contamination. Clearly, indirect reduction of Np in the natural environment would still be possible and different organisms could give different outcomes. For example, both the dissimilatory iron-reducing bacterium Shewanella putrefaciens and a mixed consortium of sulphate-reducing organisms have been shown to reduce Np(V) effectively [77,90]. There is thus a need to understand both reaction mechanisms and microbial community structure in applications of in situ bioremediation processes.
2.2 In situ bioremediation
As mentioned previously, uranium is the predominant problem in radioactively contaminated land. As the volumes of uranium mine waste are so large and, in some cases, the material is so inaccessible, the potential costs of remediation by conventional means are huge and the circumstances are particularly attractive for bioremediation. While the underlying principles of bioremediation have been clear for well over 10 years [91], it has taken some time to progress to practical demonstrations. Thus, we have moved from model systems (single organisms in pure culture) to proof-of-concept (laboratory studies of natural soils and sediments containing mixed microbial communities) and, more recently, to field-scale studies. Here, we review some of the recent literature relating to field trials and discuss some of the uncertainties and questions which remain to be answered.
2.2.1 Application in the field
The principle of biostimulation is straightforward – in response to addition of electron donor (e.g. glucose, ethanol, acetate) to the subsurface, a range of bioreduction processes will be triggered, including the reduction (direct or indirect) of radionuclides from high oxidation state, mobile forms to low oxidation state, immobile forms. These processes are being used at field sites such as Old Rifle, Colorado and the Field Research Center (FRC), Oak Ridge, Tennessee.
The Old Rifle site is a redundant uranium ore processing facility at which large piles of mill tailings are leaching uranium into the subsurface and uranium plumes in groundwater are migrating towards the Colorado River [92]. Acetate was injected into the subsurface from an array of wells, leading to a decrease in dissolved uranium concentrations down-flow within 9 days and, by 50 days, dissolved uranium concentrations had fallen from typical values of 0.8–1.5 μM to <0.18 μM at some locations. In the early stages of treatment, Geobacter species were dominant but, as the experiment continued, the system switched to sulphate reduction and this was associated with an increase in dissolved uranium concentrations. Thus, to sustain effective uranium removal from groundwater, it appears to be advantageous to poise the system in Fe(III)-reducing conditions. A subsequent study [93] showed that heterogeneity was a dominant control in in situ treatment, with local differences in hydrology leading to uneven distribution of electron donor and hence of Geobacter activity and uranium reduction.
At sites such as the FRC which have been used in the processing of nuclear materials, subsurface nitrate concentrations are often very high and this can complicate matters in comparison with former uranium mining and ore processing sites [94]. For example, FRC groundwater can contain 50–150 mM NO3−. Wu et al. [95,96] found it necessary to flush excess mobile nitrate, aluminium and calcium from the groundwater before commencing U(VI) reduction, while Luo et al. [97] removed mobile nitrate over a period of months, then maintained the system in conditions where in situ nitrate removal occurred. Only after this preliminary treatment was U(VI) reduction achievable [96]. By contrast, Istok et al. [98] identified variable responses. On some occasions, both uranium and technetium were removed from solution, although some reduced U(IV) reoxidised later and dissolved concentrations increased again. In other locations, technetium was readily removed from solution but dissolved uranium concentrations were unaffected. Substantial variability in FRC sediment biogeochemistry was also observed in laboratory microcosms [99]. Likewise, Shelobolina et al. [100] found that biological reduction of nitrate, stimulated by acetate amendment, caused a rise in pH and loss of UO22+ from solution, but through hydrolysis and precipitation, rather than through reduction.
2.2.2 The role of surfaces and solid phases
Solid phases are also important in determining the outcome of in situ treatments. For example, Jeon et al. [101] showed that the nature of Fe(III) oxide surfaces, which could take up UO22+ from solution, was variable and that anthraquinone disulfonate (AQDS), an electron shuttle, speeded up reduction of both UO22+ and Fe(III). The rate of bacterial UO22+ reduction has also been shown to be intimately related to the nature (structure, surface area) of the hydrous Fe(III) oxides involved [102,103], while Ortiz-Bernad et al. [104] found that UO22+ adsorbed on mineral surfaces (presumed to be Fe(III) hydrous oxides and clays) was not susceptible to microbial reduction.
2.2.3 Persistence
While natural UO2, the mineral pitchblende, is relatively robust in near-surface conditions, as can be seen from studies of exposed uranium mineralization (Needle's Eye, Pocos de Caldas), the products of bioreduction are, at least initially, rather different. The first-formed material has a very small particle size [105], is often poorly crystalline or amorphous, and is a hydrated phase. After precipitation, hydrous oxide phases continue to evolve both chemically and structurally so that aging effects, as previously discussed for Pu(IV) colloid and studied in detail for Fe(III) phases [106], are important. Thus, in the situation where active manipulation of the subsurface biogeochemistry has ceased, it is important to understand the potential for further redox reactions which may or may not be associated with re-establishment of oxidising conditions and which could lead to redissolution of the precipitated contaminants.
Thus, Wan et al. [107] showed, using long-term (17 months) column experiments that, even though the system remained reducing as a whole, biogenic U(IV) could be reoxidised and solubilized. They proposed that, even in a reducing system, Fe(III) and Mn(IV) could persist and act used as TEAs in U(IV) oxidation. Nitrate can similarly be used to promote reoxidation of U(IV) [98,108–110] and the redox transformations of nitrite are closely coupled with those of Fe and hence U [109]. By contrast, at an abandoned uranium mine (Midnite, Washington), enzymatically reduced uranium persisted in the sediments in spite of repeated periods of drying [111].
It has been demonstrated clearly that technetium can be effectively reduced in sediment systems by microbial activity [98,112,113]. Tc reduction can be both indirect, mediated by biogenic Fe(II) [114,115], and direct [115]. While, in most studies, Tc reduction is associated with loss from solution, Wildung et al. [113] found that dissolved Tc concentrations decreased less than expected, and attributed this to the formation of a soluble complex of reduced Tc. The behaviour of technetium on reoxidation is not so clearly understood. Istok et al. [98] found that the reduced form of Tc was stable on the re-establishment of oxic conditions. By contrast, Burke et al. [116] found that, with air as oxidant, ca. 50% of Tc was reoxidised to TcO4− and released to solution whereas, with NO3− as oxidant, even though substantial quantities of TcO4− were again formed, <10% of the Tc inventory was released to solution.
3 Conclusions
The environmental behaviour of radionuclides is dominated by their chemical speciation (oxidation state, complex form). The actinide elements can exhibit very complex speciation chemistries, as illustrated by Pu, which can exist in multiple oxidation states simultaneously and, as Pu(IV), undergoes complicated hydrolysis reactions. Understanding the chemistry is of fundamental importance for predicting and controlling radionuclides in the environment; anything that impacts their chemical speciation will significantly affect their environmental behaviour. Microorganisms can alter both the complex form and oxidation states of a range of radionuclides. Microbially produced ligands can complex radionuclides and lead to either increased mobility of radionuclides in the environment, for example through leaching from insoluble phases, or reduced mobility through precipitation. The focus of many recent studies has been microbial redox transformations, with most investigations concentrating on U(VI). Microbes have been found to mediate redox transformations of U(VI), Np(V) and Tc(VII), either through direct enzymatic mechanisms or indirectly through the formation of other redox active chemical species (e.g. Mn(II) or Fe(II)). In the case of uranium, soluble, mobile U(VI) (as UO22+) is reduced to insoluble, immobile U(IV) (as UO2) and in situ investigations of uranium-contaminated sites have sought to stimulate these transformations (through the addition of an electron donor such as acetate), to act as a possible bioremediation strategy [92,93]. However, the geochemical environment at such sites can affect microbial redox transformations and the reduced products. Mineral phases can limit the bioavailability of U(VI) for microbial reduction [104], whilst the presence of other redox active species can inhibit U(VI) reduction by acting as competing electron acceptors, or can re-oxidise the biogenic U(IV) [94,98,108–110]. A thorough knowledge of these microbial transformations and their role in the biogeochemistry of redox active radionuclides is needed to understand the environmental behaviour of these radionuclides and to develop bioremediation strategies to limit their migration.