

1 Introduction
Modified tetraazamacrocycles bearing coordinative pendant arms are continuously of growing interest because of their potentialities in medicinal and biological applications that exploit the large ability of these ligands to form specially inert metal complexes [1–3]. The reinforced macrocycles, constrained by an ethylene or propylene bridge between two adjacent (side-bridged ligand [4]) or opposite nitrogen atoms (cross-bridged ligand [5,6]) were also investigated. This particular class of ligands exhibits unusual acid–base properties [6,7], redox behaviour [8,9] and has revealed a particular predisposition to give kinetically inert metal complexes [9,10] which motivated different groups to investigate their coordinating properties [11–15]. Moreover, in a recent paper Archibald and coworkers presented the synthesis of a side-bridged configurationally restricted analogue of the bis-cyclam AMD3100 and described the high HIV inhibitors properties of its zinc complex [16].
We previously reported a facile route to the synthesis of trans-alkylated bis-macrocycles and of side-bridged bis-macrocycles using the bis-aminal cyclen derivative as key-intermediate [17–19]. We here report the facile route to new pendant arms cross-bridged bis-cyclens and their characterization as proton-sponge structures. Their properties are evidenced from NMR, crystal structure, and potentiometric data.
2 Experimental
2.1 General
All reagents were of commercial quality and solvents were dried using standard procedures. Elemental analyses were performed at the “Service de microanalyse”, CNRS, 91198 Gif-sur-Yvette, France. Mass spectrometry analyses were performed at the “Centre régional de mesures physiques de l'Ouest”, Rennes, France. 1H and 13C NMR spectra were recorded on Bruker AC300 and Bruker AMX400 spectrometers (respectively, 75.47 and 100.62 MHz for 13C).
2.2 X-ray investigations
Single-crystal X-ray diffraction data were collected by François Michaud (Université de Bretagne occidentale) at 170 K on an X-CALIBUR-2 CCD 4-circle diffractometer (Oxford Diffraction) with graphite-monochromatized Mo Kα radiation (λ = 0.71073). Analysis of compound 5b: colourless rod-shape crystals were obtained from an evaporated aqueous solution. Crystal data and structure refinement for 5b are summarized in Table 1. Unit-cell determination and data reduction, including interframe scaling, Lorentz, polarization, empirical absorption and detector sensitivity corrections, were carried out using attached programs of Crysalis software (Oxford Diffraction) [20]. Structure was solved by direct method and refined by full-matrix least squares method on F2 with, respectively, SIR92 [21] and Shelxl 97 [22] suites of programs. The hydrogen atoms were identified at the last steps and refined under geometrical restraints and isotropic U-constraints [23].
Crystal data and structure refinementa for 5b
| Empirical formula | C32H64Br2N8O4 |
| Formula weight (g mol−1) | 784.74 |
| Molecular symmetry | C2 |
| Sample shape | Truncated prism, axis [010] |
| Sample dimensions (mm) | 0.14 × 0.10 × 0.10 mm |
| Crystal system/space group | Monoclinic, C2/c |
| Z | 4 |
| a (Å) | 25.353(2) |
| b (Å) | 8.8764(8) |
| c (Å) | 16.0860(14) |
| α = γ (°) | 90 |
| β (°) | 95.934(7) |
| v (Å3) | 3600.6(5) |
| T (K) | 170(2) |
| λ (Å) | 0.7107 |
| μ (mm−1) | 2.299 |
| Dx (Mg m−3) | 1.448 |
| Measured reflections | 16 621 |
| Unique reflections | 3657; 2998 with I > 2σ(I) |
| F(000) | 1656 |
| Θ | 3.15° < θ < 26.37° |
| Rint. | 0.0451 |
| h | −31 → 31 |
| k | −11 → 11 |
| l | −20 → 19 |
| R1 [I > 2σ(I) and all data] | 0.0394 and 0.0530 |
| wR2 [I > 2σ(I) and all data] | 0.0926 and 0.0994 |
| S | 1.055 |
| w [I > 2σ(I)] | 1/[σ2 + (0.0557P)2]b |
| Δρmax (eÅ−3) | 0.606 |
| Δρmin (eÅ−3) | −0.237 |
a Refinement on all F2, parameters, restraint, data, rejected, with I > 2σ(I).
b P = (Fo2 + 2Fc2)/3.
2.3 Potentiometric investigations
Potentiometric measurements were performed in a jacketed cell thermostated at 25.0 °C, kept under inert atmosphere of purified argon, using an automatic titrator (Metrohm, DMS Titrino 716) connected to a microcomputer. The free-hydrogen concentrations were measured with a glass-Ag/AgCl combined electrode (Metrohm) filled with 0.1 M NaCl. The electrode was calibrated in order to read −log [H+], designated as p[H], by titration of a small quantity of diluted HCl by standardized NaOH at 0.10 M ionic strength and 25 °C (and determining the equivalent point by the Gran's method) followed by adjustment of the electrode parameter by using the program GLEE so as to minimize the calculated p[H] vs. observed values. log Kw for the system, defined in terms of log([H+][OH−]), was found to be −13.78 at the ionic strength employed and was settled as constant during refinements [24]. NaCl was employed as supporting electrolyte to maintain the ionic strength at 0.10 M. Potentiometric measurements obtained from NaOH titration of HCl solutions containing azaligands were made at about 1 mM in concentration and ionic strength μ = 0.10 M (NaCl). Each titration makes use of at least 10 points per neutralisation of a hydrogen ion equivalent and titrations were repeated till reaching a satisfactory agreement. A minimum of three sets of data was used in each case to calculate the overall stability constants and their standard deviations. The confidence interval based on the standard deviations obtained for the different protonation constants is ±0.02 and the range of accurate p[H] measurements was considered to be 2–12 (see Table 2). Equilibrium constants and species distribution diagrams were calculated by using the programs HYPERQUAD 2003 [25]. The stability constants Klh were noted with respect to ternary species LlHh where l and h are, respectively, the stoichiometric number of L (ligand) and H (proton).
Logarithm of protonation constants of related ligands
| Equilibrium | 5a | 5b |
| L + H = LH | >12 | >12 |
| LH + H = LH2 | 10.68 (2) | 10.54 (2) |
| LH2 + H = LH3 | 8.97 (2) | 8.81 (2) |
| LH3 + H = LH4 | 4.58 (2) | 4.31 (2) |
| LH4 + H = LH5 | <2: 1.93 | <2: 1.71 |
2.4 Syntheses and spectroscopic data
Cyclen-glyoxal 1 and the corresponding mono-alkylated ammonium salt 2 were synthesized according to experimental conditions detailed elsewhere [14,15]. The first one was quantitatively obtained by direct condensation of glyoxal with cyclen; the subsequent addition of the electrophile to a solution 0.75 M (THF) of cyclen-glyoxal at room temperature led to a mono ammonium salt in 88% of yield. Alkylation was performed with ethyl bromoacetate for 3. The dimerisation with a bis-electrophile was carried out in CH3CN with αα′-dibromo-meta-xylene (compound 3a, yield: 84%) and αα′-dibromo-para-xylene (compound 3b, yield: 89%) [18].
2.4.1 Synthesis of diesters 4a–b
In a typical procedure, a large excess of NaBH4 (24 equiv) was added in small portions over 1 h to a stirred solution of polyammonium salts in absolute ethanol (1 g in 60 mL). The mixture was allowed to stir at room temperature for one week. After cooling at 0 °C and HCl (2 M) addition until pH ∼ 3–4, the resulting mixture was evaporated to dryness. The resulting white solid was then dissolved in a small quantity of water and potassium hydroxide pellets were added until basic medium. The aqueous phase was then evaporated to dryness and the residue was extracted with chloroform (3 × 50 mL). The combined organic phases were finally evaporated to give compounds 4a–b (R = CH2COOEt) in 78% of yield for 4a (0.590 g) and 81% for 4b (0.630 g).
2.4.2 Synthesis of diols 5a–b
The reduction of the polyammonium salt was carried as described in previous procedure, excepted the solvent used here which is 95% ethanol instead of absolute ethanol for the polyammonium salt solution. After extraction with chloroform (3 × 50 mL) in order to eliminate the remaining diester (around 10 %), the aqueous phase was evaporated and absolute ethanol was added to the white residue. The mixture was stirred under reflux and filtered. Evaporation of ethanol gave 5a–b (R = CH2CH2OH) in 90% of yield for 5a (0.650 g) and 88% for 5b (0.730 g).
Finally compounds 5a–b were dissolved in a small amount of water (5 mL) and eluted through a column packed with an Dowex 1X2-200 (OH− form) anion exchange resin. After elution with water (100 mL) and solvent evaporation, the dimeric ligands 5a–b were obtained as their mono-bromhydrate forms, respectively, in 91% (0.591 g) and 89% (0.649 g) of yields.
2.4.3 Mono-alkylated ammonium salt 2
1H NMR (300 MHz, D2O, 298 K): δ = 1.35 (t, 3H), 2.47–2.88 (m, 2H), 2.91–2.99 (m, 4H), 3.20–3.36 (m, 4H), 3.58–3.64 (m, 2H), 3.66–3.70 (m, 1H), 4.10–4.22 (m, 4H), 4.36 (qd, 2H), 4.40–4.50 (m, 1H), 4.79 (s, 2H) ppm. 13C NMR (100 MHz, D2O, 298 K): δ = 16.2 (CH3), 46.8, 50.5, 51.1, 54.1, 60.0, 62.2 (CH2 α), 66.7 (OCH2), 67.3, 74.4, 74.5 (CH2 α), 87.3, 87.5 (CHaminal), 168.0 (CO) ppm. FAB-MS: m/z 361.1 [M + H]+. Found: C 44.28, H 7.00, N 14.61%. Calc. for C14H25N4O2Br·H2O: C 44.33, H 7.18, N 14.77%.
2.4.4 Tetra-ammonium dimer 3a (R = CH2COOEt, meta)
13C NMR (100 MHz, D2O, 298 K): δ = 18.7 (CH3), 48.2, 51.6, 52.3, 59.9, 60.0, 62.0, 62.9, 65.7, 66.4 (CH2 α) 66.5 (CH2Ph), 69.5 (OCH2), 83.3, 84.7 (CHaminal), 133.3, 136.6, 140.6, 141.0 (Ar), 170.1 (CO) ppm. FAB-MS: m/z 987.5 [M + H]+. Found: C 43.01, H 6.29, N 10.91%. Calc. for C36H60N8Br4O4·H2O: C 43.04, H 6.02, N 11.15%.
2.4.5 Tetra-ammonium dimer 3b (R = CH2COOEt, para)
13C NMR (100 MHz, D2O, 298 K): δ = 16.2, 45.7, 49.0, 49.8, 57.7, 59.6, 60.5, 63.0, 64.0, 67.0, 80.6, 83.1, 132.2, 167.6 ppm. FAB-MS: m/z 987.5 [M + H]+. Found: C 43.29, H 6.14, N 11.31%. Calc. for C36H60N8Br4O4·H2O: C 43.04, H 6.02, N 11.15%.
2.4.6 Di-alkylated dimer 4a (R = CH2COOEt, meta)
1H NMR (300 MHz, CDCl3, 298 K): δ = 0.91 (t, 3H, CH3), 2.60–2.85 (4 t, 16H, CH2α), 2.97 (t, 4H, CH2 bridge), 3.28 (s, 2H, NCH2CO), 3.56 (s, 2H, CH2Ph), 3.80 (qd, 2H, COCH2), 6.89 (t, 1H, CHAr), 6.94 (d, 1H, CHAr), 7.27 (s, 1H, CHAr), 11.74 (s, 2H, NH+) ppm. 13C NMR (100 MHz, CDCl3, 298 K): δ = 13.4 (CH3), 46.1, 50.1, 51.4, 54.6 (CH2α), 55.0 (OCH2), 55.1, 57.4 (CH2α), 59.8, (CH2Ph), 133.3, 136.6, 140.6, 141.0 (Ar), 170.1 (CO) ppm. Mp: 132–135 °C. Found: C 56.31, H 8.55, N 14.46, Br 6.32, Cl 6.52%. Calc. for C36H64N8O4Br0.6Cl1.4: C 56.12, H 8.37, N 14.54, Br 6.22, Cl 6.44%.
2.4.7 Di-alkylated dimer 4b (R = CH2COOEt, para)
1H NMR (300 MHz, CDCl3, 298 K): δ = 1.11 (t, 3H, CH3), 2.34–2.80 (4 t, 16H, CH2α), 3.00 (t, 4H, CH2 bridge), 3.33 (s, 2H, NCH2CO), 3.49 (s, 2H, CH2Ph), 4.21 (qd, 2H, COCH2), 7.26 (s, 2H, CHAr), 11.63 (s, 2H, NH+) ppm. 13C NMR (100 MHz, CDCl3, 298 K): δ = 14.2 (CH3), 46.7, 51.2, 52.3, 55.2 (CH2α), 56.0 (OCH2), 56.4, 57.9 (CH2α), 60.7 (CH2Ph), 129.0, 137.6 (Ar), 171.5 (CO) ppm. Mp: 136–138 °C. Found: C 55.33, H 8.41, N 14.26, Br 8.39, Cl 5.32%. Calc. for C36H64N8O4Br0.8Cl1.2: C 55.48, H 8.28, N 14.38, Br 8.20, Cl 5.46%.
2.4.8 Di-alkylated dimer 5a (R = CH2CH2OH, meta)
1H NMR (300 MHz, D2O, 298 K): δ = 2.55–2.90 (5t, 20H, CH2α), 2.93 (t, 2H, CH2α arm), 3.49 (t, 2H, CH2OH), 3.64 (s, 2H, CH2Ph), 7.07 (d, 1H, Ar), 7.14 (d, 1H, Ar) ppm. 13C NMR (100 MHz, CDCl3, 298 K): δ = 56.1, 57.2, 57.5, 57.8, 58.3 (CH2α), 59.5 (CH2α arm), 60.2 (CH2OH), 60.7 (CH2Ph), 127.1, 127.6, 128.2, 139.9 (Ar) ppm. Mp: 144–147 °C. Found: C 52.63, H 9.01, N 15.16, Br 7.89, Cl 6.02%. Calc. for C32H60N8O2Br0.7Cl1.3·2H2O: C 52.87, H 8.87, N 15.41, Br 7.69, Cl 6.34%.
After exchange resin column: Found: C 54.78, H 8.82, N 15.65, Br 10.99%. Calc. for C32H59N8O2Br·2H2O: C 54.61, H 9.02, N 15.92, Br 11.35%.
2.4.9 Di-alkylated dimer 5b (R = CH2CH2OH, para)
1H NMR (300 MHz, D2O, 298 K): δ = 2.51–2.95 (5t, 20H, CH2α), 2.99 (t, 2H, CH2α arm), 3.51 (t, 2H, CH2OH), 3.72 (s, 2H, CH2Ph), 7.28 (s, 2H, Ar) ppm. 13C NMR (100 MHz, D2O, 298 K): δ = 49.5, 53.9, 54.6, 58.7, 58.9, 59.0 (CH2α), 61.6 (CH2OH), 62.4 (CH2Ph), 132.4, 140.9 (Ar) ppm. Mp: 141–144 °C. Found: C 52.00, H 8.91, N 15.06, Br 10.01, Cl 5.11%. Calc. for C32H60N8O2Br0.9Cl1.1·2H2O: C 52.23, H 8.77, N 15.23, Br 9.77, Cl 5.30%.
After exchange resin column: Found: C 54.64, H 8.65, N 15.95, Br 10.73%. Calc. for C32H59N8O2Br·2H2O: C 54.61, H 9.02, N 15.92, Br 11.35%.
2.4.10 Crystals of di-alkylated dimer 5b (R = CH2CH2OH, para)
Mp: 136–139 °C. Found: C 49.09, H 8.45, N 14.06, Br 20.69%. Calc. for C32H60N8O2Br2·2H2O: C 48.98, H 8.22, N 14.28, Br 20.37%.
3 Results and discussion
3.1 Synthetic route to studied ligands
Starting from cyclen-glyoxal 1, the route to constrained bis-cyclens consisted of three easy-to-run steps (Scheme 1). Cyclen-glyoxal 1 was quantitatively obtained by direct condensation of glyoxal with cyclen [17–19]; the subsequent mono-alkylation, performed by addition of the electrophile to a solution of 1, led to compound 2 in good yields. The bis-macrocyclic ligands were obtained by reaction with a bis-electrophile (αα′-dibromo-meta-xylene and αα′-dibromo-para-xylene) in CH3CN as previously reported [17] to lead to 3a–b in high yields. The reductive cleavage of 3a–b with NaBH4 in ethanol 95% gave in good yields the bis-N-hydroxy-ethyl (R = CH2CH2OH) 5a–b derivatives as the main product of the reaction. It is interesting to note that when the reductive cleavage was carried out in absolute ethanol, the ethyl ester cross-bridged constrained bis-macrocycle 4a–b was obtained as only product. The diesters and diols 4a–b and 5a–b were obtained as their diprotonated bromhydrates and chlorhydrate mixture derivatives, as indicated by elemental analysis. Pure crystals of 5b·2HBr were obtained from the mixture by recrystallisation in dilute (0.5 M) KOH solution.
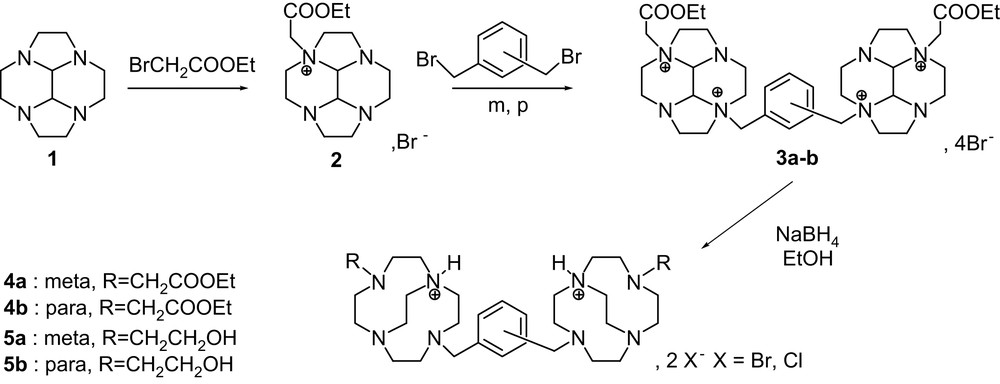
Pendant armed cross-bridged bis-cyclen synthesis.
3.2 Structure of constrained ligands
3.2.1 NMR studies
Evidence of the structure of the macrobicyclic products was also obtained from 13C, 1H NMR spectra, microanalyses and from X-ray crystallography of 5b. In aprotic solvent (unfortunately diol 5a–b derivatives are only soluble in protic solvents), the 1H NMR spectra of the diesters 4a–b exhibited a proton signal at around 11.7 ppm, integrated for 2 protons corresponding to an ammonium group NH+, in rapid exchange in each constrained cyclen subunits, as indicated by the symmetry of the overall spectrum (Fig. 1: with 4a in CDCl3). A similar NMR behaviour was previously observed in some strong basic analogous monomeric cross-bridged azamacrocycles [6] and reinforced polyazamacrocyclic compounds [19,26] in which such a signal was, respectively, detected at around 9–10 ppm and 12 ppm.
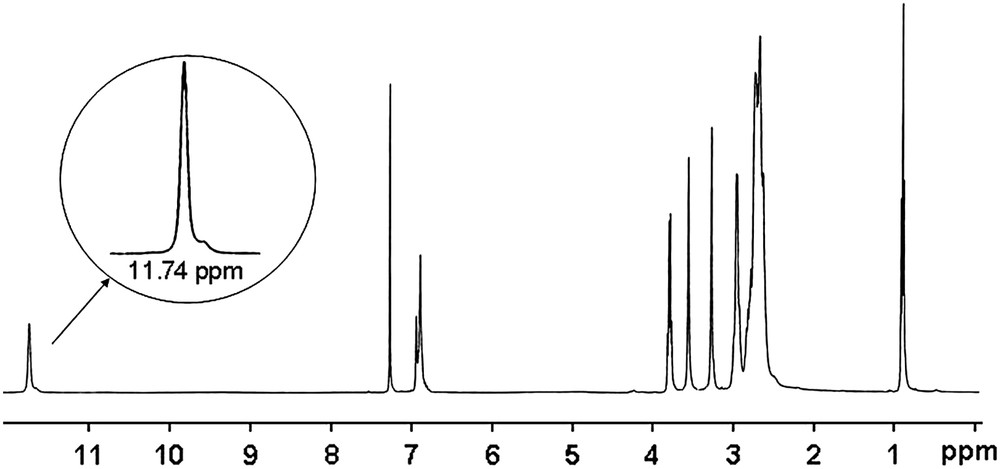
1H NMR (300 MHz, CDCl3, 298 K) of 4a.
3.2.2 Crystal structure
Crystallographic data for 5b are given in Table 1. The ORTEP representations in Fig. 2 depict a dimeric structure in a C2 symmetry, the C2 axis being in the same plane as the phenyl moiety. One can observe that in solid state, the bromine anion is not situated inside the cavity of the two macrocyclic moieties. The two cyclen cores are situated in a face-to-face conformation: the average plane of the two cycles is almost parallel. The distance between the two macrocyclic centres determined by the four nitrogen atoms of each cyclen is 7.98 Å, pointing the proximity of the two cavities. The pendant arm of each macrocycle is directed towards the opposite cycle; thus, these two arms are situated in two parallel axes with a distance between the two oxygen atoms of each alcohol function of 4.40 Å. In the solid state, the dimeric structure defines a cylindrical cavity delineated by the two cycles, the two arms and the xylenyl linker. The ethylene bridges are localized outside each delineated macrocyclic cavity, generating a strongly constrained conformation. The two protons are localized on the N2, N′2 nitrogen atoms to respect the charges repartition in the structure. In addition, the relatively weak distance between the oxygen atoms and the ammonium sites suggests the possibility of hydrogen bonding, which locks the two cyclen moieties in the face-to-face conformation. This configuration is very atypical for this kind of open bis-macrocycles: generally for diprotonated tetraaza-dimers, the two macrocyclic cores are situated as far as possible each one from the other [18].
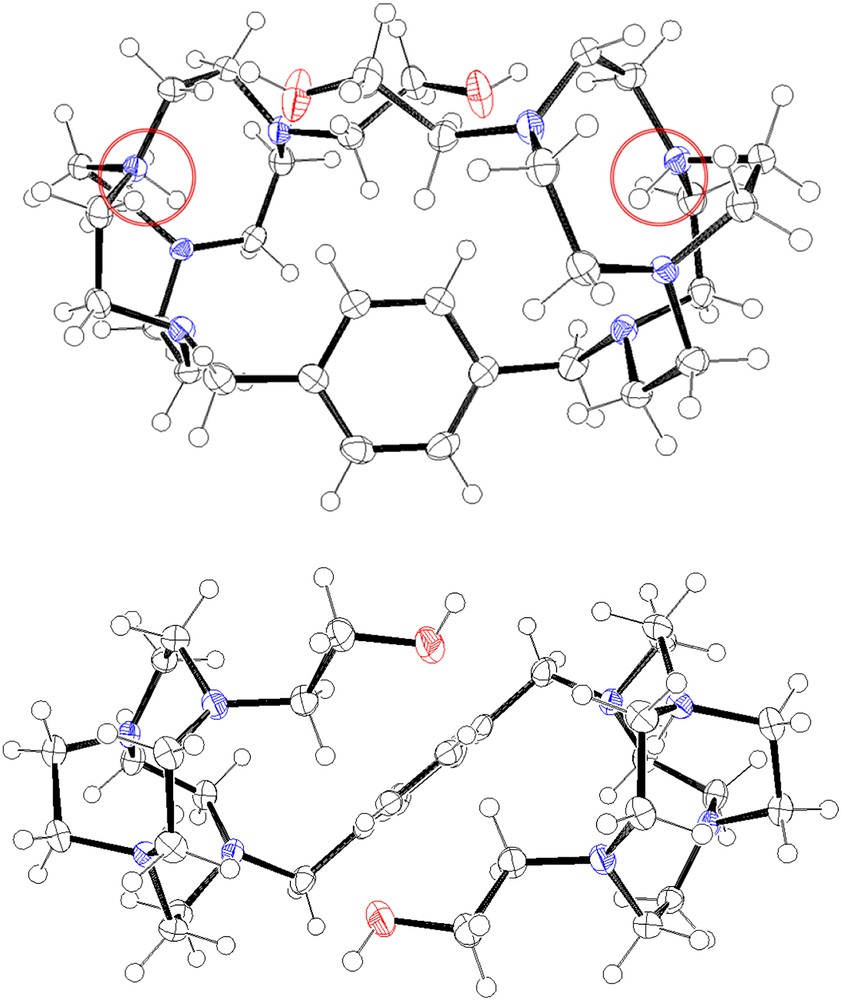
ORTEP views of 5b depending on two axes, 50% probability ellipsoids.
3.2.3 Protonation constants
In order to emphasize the behaviour of the ligands by analytical evidence, we performed potentiometric studies for diprotonated 5a–b (4a–b are hydrolyzed in acidic medium). Potentiometric measurements were acquired from NaOH titration of HCl solutions containing the azaligands. The logarithm of the stepwise protonation constants for each studied ligands, obtained from the mathematical treatment of their respective potentiometric titration data is presented in Table 1. The two ligands exhibit a first protonation constant which is one of the very strong base and cannot be appreciated by the potentiometric methodology in the investigated p[H] range. Four other protonation constants could be measured: two correspond to strong to moderate bases; one behaves as a weak acid and one as a strong acid. The other protonation constants are too low to be detected in the investigated p[H] range. Species distribution diagrams for the 5a and 5b ligands as a function of p[H] and deduced from protonation constants are presented in Fig. 3. They show the predominance of LH33+ in neutral domain and the high level of protonation of the two ligands all along the p[H] scale, even in basic medium, since LH+ are the last species identified at p[H] 12.
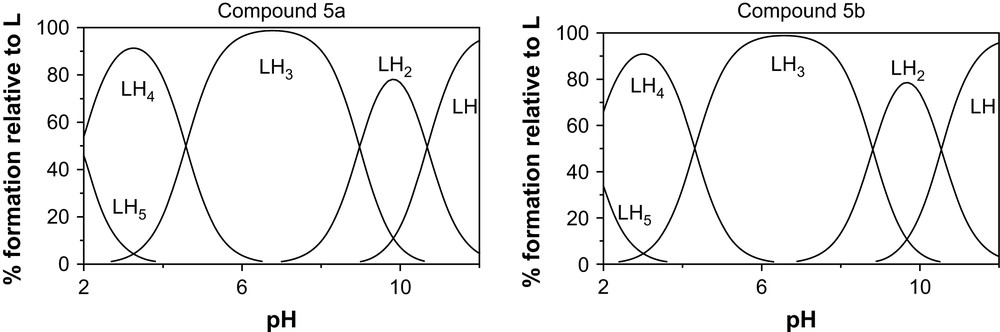
Species distribution diagrams for the 5a and 5b ligands as a function of p[H].
When compounds 5a–b were dissolved in water and eluted through a strongly basic anion exchange resin (Dowex 1X2-200, OH− form), after elution and water evaporation, the dimeric ligands were obtained as their mono-protonated forms, indicating that the first basicity is stronger than the second.
When the mono-protonated forms of 5a–b were analyzed, the titration curves of their acidic solution by NaOH presented some difference with the ones obtained from the diprotonated forms (Fig. 4).
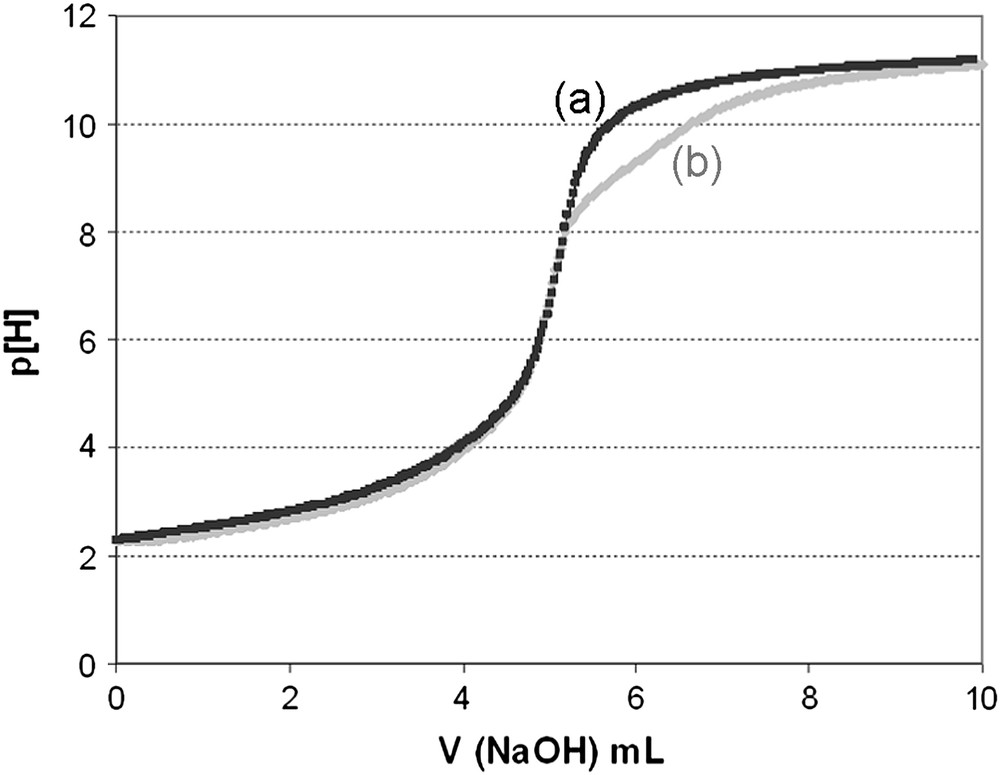
Potentiometric curve titration of 5b (mono-protonated form) before (a) and after (b) refluxing the solution.
However, when the acidic solutions of mono-protonated salts were heated to reflux for a moment before potentiometric measurements, subsequent titration curves became identical to the ones obtained from diprotonated 5a–b. This is consistent with a fast external protonation of the free macrocyclic moiety, followed by a slow insertion of the proton inside the macrocyclic bowl-cavity. Such behaviour concerning comparative ligands as bis-adamanzane was already mentioned by Springborg et al. [26].
4 Conclusion
In summary, this efficient three-step synthesis has yielded a new bis-cyclen family in which the distance between the centres of the two macrocyclic units can be modulated according to the linker employed. Neither high dilution conditions, nor fastidious protection step work-up were needed, and the compounds were obtained in good overall yields. Moreover, this ligand family of cross-bridged bis-cyclens possessing functionalized pendant arms could be easily extended to other derivatives by changing the first alkylation step of cyclen-glyoxal 1 or by reaction of the terminal function borne by the alkyl chain. The cage-like structures of these constrained macropolycycles are expected to present interesting properties in metal coordination induced by the two reinforced cycles and their proton-sponge behaviour. Moreover, pendant arms macrocyclic ligands are known to equilibrate with metal ion much faster than the parent macrocycle [27]. Following on this, it becomes of interest to investigate the thermodynamic and kinetic properties [28] of their metal and anion complexes. These features are under investigation.
Acknowledgements
This research work was supported by the “Ministère de l'Éducation nationale et de la Recherche” and by the “Centre national de la recherche scientifique”.