

1 Introduction
In the last three decades, biodegradable polymers have received increasing attention as resorbable biomaterials as well as commodity thermoplastics. Thanks to their biodegradability, biocompatibility and ready availability from inexpensive renewable resources, polylactides (PLAs) are at the forefront position in this field [1]. PLAs can be directly prepared from lactic acid by polycondensation under azeotropic distillation conditions, but ring-opening polymerization (ROP) of lactide is usually preferred, since it gives access to higher molecular weights and lower polydispersities, and allows the preparation of block copolymers (Scheme 1). As recently reviewed [2], the numerous well-defined complexes investigated for lactide polymerization over the last 15 years have contributed significantly to a better understanding of the factors that govern the polymerization, and spectacular improvements have thereby been achieved in terms of activity and productivity as well as of stereocontrol.
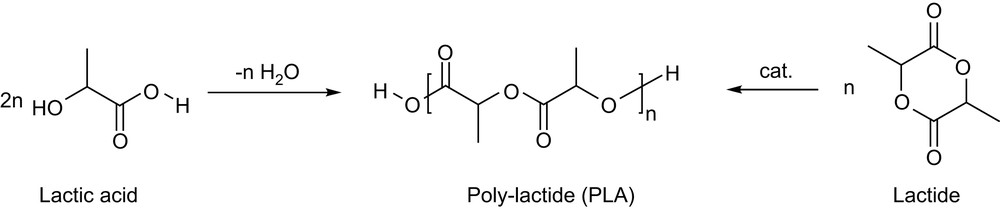
Preparation of PLAs by polycondensation or ring-opening polymerization (ROP).
In addition, various metal-free systems have been developed over the last five years for the living polymerization of lactide, following the pioneering contribution of Hedrick and Waymouth. The first part of this review focuses on these recent achievements and concerns both organocatalytic and enzymatic approaches. The nucleophilic, cationic and bifunctional systems will be presented from both polymerization and depolymerization viewpoints. The second part of the review concerns the heterocyclic monomers that give access to poly(α-hydroxy acids) such as PLAs. The two complementary approaches recently reported in the literature will each be discussed: substituted 1,4-dioxane-2,5-diones and synthetic equivalents for 1,4-dioxane-2,5-diones.
2 Metal-free catalysts for the controlled preparation of poly(α-hydroxy acids)
2.1 Nucleophilic polymerization
2.1.1 General considerations
Over the last 10 years, enzymes, amines, phosphines and carbenes have all proved efficient nucleophilic transesterification catalysts for the ROP of lactide. Their mode of action is not precisely known, but the polymerization most probably occurs through an activated-monomer mechanism [3,4] involving a transient lactide–catalyst complex 1 (Scheme 2). For lipases, a serine moiety has been suggested as the active nucleophilic site and thus, the enzyme-activated monomer 1 would be an acyl–enzyme complex. The protic initiator ROH that is required with all of these nucleophilic catalysts (typically water for lipases and alcohols for organocatalysts) would then react with this lactide–nucleophile (Nu) complex to form the ring-opened adduct 2 with simultaneous liberation of the nucleophilic catalyst. Chain propagation would proceed analogously by reaction of the ω-hydroxyl group of 2 with an activated monomer 1. Accordingly, the nature of the α-chain-end of the resulting polylactide, namely an acid or ester functionality, inherently depends on the initiating protic agent. This polymerization pathway differs fundamentally from that with metal complexes. Indeed, the nucleophilic catalyst only activates the monomer towards ring opening, whereas the metal complex activates the monomer, initiates the polymerization and remains bound to the growing chain.

Schematic mechanism for the nucleophilic ROP of lactide (Nu = enzyme, amine, phosphine or N-heterocyclic carbene, ROH = the initiating protic agent or the growing polymer chain).
2.1.2 Enzymes
The discovery of enzymes which are active in organic media at high temperatures made biocatalysis a valuable tool in organic [5] and polymer chemistry [6], including ring-opening polymerization [4,7]. In particular, various commercially available lipases proved to be active for the ROP of lactide [8,9]. The enzyme origin has a significant influence on the monomer conversion and the molecular weight of the resulting polymer (Table 1). Even with Pseudomonas cepacia lipase PS showing the highest catalytic activities, rather harsh conditions (7–14 days in bulk at 80–130 °C) were necessary to achieve high monomer conversions, but Mw as high as 270,000 g mol−1 with Mw/Mn 1.1 can be eventually reached [9]. Although the enzyme and oligomers are easily removed from the resulting PLA upon workup, the isolated yields are fairly small (<16%). The formation of these oligomeric fractions, along with the observed induction periods are characteristic features of lipase-induced polymerizations. Immobilization of PS on Celite [9] resulted in significant improvements with respect to the lower required enzyme concentration and higher molecular weights (by about 3-fold) of the resulting polymers but at the expense of the polydispersities (>4). Similarly, immobilized Candida antartica (Novozym 435) was found to become active for the ROP of l-lactide at 100 °C in bulk but only oligomers (Mw about 2440 g mol−1) with high polydispersities (Mw/Mn 2.63) were obtained after 10 days [10].
Lipase-catalyzed ROP of d,l-lactide over 7 days
| Enzyme | %wt | T (°C) | Conv. (%) | Yield (%) | Mw (g mol−1) | Mw/Mn | Ref. |
| PPL | 3 | 80 | 37 | 29 | 7500 | 1.2 | [9] |
| Pro K | 3 | 80 | 40 | 40 | 1900 | 1.2 | [9] |
| PS | 3 | 80 | 7 | 3 | 27,000 | 1.2 | [9] |
| PS | 3 | 130 | 100 | 16 | 270,000 | 1.1 | [9] |
| PSa | 5 | 100 | 96 | nd | 8900 | 2.5 | [9] |
| IM-PSa | 0.24 | 100 | >99 | nd | 26,000 | 4.0 | [9] |
| IM-CAa,b | 6 | 100 | 91 | nd | 2440 | 2.6 | [10] |
a l-Lactide was used.
b After 10 days.
Copolymerization of lactide and glycolide is also of major interest in drug delivery formulations for the modulation of the polymer degradation rate. Duchateau and coworkers recently investigated [11] the properties of both free and immobilized P. cepacia with a 4/1 lactide/glycolide molar ratio. Interestingly, an in-house method based on MALDI-TOF mass spectrometry provided not only the end groups and chemical composition but also the microstructure of the resulting copolymers. Accordingly, linear polymers are essentially obtained at 100 °C, whereas cyclic random copolymers are the main products at 130 °C (Scheme 3).
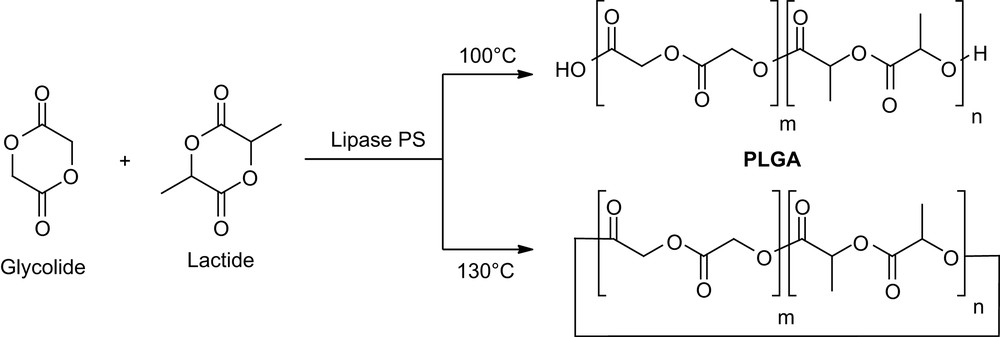
Copolymerization of d,l-lactide and glycolide (4/1) by Lipase PS.
Copolymerization of lactide and 3(S)-isopropylmorpholine-2,5-dione (IPMD) (Scheme 4) has also been reported using porcine pancreatic lipase type II (PPL) [12]. Random copolymers of various compositions were obtained after 6 days at 100 °C. Whatever the mole fraction of lactide in the feed (from 100 to 15%), its conversion remains unchanged at about 85%, while the conversion of IPMD increases (up to 66%) with its mole fraction. Rather large amounts of low molecular weight materials are formed but the high molecular weight fractions (Mw about 10,000 g mol−1, yields from 13 to 42%) exhibit very low Mw/Mn (<1.05).
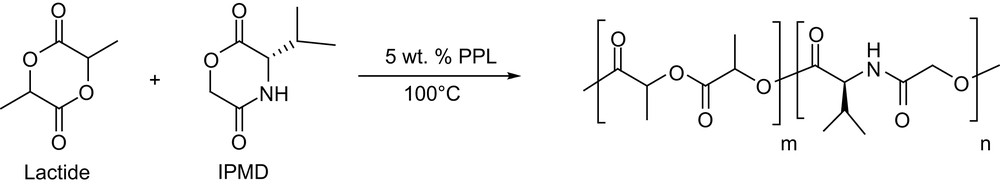
Copolymerization of lactide and IPMD by porcine pancreatic lipase type II.
Morse and coworkers recently reported the biocatalytic ROP of lactide using silicatein enzymes. These protein filaments consist of three individual species [13]. The most abundant one, referred to as silicatein α (up to 70% by weight), is highly similar to papain-like proteases except that the active site cysteine is replaced by a serine. Lactide was polymerized under mild conditions (16 °C, PBS) with both silicatein native filaments and a recombinant expression of silicatein-α at the external surface of Escherichia coli cells [14]. After incubation for 16 h, the accumulation of polymeric products at the filament and cell surface was apparent from scanning electron microscopy. Although the intimate contacts between the polymers and the reactive surfaces hampered the achievement of high yields and therefore the complete characterization of the resultant PLA, the presence of ester groups at the filament surface was deduced from X-ray photoelectron spectroscopy (XPS) and the amorphous character of the polymers was established by microscopic SAED (selected area electron diffraction) analyses.
Besides ROP, the enzymatic polycondensation of lactic acid was also investigated in various organic solvents [15]. Significantly higher conversions of lactide were obtained with less polar solvents (67% in decane and chloroform vs. 20% in ethyl acetate after 2 days at 37 °C). Notably, Wang et al. recently demonstrated the beneficial influence that chemical modification may have on enzyme activities in polycondensation. Accordingly, two decanoyl-chloride-modified enzymes (an alkaline protease from Bacilllus sp. and Novozyme 435) were found to be more soluble in organic solvents than the corresponding pegylated enzymes previously described [16]. As a result, activities up to 22 times higher, compared to native enzymes, were observed in chloroform or THF for the transesterification of ethyl lactate at 50 °C. So far, only low molecular weights PLA (Mw < 2000 g mol−1) have been produced regardless of the differences in enzyme activity, but chemical modification should be definitely considered a promising way to improve enzyme activities.
This recent progress clearly demonstrates the feasibility and potential of enzyme-catalyzed lactide polymerization in organic solvents, but the practical application of this approach still requires further improvement [17]. Enzymes are not yet capable of competing with metal-based initiators or organocatalysts in terms of reaction time and polymerization control.
2.1.3 Organocatalysts
Nucleophilic catalysts (tertiary amines, phosphines and N-heterocyclic carbenes) have been shown to promote the polymerization of strained cyclic esters such as lactide in a controlled fashion. The living character of the nucleophilic polymerization has been established for all organocatalysts by a linear correlation between the molecular weight of the polymer and the monomer conversion. As a consequence, the degree of polymerization closely tracks the initial monomer-to-initiator ratio and polylactides of controlled molecular weights and narrow distributions are obtained. In addition, PLA featuring various ester groups at the α-chain-end can be prepared using the corresponding primary or secondary alcohol as the initiator.
2.1.3.1 Pyridines
4-Aminopyridines were the first nucleophilic organocatalysts to be used for the polymerization of lactide. DMAP and PPY (Fig. 1), used in equal quantities to that of the initiator, were found to be highly active [3]. High monomer conversions were obtained for monomer to initiator ratios up to 140 both in dichloromethane solution (∼1.4 M, 35 °C, a few days) and in bulk (135 °C, a few minutes). Notably, prolonged heating in dichloromethane do not induce detectable changes in molecular weight and polydispersity, indicating that undesirable side reactions are less operative with pyridine catalysts than with metal complex initiators. Interestingly, a polystyrene-immobilized form of DMAP, which can be easily removed by filtration, was reported to give comparable molecular weights, polydispersity indexes and even polymerization kinetics.
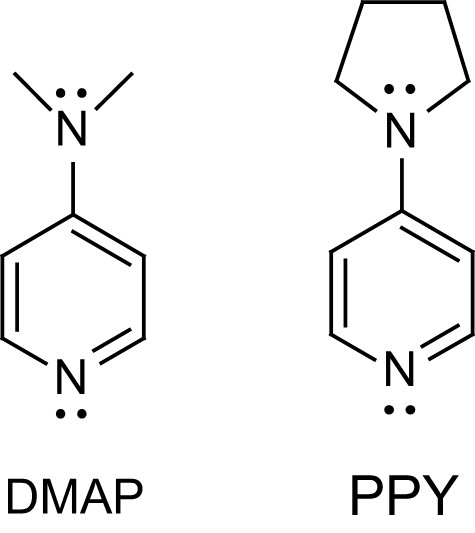
Structure of 4-dimethylaminopyridine DMAP and 4-pyrrolidinopyridine PPY.
DMAP has also been reported to catalyze efficiently the condensation of lactic acid oligomers in the presence of dicyclohexylcarbodiimide (DCC) (Scheme 5). This approach, first reported in dichloromethane [18], has been recently extended to supercritical carbon dioxide [19]. With this “green” medium, the residual DMAP and DCC can be easily removed from the resulting PLLA samples by supercritical fluid extraction, but a subsequent dissolution/filtration sequence remains necessary to eliminate the dicyclohexylurea produced. Despite their insolubility in scCO2, PLLA with molecular weights as high as 13,500 g mol−1 with Mw/Mn of 1.56 could be achieved at 80 °C and 3500 psi. This result further highlights the potential of scCO2 as an alternative reaction medium for the ROP of lactide, with no apparent deleterious effects compared to the use of dichloromethane.
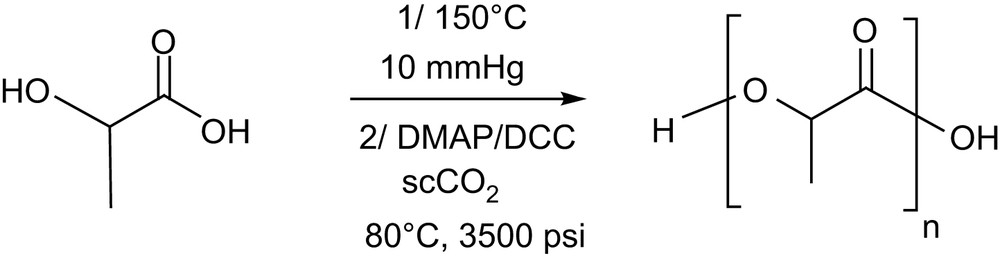
Synthesis of PLAs by condensation of lactic acid oligomers in scCO2.
2.1.3.2 Phosphines
Although phosphines also catalyze the ROP of lactide, they have proved to be much less active than 4-aminopyridines [20]. As anticipated, the substitution pattern of the phosphines strongly affect their activity, but even the most active phosphine catalyst P(n-Bu)3 required rather high temperatures (94 °C in toluene solution, 135 °C in bulk) to achieve high monomer conversions, and catalyst-to-initiator ratios higher than 1 result in broader molecular weight distributions (Mw/Mn 1.3–1.5). In addition, prolonged heating also induced polydispersity broadening. The higher contribution of adverse transesterification reactions for phosphines compared with pyridines most probably results from the higher polymerization temperatures.
2.1.3.3 Carbenes
Over the last 10 years, stable carbenes [21] have led to spectacular achievements both in organometallic catalysis and organic synthesis [22]. The promising results obtained with pyridines and phosphines then motivated the investigation of N-heterocyclic carbenes as organocatalysts for lactide ROP. The representative imidazol-2-ylidene IMes (Fig. 2) proved to be extraordinarily active, with polymerization of up to 200 equiv of lactide being typically achieved in less than 1 h at room temperature to afford PLA of high and controlled molecular weights [23,24].
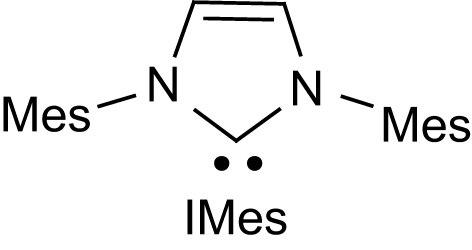
Structure of the representative N-heterocyclic carbene IMes (Mes = 2,4,6-Me3C6H2).
Care must be taken with the catalyst-to-initiator ratio and to the monomer concentration in order to achieve precisely controlled and narrowly distributed molecular weights. Cat/I ratios ranging from 0.25 to 1.5 and 1 M lactide solutions were found to be optimal for DP > 100, leading to Mw/Mn lower than 1.15, whereas much lower Cat/I ratios of about 0.01–0.02 and 2 M lactide solutions were preferred to obtain oligomers (DP ≈ 10) with low Mw/Mn (<1.16). Notably, this NHC-catalyzed approach can be carried out not only in dichloromethane, but also in tetrahydrofuran and toluene, without significant modification of the polymerization rate.
Work then aimed at extending the catalyst pool, and the substitution pattern of the N-heterocyclic carbene was broadly varied in order to reach a basic understanding of the steric and electronic effects [25]. At that stage, the extreme moisture sensitivity of NHCs prevented a rapid screening so that most of the investigated catalysts 3 were formed in situ via deprotonation of the corresponding azolium salts (Scheme 6) [24]. Imidazol-2-ylidenes and imidazolin-2-ylidenes proved to be significantly more active than thiazol-2-ylidenes. As expected, sterically demanding and electron-withdrawing substituents were found to be less favorable [24], while the saturated/unsaturated nature of the NHC backbone has no appreciable influence. As for IMes, careful control of the Cat/I ratio and lactide concentration is needed in order to obtain controlled polymerization, given that the optimal conditions are apparently rather carbene-dependent.

Structure of the N-heterocyclic carbenes investigated as preformed or in situ-generated nucleophilic catalysts for the ROP of lactide.
Polymer-supported thiazolium and imidazolium precatalysts were also effective, but with some restrictions (limited molecular weights, higher Mw/Mn), probably associated with their low solubility [24]. Ionic liquids (ILs) such as the readily available 1-ethyl-3-methylimidazolium tetrafluoroborate [emim][BF4] are much more promising, not only as environment-friendly solvents, but also as NHC reservoirs. Although the precipitation of the polylactide in neat [emim][BF4] makes the conversion incomplete (ca. 50%), much better results were obtained with THF/[emim][BF4] biphasic systems. Indeed, in situ deprotonation of a small amount of [emim][BF4] with potassium tert-butoxide generated the corresponding imidazol-2-ylidene, which migrates to the THF phase and initiates there the ROP of lactide. Once the polymerization is complete, the imidazolium precatalyst can be easily regenerated and returned to the IL phase by addition of a tertiary ammonium salt. The produced polymer is not at all soluble in the IL phase, and is therefore easily and quantitatively recovered from the THF phase (Scheme 7). The feasibility of recycling the ionic liquid was demonstrated with four successive polymerization runs. The practical interest of this phase-transfer process has been substantiated with the preparation of high molecular weight PLAs (Mw > 24,000 g mol−1) with relatively low Mw/Mn (1.4) in >95% yield within 10 min.

Generation of a NHC catalyst from an ionic liquid reservoir for the ROP of lactide (ROH = initiator or propagating chain).
Alternative methods for generating the NHC catalyst in situ were then investigated, especially from thermally activable NHC adducts. The imidazolidines 4–6 (Fig. 3), resulting formally from arene addition to imidazolin-2-ylidenes HIMes and HIPh, were efficiently prepared from the corresponding diamines and aldehydes. The decomposition temperatures of adducts 4–6 were evaluated by modulated differential scanning calorimetry (MDSC) and thermal gravimetric analysis (TGA) measurements. Accordingly, derivative 4 was found to begin to decompose in the solid state at 80 °C, a significantly lower temperature than that required to activate 6 (130 °C) and 5 (165 °C). Polymerization experiments carried out with 100 equiv of lactide in the presence of benzyl alcohol as the initiator established that the lower the thermal stability of the precatalyst, the higher its activity (conversion of 80% was achieved after 3 h at 65 °C with 4 while only 68% conversion could be achieved with 5 after 12 h at 144 °C). The known chloroform adduct 7 led to very similar results in ROP of lactide than 4, in agreement with a comparable thermal stability in the solid state. Unlike 7, the pentafluorophenyl adduct 4 was found to be stable in solution at room temperature, and was therefore the preferred carbene source [26].

NHC adducts used as precatalysts for ROP of lactide.
From a practical viewpoint, however, the use of imidazolidine adducts such as 4–7 is limited by lactide racemization (possibly observed during polymerization) so that alternative delivery agents for NHC were sought. Taking into account that NHC can be efficiently transferred from silver(I) to transition metals, Waymouth and Hedrick evaluated the propensity of compound 8 (Fig. 4), an ionic liquid at room temperature, to promote the ROP of lactide [27]. Thermal decomposition of this precursor occurs at about 90 °C and the polymerization is optimally carried out at 60 °C in toluene, leading to narrow molecular weight distributions (Mw/Mn 1.12) without detectable monomer epimerization.
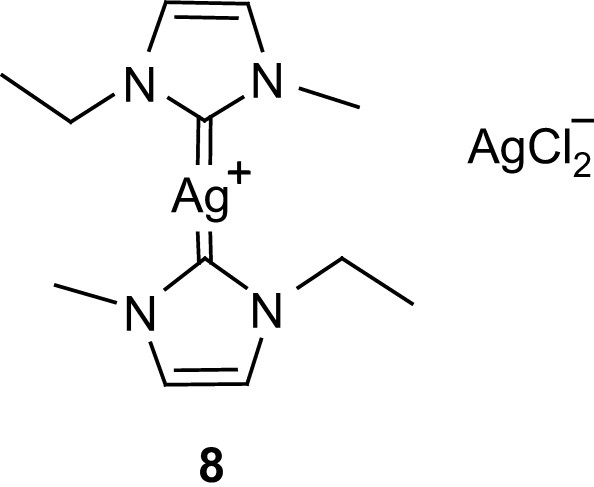
Carbene silver complex.
Even more spectacular results were recently reported with alcohol adducts of NHCs [28,29]. These studies were stimulated by the far diminished reactivity observed for the triazol-2-ylidene 9 (Scheme 8) compared to imidazol-2-ylidenes in the ROP of lactide in the presence of methanol at room temperature. The propensity of carbene 9 to form stable adducts with alcohols was hypothesized to attenuate its reactivity towards polymerization. Taking advantage of the reversible character of this O–H insertion reaction, the polymerization was carried out at 90 °C with various alcohols as initiators. A temperature switch could be operated by alternating high (90 °C) and low temperatures (20 °C) either activating or deactivating the ROP of lactide. Accordingly, the formation of a dormant form of the catalyst with the initiating/propagating alcohol allows precise control of the concentration of the active species, thereby limiting the adverse side reactions. This system is reminiscent of those recently developed for controlled radical polymerization, and was applied to the preparation of block as well as dendritic star copolymers [28]. Variation of the NHC structure allowed further improvement of this approach and culminated with the development of alcohol adducts that function as “all-in-one” catalyst/initiators under mild conditions [29]. Indeed, both primary and secondary alcohol adducts of the imidazolin-2-ylidene 10 were found to be stable solids that readily and reversibly dissociate in solution at room temperature. Under these conditions, the ROP of 100 equiv of lactide (0.7 M) requires less than 15 min and affords well-controlled PLAs. By comparison with that observed for free NHCs, Mw/Mn range from 1.16 to 1.30 and slightly broader distributions were obtained with increased polymerization times, due to adverse transesterification reactions [29].
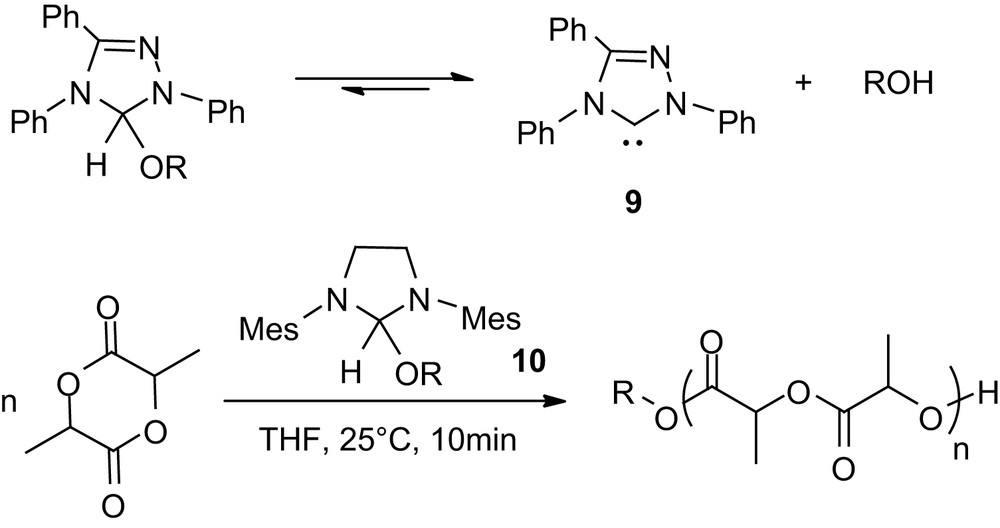
Alcohol adducts of carbenes: single components catalyst/initiator
Intrigued by the possibility of using even simpler “all-in-one” catalyst/initiators, Waymouth and Hedrick also investigated the behaviour of the related, commercially available, tert-butoxybis(dimethylamino)methane 11 (known as Bredereck's reagent) and tris(dimethylamino)methane 12 (Scheme 9). PLAs of controlled molecular weights and narrow distributions were actually accessible with or without initiating alcohol. However, these reagents proved to be far less reactive than the imidazolin-2-ylidene adducts, a few hours at 70 °C being required to achieve high monomer conversions. Under these conditions, an anionic mechanism involving heterolytic C–O cleavage is likely to compete with the initially envisioned O–H α-elimination providing the bis(dimethylamino)carbene [30].

ROP of lactide by Bredereck-type reagents.
In addition, Arnold and coworkers recently reported noticeable activities towards ROP of lactide for derivatives 13 and 14 that combine the imidazol-2-ylidene moiety with amine and alcohol functionalities, respectively [31]. High molecular weight PLAs could be prepared in a few minutes at room temperature (M/I/cat 100/0/1), but rather broad distributions (Mw/Mn 1.29–1.46) were obtained. Interestingly, a stoichiometric reaction of 13 with lactide revealed the presence of two competitive pathways involving either alkoxide-induced O-alkyl cleavage or NHC-induced O-acyl cleavage of the monomer (Scheme 10).
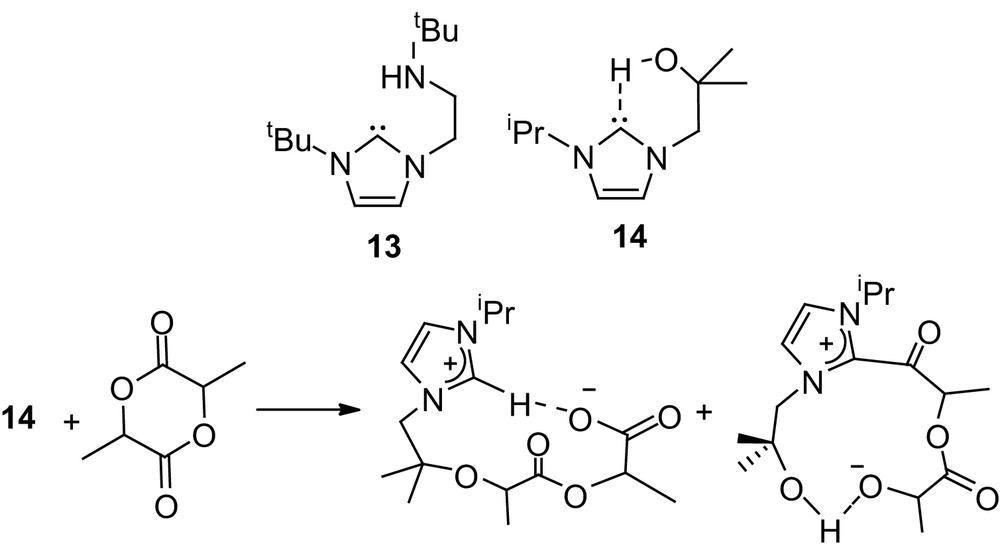
Alcohol- and amine-functionalized NHCs as nucleophilic catalysts for the ROP of lactide.
2.1.3.4 Stereoselective ROP
Besides all these efforts aiming at improving the efficiency of nucleophilic organocatalysts in terms of activity and polymerization control, preliminary studies have also been carried out in stereoselective polymerization. Due to their low activity and the ensuring rather drastic polymerization conditions, chiral phosphines (such as Josiphos or Me-Duphos) do not induce any stereosequence enrichment upon polymerization of d,l-lactide [20]. In contrast, promising results have been obtained with NHCs, in line with their much higher activity. Hillmyer and Tollman first reported the formation of isotactic enriched PLAs upon ROP of d,l-lactide with three imidazol-2-ylidenes, Pm (the probability of meso linkage between monomer units, assuming negligible transesterification) around 0.55 being obtained at room temperature whatever the substitution pattern of the NHC [32]. Quite strikingly, the related [(IMes)Zn(OBn)2]2 complex affords under similar conditions heterotactic enriched PLAs via a coordination–insertion mechanism. Note also that the high activity of IMes allows the polymerization to proceed at −20 °C, which results in a significant increase of the tacticity (Pm = 0.75). Further improvements were recently achieved by Waymouth and Hedrick with sterically encumbered NHCs 15 and 16 [33]. Accordingly, the achiral catalyst 15 (Fig. 5) provided isotactic PLAs with Pm as high as 0.90 from d,l-lactide at −70 °C, and highly enriched heterotactic PLAs (Pm 0.83) from meso-lactide at −40 °C. The phenyl groups in the carbene backbone are supposed to influence the steric hindrance around the catalytic site and thereby to maximize the chain-end stereocontrol. Note also that identical isotacticities were observed from d,l-lactide using (R,R)-16 and rac-16, demonstrating the predominant role of the steric congestion of the catalytic site over its chirality.
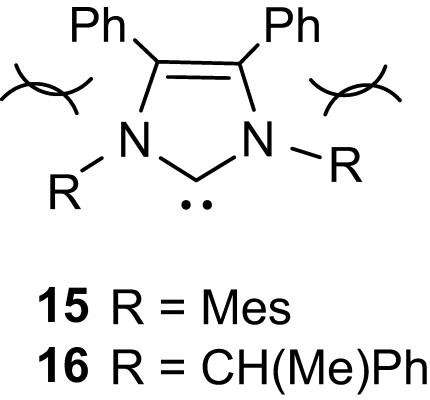
Sterically encumbered NHCs.
These results show that N-heterocyclic carbenes are very active nucleophilic catalysts for the controlled ROP of lactide [25]. The variation of the framework and of the substituents allowed testing of the stability and reactivity of the carbene. In situ generation from the imidazolium salts or from thermolysis of stable adducts liberating the carbene, or even the initiator, now give access to stable, tunable and isolable precatalysts at room temperature. First examples of NHC-catalyzed stereoselective polymerizations were also reported. This catalyst platform should lead to further optimization by adjusting the time and temperature of reaction to control the versatile formation of narrowly dispersed products.
2.2 Cationic polymerization
The use of acids to promote the ROP of lactones has only been scarcely investigated until recently. The ability of trifluoromethanesulfonic acid (HOTf) and methyl trifluoromethanesulfonate (MeOTf) to promote the cationic ROP of lactide and glycolide was demonstrated by Kricheldorf et al. in the late 1980s [34]. The polymerization rates were significantly higher in nitrobenzene than in chlorinated solvents, with 50 °C being found to be the optimum reaction temperature. According to 1H NMR, polymers with methyl ester end groups were obtained with MeOTf as initiator. Optical rotation measurements revealed that samples of 100% optically active poly(l-lactide) were obtained from l-lactide with both HOTf and MeOTf. On this basis, the authors proposed that the chain growth proceeds via alkyl–oxygen bond cleavage and involves ring-opened trifluoromethanesulfonates as intermediates (Scheme 11). These results demonstrated the feasibility of the cationic route to PLAs, but the polymerization was far from living even under these conditions, polymer samples of similar viscosities being obtained whatever the initial monomer-to-initiator ratio.

Proposed pathway for the cationic ROP of lactide (R = H, Me or growing polymer chain, Tf = CF3SO2).
Inspired by the work of Endo and Jérôme on δ-valero- and ɛ-caprolactone [35], we recently demonstrated that controlled cationic polymerization of lactide can be achieved in solution at room temperature combining HOTf with a protic reagent ROH [36,37]. With this system, lactide is polymerized in dichloromethane within a few hours for [M]0 = 1 M and M/ROH ratios up to 100. The critical influence of the protic initiator is illustrated by the much lower conversions observed with HOTf alone. In marked contrast with that observed for δ-valero- and ɛ-caprolactone, weaker acids such as HCl·Et2O or CF3CO2H proved completely inactive towards lactide at room temperature. Water and primary or secondary alcohols used as initiators are quantitatively incorporated in the polymer chain as an acid or ester chain-end. The presence of only even lactate units in the electron-ionization mass spectra of the resulting polymers demonstrated that transesterifications do not occur to a significant extent during the polymerization [38]. Moreover, the production of perfectly isotactic l-PLAs from l-lactide indicates that despite its strong acid character, HOTf does not induce noticeable epimerization of the stereogenic carbon in these conditions [36].
The controlled character of the polymerization was supported by the following evidence: (i) the molecular weights Mn of the resulting PLAs (up to 16,000 g mol−1) were found to increase linearly with the monomer-to-initiator ratio, (ii) the Mn values of the growing polymer also vary linearly with the monomer conversion. In addition, the living character of the polymerization was substantiated by a second-feed experiment and kinetic studies revealed a first-order dependence on lactide with a kobs of 6.8 × 10−3 min−1 at room temperature. Accordingly, HOTf clearly does not compete with NHCs in terms of activity, but slightly surpasses DMAP, for which polymerization requires longer reaction times, even at 35 °C.
All these data strongly support an activated monomer cationic polymerization [39]. Accordingly, HOTf would protonate one exo oxygen of the lactide and ring opening via acyl cleavage would then result from the nucleophilic attack of the initiating protic agent or propagating alcohol ROH (Scheme 12).

Proposed activated monomer pathway for the cationic ROP of lactide (ROH = the initiating protic agent or the growing polymer chain).
The practical interest for such cationic ROP of lactide is further increased by the ready availability and ease of removal of HOTf. Supported versions of such cationic ROP have also been recently investigated [40]. Acidic resins such as Amberlyst®15 proved to be active in refluxing dichloromethane, but so far decreased polymerization rates prevent the preparation of high molecular weight polymers.
In principle, the cationic route can also be applied to the polycondensation of lactic acid, although efficient water removal is essential if high molecular weights are to be obtained. Ju et al. recently took advantage of the acidic character of silica gel to reach high conversion of l-lactic acid (>93%) after 120 h at 37 °C, but the Mn values remained fairly low (>1200 g mol−1) [41].
2.3 Bifunctional organocatalysts
2.3.1 (Thio)urea/amine associations
Inspired by the spectacular achievements reported in organic synthesis through hydrogen-bonding catalysis [42], Waymouth and Hedrick investigated the thiourea–amine catalyst 17 [43] as a bifunctional catalyst for the ROP of lactide [44] (Fig. 6). Using 4-pyrene butanol as the initiator, polymerization actually occurs at room temperature in CH2Cl2 and the monomer conversion reached 97% after 72 h for a M/I/cat ratio of 100/1/5 and [M]0 = 1 M. The living character and the initiation efficiency were established by 1H NMR and GPC analyses and corroborated by a chain-extension experiment. Catalyst 17 was found to be extraordinarily selective for polymerization relative to transesterification, with very low Mw/Mn (1.05) being obtained even after extended reaction times. Moreover, 13C NMR and DSC analyses indicated that no racemization had occurred in the polymerization of the enantiomerically pure l-lactide.
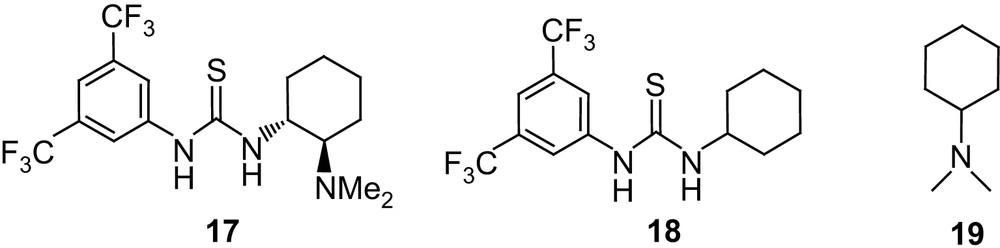
Structures of the thiourea–amine catalyst 17, thiourea 18 and amine 19.
By analogy with that proposed by Berkessel et al. for the dynamic kinetic resolution of azalactones [45], a bifunctional activation was postulated for this ROP. Hydrogen bonding to the thiourea group would thus activate the carbonyl group of the monomer, while the basic amine group of the catalyst would facilitate the nucleophilic attack of the initiating/propagating alcohol (Scheme 13). In line with this hypothesis, the concomitant presence of the hydrogen-bonding and amine moieties was found to be critical, no polymerization being observed under the same conditions using 18 or 19 alone. The critical role of hydrogen bonding was also apparent from a strong solvent dependence: polymerization occurs with 17 in chloroform, dichloromethane and toluene but not in hydrogen-bonding solvents such as tetrahydrofuran and dimethylformamide.

Mode of action of bifunctional catalyst 17 in the ROP of lactide.
Although catalyst 17 provides unprecedented control over the ROP of lactide, its activity is far below that of NHCs, so that long reaction times (48–72 h) are required under typical conditions. This stimulated a rather comprehensive optimization of the catalyst structure, not only by varying the linkage between the thiourea and amine functionalities (compounds 20, 21), but also by using two separate compounds (22–28 and 29–31) [46] (Fig. 7). The “two-in-one” systems 20 and 21 proved to be far less active than 17, the increased flexibility of the alkyl tether presumably inducing an intramolecular interaction between the hydrogen bonding and basic groups leading to deactivation. Further investigations demonstrated that the thiourea and amine do not have to be assembled in a “two-in-one” system to promote efficiently the ROP of lactide. Accordingly, the two partners could be varied over a broader range of structures, and significantly higher catalytic activities could be obtained through careful optimization of steric and electronic effects. Comparing thioureas 18 and 22–25 highlights the critical role of the electron-withdrawing substituents in increasing the acidity of the protons. Replacement of the cyclohexyl group for a second aryl moiety did not bring further improvement (derivative 26 behaves similar to 18), but demonstrated the detrimental effect that steric congestion has (compound 27 gives much slower polymerization rates). Among the tertiary amines and diamines tested, 19 and TMCHD 30 were found to be similarly efficient. The best results were obtained with sparteine, which could be used at lower loadings and allowed significant shortening of the polymerization times (by up to 25-fold). Accordingly, for a M/I/18/sparteine ratio of 100/1/2.5/1.5, complete monomer conversion was achieved within only 2 h at room temperature (for a monomer concentration of 2.8 M), affording high molecular weight (Mn 22,300 g mol−1) and narrowly distributed (Mw/Mn 1.05) PLAs. In marked contrast with the results observed with the “two-in-one” catalyst 17, stereoerrors in poly(l-lactide) could be detected and quantified by homonuclear decoupled 1H NMR when separated thiourea and amine were used. These stereoerrors were demonstrated to result from monomer epimerization, the extent of which is only 4% for the lead combination 18/sparteine. In addition, preliminary investigations suggest some isotactic enhancement attributable to chain-end control in the ROP of rac-lactide (Pm up to 0.77 at room temperature). The efficiency and functional-group tolerance of such thiourea–amine bifunctional catalysts have been substantiated by the preparation of various end-capped PLAs (using thiols, silanols or primary amines as the initiator) as well as block copolymers [using dual-headed initiators and tandem NMP (nitroxide mediated polymerization)-ROP and RAFT (reversible addition–fragmentation chain transfer)-ROP] [46].

Thiourea–amine catalysts.
2.3.2 Amidines and guanidines
Following the successful development of NHCs and thiourea–amines, Waymouth and Hedrick have recently investigated the commercially available guanidine 1,5,7-triazabicyclo[4.4.0]dec-5-ene TBD [47], a strongly basic catalyst that has been applied to numerous organic reactions, including transesterifications [48] (Fig. 8).
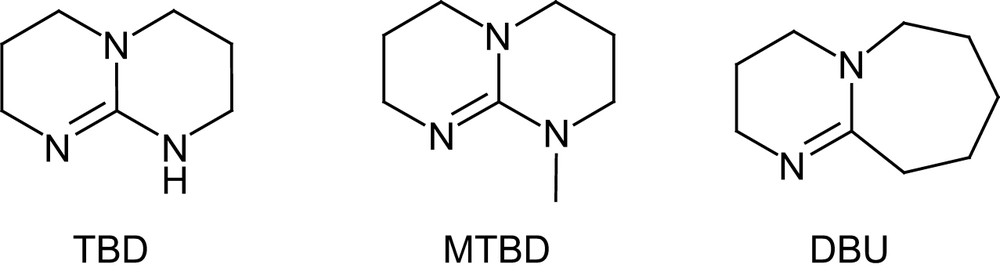
Structures of TBD and MTBD catalysts and DBU.
TBD proved to be more active for the ROP of lactide than any other organocatalyst, even competing with the most efficient metal systems. Indeed, for a M/I/cat ratio of 500/1/0.5 and [M]0 = 1 M, complete polymerization was achieved in only 1 min at room temperature in dichloromethane. The controlled character of this ultra-fast polymerization was supported by a linear increase in molecular weight with conversion, and end-group fidelity was classically established by simultaneous refractive index and UV absorbance-monitored GPC using 4-pyrene butanol as an initiator. Compared with thiourea–amine catalysts, slightly higher Mw/Mn values (1.11–1.19) were obtained. The molecular weight distributions further broaden upon prolonged reaction times, a similar feature, albeit to a lesser extent, to that observed with NHCs [25]. To prevent these adverse transesterification reactions, the guanidine catalyst can be quenched by addition of benzoic acid. Given its strong basicity in organic solvents ( in MeCN) [49], TBD could be anticipated to deprotonate or at least activate the initiating/propagating alcohol and promote thereby a pseudo-anionic ROP. However, higher polymerization rates were observed for TBD in benzene and dichloromethane relative to tetrahydrofuran and dimethylformamide. In addition, both the N-methylated analog of TBD (MTBD, see Fig. 8) and amidine DBU were found to be much less active under the same conditions, despite only slightly lower basicities ( respectively). These comparisons suggest that pseudo-anionic ROP of lactide is indeed operating for MTBD and DBU, but that a more sophisticated mode of activation must be involved with TBD. Taking into account the capability of bicyclic guanidines to function as bifunctional catalysts, as demonstrated by Corey and Grogan [50], a dual mechanism was proposed for ROP. After nucleophilic ring opening of the monomer by the imine nitrogen, the adjacent acidic proton would be ideally suited to proton transfer, which would liberate a new basic nitrogen capable of activating the incoming alcohol (Scheme 14). In marked contrast with most of the catalysts reported so far, TBD depicted high selectivity for lactide over other lactones (such as δ-valerolactone, ɛ-caprolactone and β-butyrolactone), opening the way to block copolymers. However, the competition between the polymerization of the least reactive monomer and the transesterification of the polymer backbones resulting from the most active monomer requires sequential addition of the monomers. Typically, block polyesters of high molecular weights (Mn 23,300–26,700 g mol−1) and fairly low Mw/Mn (1.17–1.21) were obtained by polymerizing δ-valerolactone or ɛ-caprolactone to completion, followed by addition of lactide.
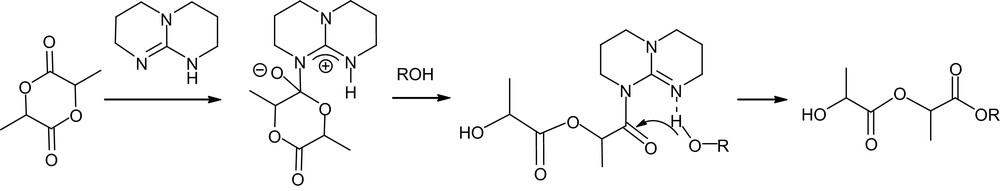
Postulated dual activation of lactide and alcohol (ROH = initiator or propagating chain) by TBD.
Combining ready availability, high activity and good control of the polymerization, the thiourea–amine and guanidine systems are undoubtedly among the most efficient catalysts for the preparation of tailored PLAs. It might also be anticipated that the structural flexibility of such bifunctional, metal-free catalysts will allow highly stereocontrolled polymerizations of rac- and meso-lactide in due course.
Finally, the tethered yttrium(III) and titanium(IV) complexes 32 and 33, featuring alkoxy- and amido-NHC ligands, respectively, are also thought to function as bifunctional catalysts [31] (Fig. 9).
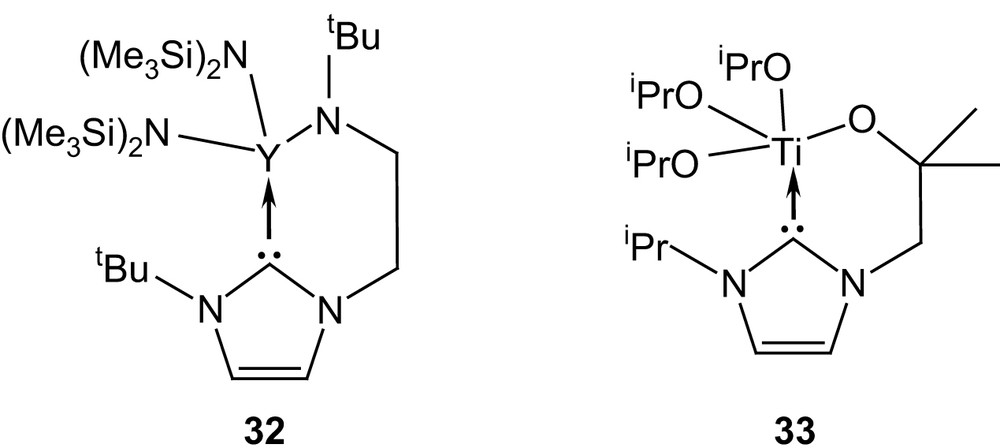
Yttrium(III) and titanium(IV) complexes of functionalized NHCs.
Both compounds display quite high activities towards the ROP of lactide at room temperature, although they do not compete with the organic systems discussed above. Labilization of the NHC ligand would allow concomitant activation of the monomer by coordination to the Lewis acid metal center and by nucleophilic attack from the carbene (Scheme 15). This mechanism, supported by the presence of imidazolium end groups on all chains (according to MALDI mass spectrometry analyses), would bridge the gap between coordination–insertion and monomer-activated mechanisms.

ROP of lactide initiated by 32.
3 Metal-free catalysts for the depolymerization-recycling of poly(α-hydroxy acids)
3.1 Controlled depolymerization of PLAs
Hedrick introduced another method for the preparation of controlled molecular weight and end-group functionalized polylactides consisting in the scission of higher molecular weight chains [51]. This strategy is based on the differing behaviour of primary vs. secondary alcohols in pyridine-catalyzed transesterification. Indeed, primary alcohols react not only with lactide, but also with acyclic esters, whereas secondary alcohols react only with lactide and are inert towards acyclic products. Accordingly, PLAs of Mn up to 100,000 g mol−1 were depolymerized with primary alcohols in the presence of DMAP or PPY as catalysts [51]. The resulting DPs are consistent with the alcohol-to-polymer ratio and polydispersity indices are in the same range as those of the initial polymers (Scheme 16). Following this strategy, more complex macromolecular architectures such as star-shaped polymers and block copolymers were prepared using pentaerythritol and monohydroxy-functionalized polyethylene oxide oligomers, respectively, as PLA depolymerization initiators.
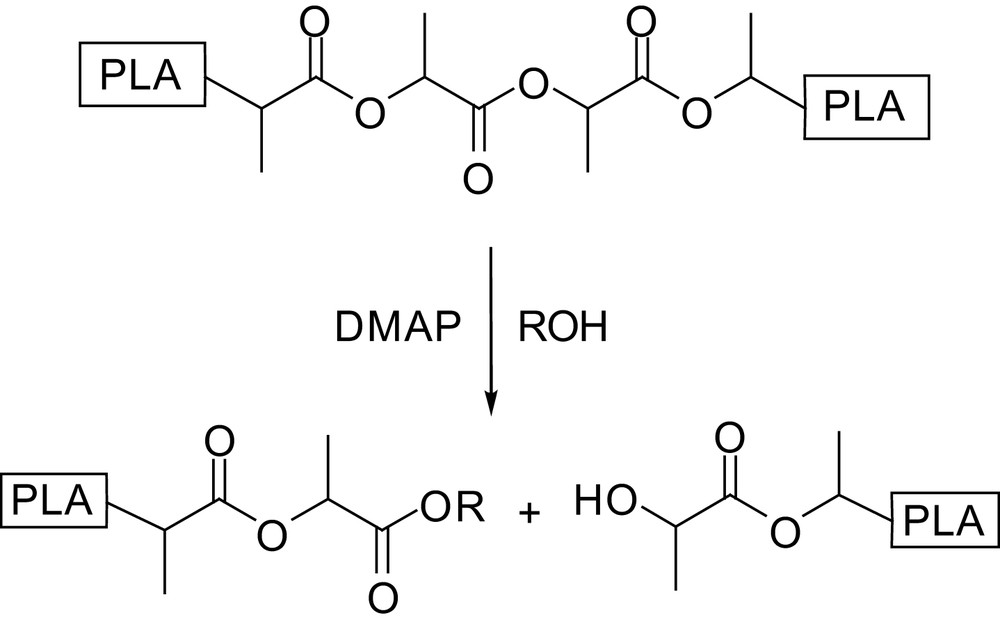
Basic principle of the DMAP-catalyzed PLA depolymerization.
3.2 Recycling of PLAs
Chain scission of PLAs has also been investigated with the objective of establishing a sustainable recycling system for lactic acid. In this regard, (hydro)thermal depolymerization of PLAs requires temperatures above 180 °C [52], but enzymatic catalysis allows it to proceed under much milder conditions [53]. Accordingly, the Rhizomucor miehei lipase, referred to as lipase RM, efficiently promotes the degradation of PDLLA (Mw 84,000 g mol−1, Mw/Mn 1.6) into cyclic oligomers (Mw 840 g mol−1, Mw/Mn 1.5) at 60 °C in a chloroform/hexane mixture, while PLLA (Mw 120,000 g mol−1, Mw/Mn 1.5) is preferentially depolymerized with lipase CA (Novozyme 435) in o-xylene at 100 °C. In both cases, large enzyme quantities (500–600 wt%) are necessary to achieve high to complete depolymerization in 24–48 h [54].
Jarerat et al. recently reported that a fungus, Tritirachium album, and two strains of actinomycetes, Lentzea waywayandensis and Amycolatopsis orientalis, exhibit PLLA degradation activities when proteins, peptides or amino acids are added into the liquid culture medium [55]. The best results were obtained with silk fibroin, an agro-industrial by-product of silk manufacture, as inducer for the depolymerase production. This provides the most efficient and mildest biological recycling process reported so far. Indeed, PLLA (2 g/L, Mn 340,000 g mol−1) was completely depolymerized within 8 h at 40 °C using 20 mg/L purified enzyme to afford oligomers (1.4 g/L) and l-lactic acid (0.6 g/L) without detectable traces of d-lactic acid. In addition, this extracellular enzymatic degradation does not require an organic solvent.
Note also that the degradation of PLLA (Mw 120,000 g mol−1) into repolymerizable oligomers has been described using a clay catalyst, montmorillonite K10 (MK10) [56]. In toluene at 100 °C for 6 h with 400 wt% MK10, only linear lactic acid oligomers (78%, Mn < 300 g mol−1) and monomeric l-lactic acid (22%) are obtained, without any racemization. From a mechanistic viewpoint, the polymer chains are most likely absorbed on the surface or between the layers of MK10, before the ester groups are attacked by the clay-bound water with the aid of catalytic protons. Addition of ethanol to anhydrous MK10 allows further improvement: it not only accelerates the reaction, but also provides ethyl ester instead of carboxylic acid end groups.
4 New monomers for the preparation of poly(α-hydroxy acids)
In order to tune and further broaden the properties of these biodegradable and bioassimilable polymers, various approaches are currently under investigation, and so the two most widely used strategies will be presented hereafter.
4.1 Substituted 1,4-dioxane-2,5-diones
The first strategy consists in the homo- and copolymerization of monomers related to lactide and glycolide, especially those featuring functional groups [57]. From a synthetic point of view, the symmetric 1,4-dioxane-2,5-dione skeleton can be easily accessed by condensation of α-hydroxy acids. However, the direct self-condensation (Scheme 17, Eq. 1a) usually requires dilute solutions to favor dimerization over oligomerization, which results in prolonged reaction times (for example, 1 week in refluxing toluene to reach 90% conversion of 3,6-dibenzylglycolide) [58]. Alternatively, the desired monomers can be obtained by cracking the corresponding oligomers in the presence of a catalyst such as ZnO (Eq. 1b), a route which mirrors the standard preparation of lactide. However, the low volatility of substituted 1,4-dioxane-2,5-diones considerably limits the practical application of this strategy, rather harsh conditions usually being necessary. Typically, such a preparation of 3,6-dihexylglycolide requires its distillation at 145 °C at 50 mTorr pressure, while that of 3,6-dibenzylglycolide requires 230 °C at 20–50 mTorr, which induces about 10% epimerization [58]. The synthesis of unsymmetrically substituted monomers is usually achieved by step-by-step condensation, starting from an α-hydroxy acid and an α-haloacyl halide (Eq. 2a). Alternatively, Hennink and coworkers recently demonstrated that cyclization of linear α-hydroxy acid dimers can be efficiently carried out with cyanuric chloride (Eq. 2b) [59], but the synthesis of the required precursors necessitated five steps and orthogonal protecting groups. With both routes 2a and 2b, the formation of the six-membered ring is inherently in competition with oligomerization, and modest to good yields (∼35–70%) are obtained depending on the substitution pattern.
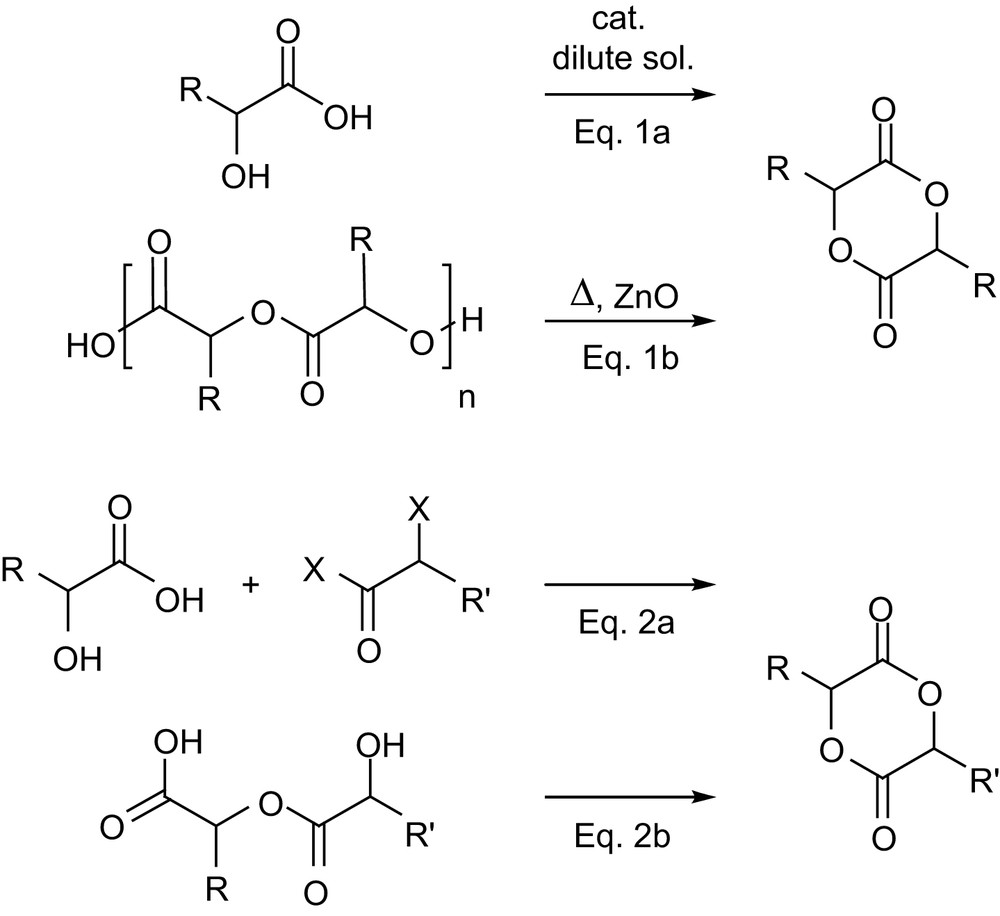
Routes to symmetrically and unsymmetrically substituted 1,4-dioxane-2,5-diones.
Various substituted 1,4-dioxane-2,5-diones have been investigated, starting with methylglycolide 34 [60,61]. Given the different reactivity of lactide and glycolide (rG/rL ≈ 10), the polymerization of the crossed dimer 34 was anticipated to occur preferentially by cleavage of the least sterically hindered acyl moiety (arrowed, Scheme 18). This unique opportunity to prepare alternating copolymers of lactide and glycolide has been recently validated with tin octanoate SnOct2 as a catalyst [61], but further developments in this direction have so far been limited by the poor availability of methylglycolide (obtained in two steps and 43% overall yield from glycolic acid and 2-bromopropionyl bromide [61]).

Preparation of poly(lactide-alt-glycolide) from methylglycolide 34.
The introduction of alkyl or benzyl groups on the poly(α-hydroxy acid) backbone should allow the modulation of their physical and chemical properties. Accordingly, symmetrically as well as asymmetrically 3,6-disubstituted glycolides 35–39 [58,62,63] (Fig. 10) have been prepared and the properties of their homopolymers, prepared in bulk with SnOct2, were investigated. As expected, the longer the alkyl group, the lower the glass transition temperature Tg (the flexible side chain acting as an internal plasticizer), whereas branched alkyl groups result in higher Tg due to hindered rotation of the polymer backbone. The introduction of a phenyl ring, from PLA to poly(benzylglycolide), does not significantly affect the Tg [58]. Preliminary investigations have also been carried out regarding the influence of the pendant group on the hydrolytic degradation of the polymers [58,63]. As anticipated, studies above Tg (with polymers in a rubber state) highlighted the hydrophobic influence of the substituents, slower degradation rates being observed for poly(benzylglycolide) and poly(hexylglycolide) compared with PLAs. In contrast, degradation studies performed under physiological conditions (37 °C and pH 7.4) demonstrated the critical role of Tg. Indeed, under these conditions, the degradation rate of the flexible poly(methylhexylglycolide) is slightly higher than that of glassy PLAs.
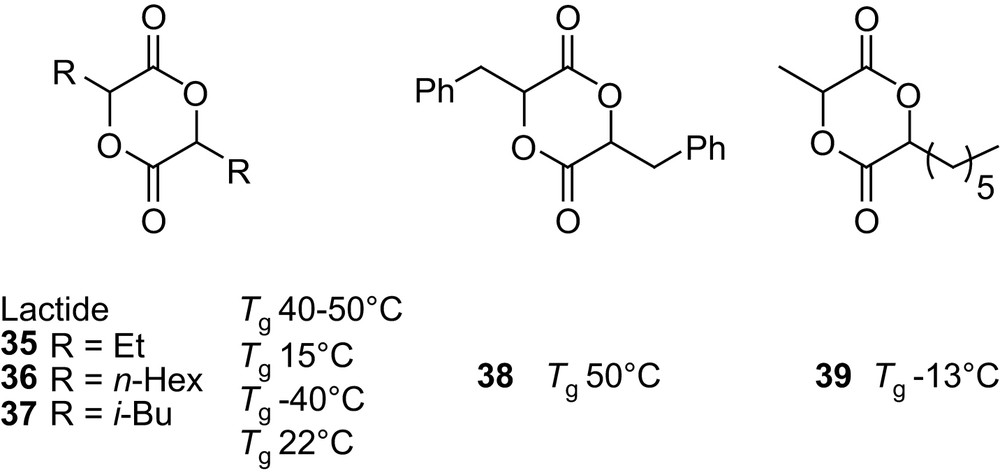
3,6-Disubstituted 1,4-dioxane-2,5-diones and influence of the pendant alkyl and benzyl groups on the Tg of the corresponding homopolymers.
Substitution of the 1,4-dioxane-2,5-dione ring also presents an opportunity to prepare polyesters featuring functional side groups. The more straightforward method is obviously the ROP of protected functionalized monomers, as pioneered by Kimura et al. with the malic acid-derived compound 40a [64] (Scheme 19). Homopolymerizations were carried out in bulk and in solution and high conversions (up to 90–95%) were achieved using SnOct2, Al(OiPr)3 or ZnEt2 as initiators. Low to high molecular weights were prepared (Mn from 2000 to 20,000 g mol−1), but rather large molecular distributions were obtained in all cases (Mw/Mn from 1.6 to 3.4). The resulting copolymers were shown by 1H and 13C NMR analyses to be perfectly alternated, as the result of the exclusive cleavage of the least sterically hindered acyl group of 40a. Quantitative removal of the benzyl protecting groups was performed by catalytic hydrogenation to afford alternated copolymers of glycolic and malic acids. Copolymerizations of 40a and of the related dodecyl-protected compound 40b with lactide have also been studied using SnOct2. The presence of pendant carboxyl groups in the poly(α-hydroxy acids) obtained after deprotection was demonstrated to increase significantly their hydrolytic degradation rates (at 37 °C and pH = 7.2), because of higher water absorption and potential autocatalytic behaviour. The interest of such pendant functional groups for further chemical modification of PLAs has also been illustrated by the introduction of a photoaffinity labelling group [65] or a cell-binding tripeptide [66]. Incorporation of a small amount (<3 mol%) of 40a in poly(β-butyrolactone) has also been reported, and after hydrogenolysis, the pendant carboxyl groups were used to induce some cross-linking upon reaction with 1,4-dibromobutane [67].
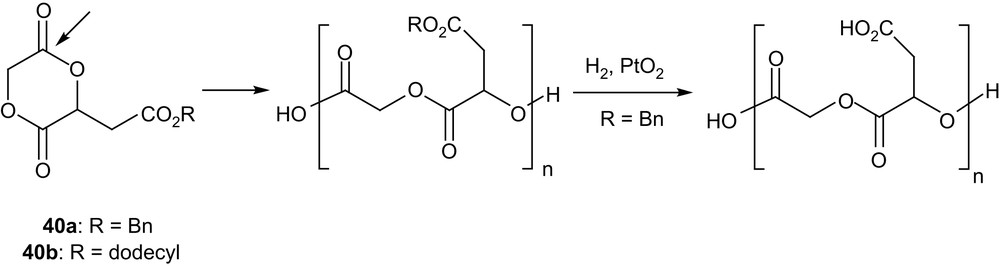
Structure of the malic acid-derived monomers 40a,b and ensuing polymers.
Steric factors were shown to play a prominent role in the polymerization of such functionalized 1,4-dioxane-2,5-diones. Indeed, only poor conversions (<29%) could be obtained in the homo- and copolymerization of the related protected malic acid dimer 41 [68].
In a similar way, the two heterodimers 42a and 42b, derived from d-gluconic acid on the one hand and glycolic or lactic acid on the other hand, were found to behave very differently in SnOct2-initiated homopolymerization (Fig. 11). Indeed, 42a provided high molecular weight polymers (20,000 g mol−1) in 83% yield, whereas only oligomers (2000 g mol−1) were obtained from 42b [69]. Both monomers could be copolymerized with lactide, affording high molecular weight but broadly distributed samples. The cleavage of the isopropylidene acetals remains to be studied to give the desired PLAs with pendant hydroxyl groups.
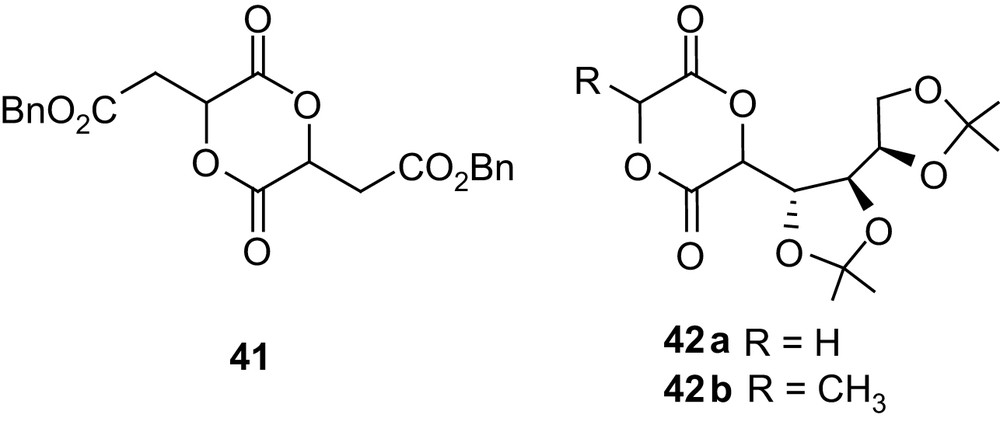
Structure of the malic acid dimer 41 and of the d-gluconic acid-derived heterodimers 42.
In this regard, Feng and coworkers investigated the heterodimer 43a of glycolic and glyceric acids [70] (Fig. 12). Its preparation was accomplished in 40% overall yield following route 2a (Scheme 17). Homopolymerizations in bulk (130–140 °C) using SnOct2 afforded high molecular weight samples (Mn up to 16,000 g mol−1 with Mw/Mn ∼ 2). As with 42b, exclusive cleavage of the least sterically hindered ester group resulted in perfectly alternated copolymers, and the benzyl protecting groups could be quantitatively removed by catalytic hydrogenation. In addition, water absorption and contact angle measurements substantiated the major modifications that pendant benzyloxy and hydroxyl groups had induced on the hydrophilicity of PLAs. Complementary results were recently reported by Hennink and coworkers for both 43a and its lactic acid analog 43b using various initiating systems and polymerization conditions [71]. The comparison of the 13C NMR spectra confirmed that homopolymerization of 43a provides perfectly alternating copolymers, whereas competitive cleavage of the two ester groups occurs for 43b, resulting in a statistical distribution. In addition, random as well as block copolymerizations of both monomers with lactide have been reported. Starting from protected lysine and glutamic acid, the related monomers 44 and 45 have also been studied recently by Collard and coworkers [72]. Polymerization studies have been performed in bulk (140 °C) with SnOct2. Accordingly, the pendant group was found to affect significantly the yields in homopolymers and the amounts of functionalized monomer incorporated in lactide copolymers (43b > 44 > 45). In addition, hydrogenolysis of the polymer samples proved convenient to deprotect the hydroxyl and carboxyl groups derived from 43b and 45, but acidic hydrolysis (with HBr/AcOH) was necessary to reveal the free amino groups derived from 44.
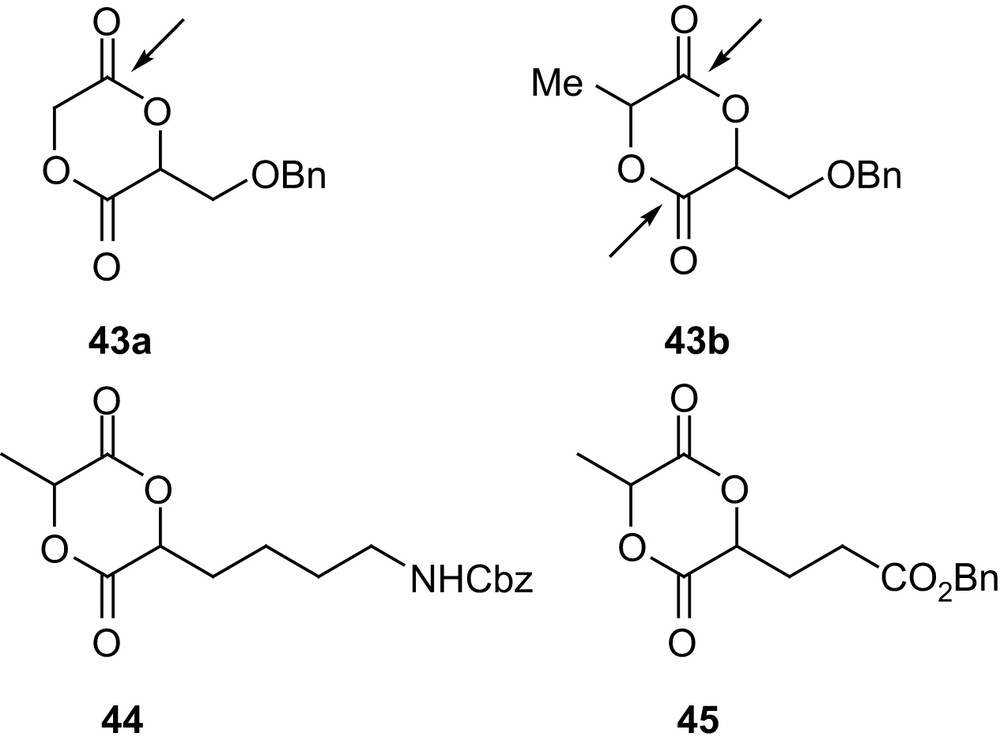
Structure of the 1,4-dioxane-2,5-diones 43a,b, 44 and 45 derived from serine, lysine and glutamic acid, respectively.
Alternatively, the functional groups may be introduced after the ROP of well-designed monomers such as 46 [73], whose allyl side groups can be subsequently transformed. The feasibility of this approach has been investigated via olefin metathesis using allylbenzene and Grubbs' second generation catalyst (Scheme 20).
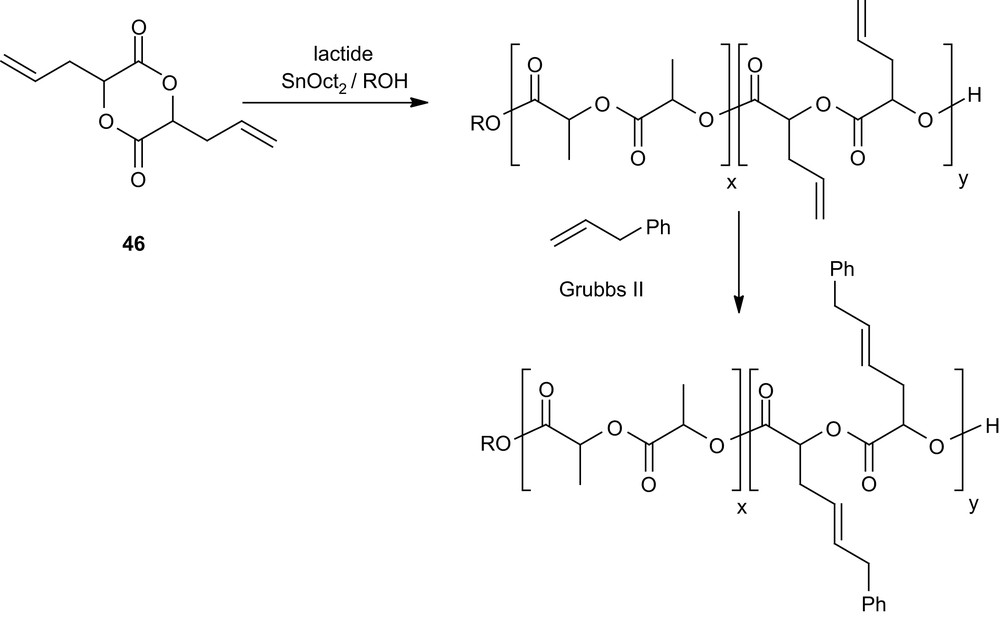
Post-polymerization functionalization of PLAs via olefin metathesis.
Recently, the introduction of fluorine atoms has also been investigated in order to modify the properties of the polyester backbone (e.g., improved chemical resistance, lower water absorption, lower surface energies and higher solubility in supercritical CO2 can be expected). Successive treatment of a preformed PLA with sodium hydride and N-fluorobenzenesulfonimide allowed about 80% of the methine groups to be fluorinated; however, the GPC data of the resulting material indicated significant degradation of the polymer chain. Alternatively, fluorination of lactide itself following the same procedure afforded the desired 2,6-difluorolactide 47 (Scheme 21) [74], albeit in only 10% yield.
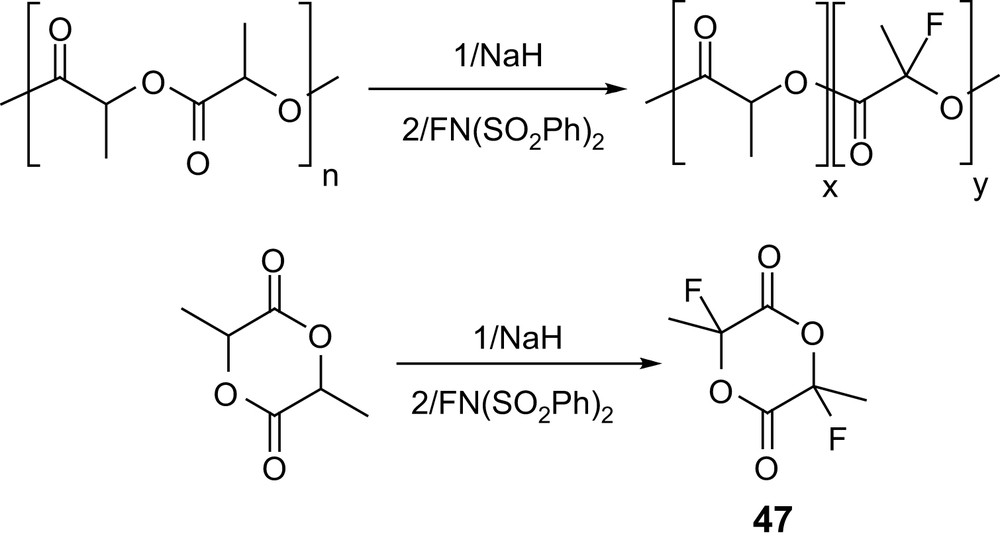
Electrophilic fluorination of PLAs and of lactide.
Finally, the crossed dimers of α-hydroxy and α-amino acids, namely morpholine-2,5-diones 48 [75,76], are clearly worthy of mention (Fig. 13). Indeed, these unsymmetrical monomers are usually obtained in better yields than the related 1,4-dioxane-2,5-diones, and the introduction of pendant functional groups is straightforward from aspartic acid, glutamic acid, lysine, serine or cysteine. Although the steric bulk of the protected side chains significantly lowers the reactivity of the 6-membered ring, copolymerization with lactide could be conveniently achieved to give polyesteramides with pendant functional groups.

Structure of morpholine-2,5-diones 48.
4.2 Synthetic equivalents for 1,4-dioxane-2,5-diones
The preceding section illustrates the broad variety of poly(α-hydroxy acids) that can be envisaged by varying the substitution pattern of lactide and glycolide, but the only moderate polymerizability of substituted 1,4-dioxane-2,5-diones remains a major limitation. Indeed, although lactide is among the rare example of polymerizable six-membered rings [77], the relief of ring strain, which results from the two planar ester moieties, provides only a small driving force for the ROP, so that highly active promoters are required if the ROP of lactide is to be carried out under mild conditions. Associated drawbacks are typically the high sensitivity of the promoters and significant amounts of undesirable transesterification reactions. In order to circumvent these limitations, activated equivalents of lactide would be highly desirable. Since α-lactones themselves are far too reactive to be used practically as monomers [78], we recently turned our attention to synthetic equivalents thereof and considered the readily available [79] 1,3-dioxolane-2,4-diones to be promising candidates [80]. The latter derivatives, so-called O-carboxy anhydrides (OCAs), can be considered oxygen analogs of the N-carboxy anhydrides (NCAs) that proved to be convenient precusors for polyamides [81]. The polymerization of lacOCA (Scheme 22) was predicted computationally to be thermodynamically much more favorable than that of lactide, the liberation of a CO2 molecule being a considerable driving force. The nucleophilically-activated ROP was found experimentally to proceed much faster and under much milder conditions with lacOCA than with lactide. Accordingly, the DMAP/ROH system that requires prolonged heating with lactide (see Section 2.1.3.1) afforded PLAs of controlled molecular weights and narrow polydispersities in a few minutes at room temperature from lacOCA (Scheme 22) [82].
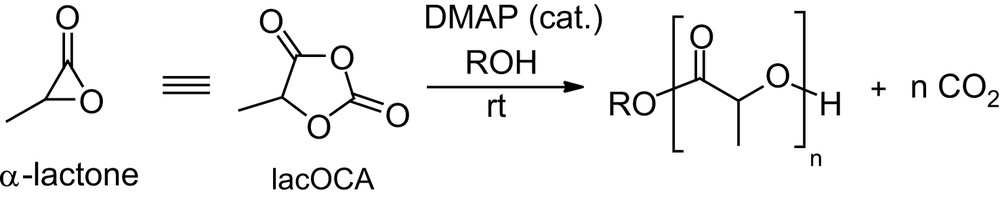
Preparation of PLAs from lacOCA.
The much higher reactivity of lacOCA, compared with lactide, and the ensuing milder polymerization conditions broaden the scope of compatible initiators to include functionalized ones. As a representative example, a PLA oligomer featuring a β-brominated ester end group was efficiently prepared in 5 min at room temperature by initiating the polymerization of lacOCA with 2-bromoethanol. In marked contrast, the related DMAP-catalyzed polymerization of lactide required 5 days at 35 °C to reach 93% monomer conversion, and was contaminated with a significant amount of pyridinium salt formation (up to 30%) (Scheme 23).

Preparation of PLAs featuring a β-brominated ester end group from lacOCA and lactide.
These results demonstrate the synthetic potential of OCAs as readily available and highly polymerizable monomers for the preparation of poly(α-hydroxy acids).
5 Conclusion
From this review, it is clear that the ROP of lactide, glycolide and related monomers has been an extremely active research field over the last 5 years. Spectacular improvements have been achieved with metal-free systems such that organocatalytic monomer activation can now really compete with the traditional metallic coordination–insertion pathway in terms of activity and polymerization control. Promising avenues of research have also emerged for the enzymatic approach, not only for the ROP of lactide but also for the PLA recycling via depolymerization. Increasing interest has also been dedicated to chemically modified poly(α-hydroxy acids). In this regard, the preparation and polymerization of various substituted 2,4-dioxane-2,5-diones have demonstrated that the polymer properties are tunable across a broad range. Poly(α-hydroxy acids) featuring pendant functional groups also appear particularly promising sustainable materials and further developments in this direction should be facilitated using O-carboxy anhydrides which are readily available, structurally modulable and highly polymerizable monomers.
Acknowledgements
We thank the CNRS, the University Paul-Sabatier, Ipsen Pharma and Isochem for financial support of our contribution in this field. Special gratitude is expressed to the research associates involved in this work: J.-B. Cazaux, A. Dumitrescu, F. Ben, O. Dechy-Cabaret, E. Marchal, M. Graullier, O. Thillaye du Boullay, C. Bonduelle and A. Alba. M.W.P. Bebbington is warmly acknowledged for helpful discussions during the preparation of this manuscript.