

1 Introduction
Nanoscaled materials are a field of growing interest because of the fundamental differences in properties between nanoscale and bulk materials [1]. Silicon nanoparticles (Si-np) are very attractive because of their semiconducting and luminescence properties which open a great interest for industrial applications in the field of electronic and also biological applications [2–4]. However, the chemistry of silicon is based on covalent bonds and the methodologies used for growing silicon particles implies very different routes from those used in the case of metals.
Usually, access to Si-np is mainly obtained via physical routes by “top-down” [5] or “bottom-up” [6] strategies. Even though these techniques allow one to obtain Si-np with a good control of composition and size, they do not permit to have a good control of the chemistry at the surface. In most cases, oxidized surfaces are reported [7–9]. The chemical strategy is the reduction of tetrachlorosilane in solution. The first report was presented by Heath [10]. The procedure was the reduction by sodium metal of a mixture of tetrachlorosilane and organotrichlorosilane at high temperature and pressure and gives Si-np, which have a surface passivated by hydrogen or octadecanyl substituents. Kauzlarich and co-workers have reported several procedures [11–18] for producing silicon nanoparticles under mild conditions in solution using reactive Zintl salts [11,12] or sodium naphthalenide [16,19] as reducing agents. Other routes involving silicon tetrachloride and magnesium [20,21] or hydrides [22–24] and also electrochemical reduction [25] were described. Tetraethoxysilane and colloidal sodium under ultrasonic waves [26] have also been reported. The variety of the reagents used demonstrates the importance of the reducing agent and the difficulty for obtaining Si-np.
Graphite intercalation compounds such as C8K are known to allow in some cases to obtain polysilanes with increased yield and higher quality than conventional routes [27–29]. They are also used to obtain polysilynes [30,31].
In this paper we report the use of C8K as a route to silicon nanoparticles presenting different surface substituents.
2 Experimental
2.1 Materials and instruments
Analytical-grade reactants SiCl4, K, H2O, MeMgCl, propan-2-ol, γ-aminopropyltrimethoxysilane and lithium diisopropylamide were purchased from Aldrich and used as received. Reactions were performed under inert atmosphere of dry argon using standard Schlenk techniques and vacuum lines (10−1 mbar). Tetrahydrofuran was dried and distilled from Na/benzophenone prior to use. Graphite powder was washed three times with ethanol and diethylether, then dried at 120 °C for 8 h under reduced pressure (10−1 mbar).
FTIR spectra were recorded on a PerkinElmer 1600 series IRFT, liquids spectra between NaCl pellets and solids spectra in dilution of 95% dry KBr. A blank KBr pellet was examined before each sample. Liquid-phase NMR spectra were recorded at room temperature on a BRUKER ADVANCE DPX 200 (1H, 13C) or a AC 200 (29Si) spectrometer. Chemical shifts are referenced in pats per million. Solid-phase NMR spectra were recorded at room temperature with a BRUKER ADVANCE DPX 300 spectrometer (13C, 29Si). Elemental analysis was performed at the “Laboratoire central d'analyse du CNRS” in Vernaison, France. Air-sensitive compounds were handled under argon and elemental analysis was done without any contact with air. UV–vis solid spectra were recorded on a UV–vis Perkin-Elmer Lambda 14. A blank BaSO4 pellet was examined before each sample. Photoluminescence spectra were recorded on a Aminco SLM 8100. TEM pictures and related SAED were recorded on a 100 kV JEOL JEM-1200EX microscope.
2.2 Synthesis of potassium-graphite C8K
C8K was prepared by reacting potassium (7.33 g, 0.19 mol) with graphite powder (18.00 g, 1.5 mol) at 150 °C for 15 min under vigorous stirring in a three-necked, flame-dried, 500-ml round-bottomed flask. The resulting product was a powder with a characteristic gold color.
2.3 Synthesis of chlorinated nanoparticles S0
SiCl4 (9.04 g, 0.05 mol) in tetrahydrofuran (50 ml) was added rapidly at 0 °C via a cannula to a stirred suspension of potassium-graphite C8K (freshly prepared from 7.33 g of potassium and 18.00 g of graphite stirred vigorously at 150 °C for 15 min). The gold powder gradually turns to black. The suspension was stirred overnight at room temperature and then refluxed for 1 h. Graphite and KCl were removed by filtration and solvent by heating in an oil bath under reduced pressure (10−1 mbar). The resulting brown oil M0 was separated under reduced pressure (10−2 mbar). At 100 °C, very viscous colorless oil H0 distilled and a brown insoluble solid S0 was isolated.
M0 (2.50 g, 34%): brown oil (found: Si, 20.9; Cl, 40.5; C, 20.2; H, 3.5; calc. for Si1Cl0.12: Si, 86.8; Cl, 13.2%), νmax/cm−1: 2940, 2868, 2798, 1460, 1380, 1301, 1269, 1243, 1200, 1100 (br), 763, 692, 572; δH (200 MHz; CDCl3; Me4Si): 0.9, 1.4, 1.7, 3.4, 3.9; δC (50 MHz; CDCl3; Me4Si): 15.8, 20.1, 26.7, 30.1, 45.1, 63.6, 70.0; δSi (40 MHz; CDCl3; Me4Si): 14.0, −10.2, −37.9, −55.8, −69.9, −78.4, −85.5.
S0 (1.50 g, 25%): brown solid (found: Si, 26.0; Cl, 19.1; C, 20.8; H, 3.5; calc. for Si1Cl0.12: Si, 86.8; Cl, 13.2%) δC (75 MHz; HPDEC; Me4Si): 13.4, 19.8, 27.1, 30.4, 63.1, 70.6; δSi (60 MHz; HPDEC; Me4Si): 14.1, −10.2, −38.8, −55.7, −69.6, −78.2, −86.2.
2.4 Synthesis of functionalised nanoparticles S1–S5
Compounds S1–S5 were synthesized following the same experimental procedure from 1 g of S0. A detailed procedure is given for S3.
A solution of propan-2-ol (1.03 g, 17 × 10−3 mol) in 20 ml tetrahydrofuran was rapidly added to 1.0 g of S0 in suspension (40 ml tetrahydrofuran) cooled at −30 °C. Once solution was back at room temperature, it was allowed to react for 30 min and then refluxed for 1 h. Pale green solid S3 was isolated by filtration, washed three times with water, ethanol and diethylether then dried at 120 °C for 2 h under reduced pressure (10−1 mbar).
- S1, reagent: H2O HPLC, 0.28 g (18 × 10−3 mol).
- S2, reagent: MeMgCl, 3.8 ml (16 × 10−3 mol).
- S4, reagent: LDA, 9.58 ml (15 × 10−3 mol).
- S5, reagent: γ-aminopropyltrimethoxysilane, 3.66 g (21 × 10−3 mol). In order to avoid hydrolysis of trimethoxysilyl function, S5 is only washed with methanol.
S1 (0.80 g, 18%): pale brown solid, mp > 300 °C (dec) (found: Si, 23.1; Cl, 2.9; C, 28.5; H, 5.1; calc. for Si1H0.12O0.12: Si, 93.2; H, 0.4%), λmax/nm: 300; νmax/cm−1: 3310, 2935, 2868, 2255, 1713, 1638, 1463, 1381, 1060 (br), 943, 880, 791, 694, 588, 429; δC (75 MHz; HPDEC; Me4Si): 14.2, 19.8, 22.5, 29.8, 38.1, 66.8, 72.1; δSi (60 MHz; HPDEC; Me4Si): −63.6, −101.9, −107.7, −129.6; photoluminescence λmax/nm: 430 (exc. 334 nm), powder XRD: no signal.
S2 (0.72 g, 16%): yellow solid, mp > 300 °C (dec) (found: Si, 22.8; Cl, 3.5; C, 32.0; H, 6.8; calc. for Si1C0.12H0.36: Si, 94.0; C, 4.8; H, 1.2%), λmax/nm: 260, 300; νmax/cm−1: 3378, 2955, 2924, 2862, 1641, 1526, 1442, 1250, 1040 (br), 840, 791, 766, 670; δC (75 MHz; HPDEC; Me4Si): 2.4, 14.6, 21.0, 26.7, 31.0, 62.8, 70.9; δSi (60 MHz; HPDEC; Me4Si): 15.3, 7.6, −20.9, −66.6, −95.9, −109.9; photoluminescence λmax/nm: 410 (exc. 334 nm); powder XRD: no signal.
S3 (0.59 g, 17%): pale green solid, mp > 300 °C (dec) (found: Si, 29.6; Cl, 1.2; C, 24.2; H, 4.7; calc. for Si1C0.36H0.84O0.12: Si, 79.8; C, 12.3; H, 2.4%), λmax/nm: 300; νmax/cm−1: 3410, 2932, 2870, 2250, 1459, 1385, 1370, 1060 (br), 880, 800, 798; δC (75 MHz; HPDEC; Me4Si): 12.6, 19.8, 26.2, 32.0, 65.4, 72.1; δSi (60 MHz; HPDEC; Me4Si): −65.3, −83.7, −103.4, −112.4; photoluminescence λmax/nm: 410 (exc. 334 nm); powder XRD: no signal.
S4 (0.50 g, 10%): yellow solid, mp > 300 °C (dec) (found: Si, 20.4; Cl, 1.4; C, 41.0; H, 6.8; N, 0.5; calc. for Si1C0.72H1.68N0.12: Si, 70.0; C, 21.6; H, 4.2; N, 4.2%), λmax/nm: 260, 330, 390; νmax/cm−1: 3350, 2934, 2853, 2798, 2113, 1458, 1403, 1366, 1180, 1070 (br), 788; δC (75 MHz; HPDEC; Me4Si): 14.1, 18.9, 27.3, 45.2, 63.7, 68.8; δSi (60 MHz; HPDEC; Me4Si): −17.7, −62.2, −101.2, −110.0; photoluminescence λmax/nm: 430 (exc. 334 nm); powder XRD: no signal.
S5 (0.45 g, 11%): yellow solid, mp > 300 °C (dec) (found: Si, 25.1; Cl, 1.4; C, 32.0; H, 6.3; N, 1.2; calc. for Si1.12C0.72H1.92N0.12O0.36: Si, 63.5; C, 17.5; H, 3.9; N, 3.4%), λmax/nm: 260; νmax/cm−1: 3424, 3390, 3036, 2937, 2880, 2027, 1616, 1506, 1462, 1191, 1131, 1043 (br), 941, 780, 696, 563, 455; δC (75 MHz; HPDEC; Me4Si): 6.1, 13.6, 21.5, 26.7, 43.7, 50.7, 62.7, 71.5; δSi (60 MHz; HPDEC; Me4Si): −16.1, −42.4, −50.1, −57.9, −65.1, −87.6, −96.2, −101.2, −109.6; photoluminescence λmax/nm: 430 (exc. 334 nm); powder XRD: no signal.
3 Results and discussion
3.1 Synthesis of silicon nanoparticles functionalised by chlorine atoms
C8K is easily obtained as a golden solid in just 15 min by heating 1 equiv of potassium with 8 equiv of graphite without any solvent [32]. This strong reducing agent, which is flammable in air, reacts stoichiometrically with the Si–Cl bonds and allows the formation of Si–Si bonds with KCl as the sole by-product.
The aim of our work was to prepare silicon nanoparticles, Si-np, by reaction of tetrachlorosilane with potassium graphite. The ratio SiCl4/C8K was chosen from stoichiometry to a slight excess in SiCl4, in order to allow further functionalization of the surface by substitution of the remaining chloride substituents (Eq. (1)).
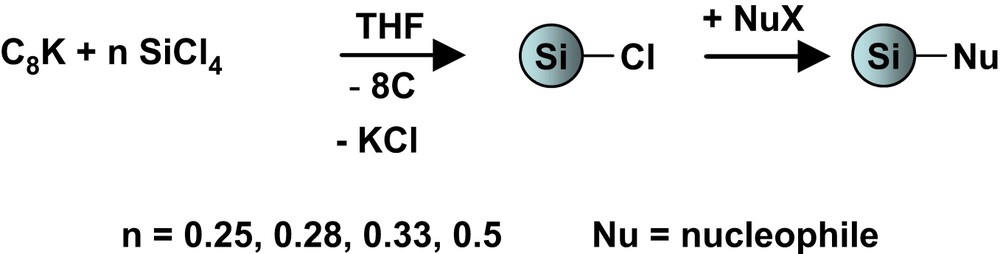
Synthesis of functionalised silicon nanoparticles.
Various ratio SiCl4/C8K were investigated in the range 0.25 < n < 0.5 (Eq. (1)). The first experiment involved the ratio n = 0.25, corresponding to a stoichiometric amount of C8K and SiCl4. This experiment permits to evaluate the extent of the reactivity of SiCl4 with C8K under such heterogeneous conditions. The reaction resulted in a full condensation of SiCl4 to produce a silicon material with no remaining Cl atoms, as confirmed by the elemental analysis of the resulting Si/graphite material, with a calculated atomic ratio of C8Si0.23 (for a theoretical value of C8Si0.25). In addition, no residual Cl or K was detected in the solid by EDX and elemental analysis. The reaction product could not be extracted from graphite since it was an insoluble solid material (as expected for Si-np). High-resolution transmission electron microscopy (HRTEM) revealed the presence of Si-np with a well-defined organization at the nanometric scale (Fig. 1). Thus, this experiment not only confirmed the stoichiometric reactivity of SiCl4 with C8K, but also showed that the resulting silicon nanoparticles presented some organization at the nanoscale, probably induced by the environment in which they were formed.

HRTEM micrograph showing lattice fringes of silicon in the graphite.
In the case of a ratio SiCl4/C8K n = 0.33 and 0.5, the products were soluble in THF and easily separated from graphite by filtration and washing. The products were isolated as yellowish light oils, most likely corresponding to polysilyne-type oligomers.
The best results were obtained for a ratio n = 0.28. The synthesis was carried out in tetrahydrofuran (THF). Graphite was removed by filtration and the solvent was pumped off under 10−1 mbar, leading to M0 as a brown viscous oil in 34% yield based on silicon. It was soluble in various solvents such as pentane, THF and chloroform. The FTIR spectrum showed the characteristic band of Si–Cl stretching at 570/590 cm−1. Additional bands at 2940, 2870, 1460, 1380 cm−1 corresponding to alkyl C–H stretching and at ∼1080 cm−1 due to C–O or Si–OR stretching were also observed. It is noteworthy that no band indicating the presence of O–H bonds were observed. The solution 1H NMR spectra showed the presence of signals at 0.9, 1.4, 1.7, 3.4 and 3.9 ppm. They were attributed to organic residues arising from a reaction of the compound M0 with THF leading to ring opening compounds. A similar observation raised from 13C NMR analysis, which showed signals corresponding to the presence of THF (∼27 and 70 ppm) and ring opening products (∼16, 20, 30 and 64 ppm). 29Si NMR exhibited signals between 12 and −86 ppm, which were assigned to silicon atoms bonded with 3 to 0 Cl atoms, respectively [33,34]. Elemental analysis led to the following stoichiometry Si1Cl1.53C2.26H4.69 in agreement with the presence of organic residues formed by reaction with THF (calculated Si1Cl0.12).
When M0 was exposed to air, the IR band corresponding to Si–Cl bonds disappeared and a broad band corresponding to O–H bonds appeared at 3260 cm−1. This observation confirmed that the Si–Cl bonds identified in the sample were readily accessible.
M0 was distilled under 10−2 mbar in order to remove the low molecular weight fraction. A viscous colorless oil H0 (14%) and a dark brown insoluble powder S0 (64%) were obtained (Eq. (2)). The ratio Cl/Si determined by elemental analysis dropped from 1.53 for M0 to 0.58 for S0, confirming that the lighter compounds, with a higher content of chloride and thus lower Si–Si reticulation, had been distilled off.

Synthesis of M0, S0, and H0.
S0 was analyzed by solid-state 13C NMR, where no signals were expected. However, as for M0, several peaks attributed to compounds formed by side reaction with THF were still observed. The solid-state 29Si NMR exhibited several signals in the region from 20 to −90 ppm. There are very few examples of Si-np 29Si NMR studies [15,35]. The size distribution of the Si-np and the numerous different chemical environment of silicon widen the signal. However, the different parts of the spectrum can be assigned to silicon atoms bonded with 3 to 0 Cl atoms, respectively [33,34] and it is noteworthy that no signal in the region from −90 to −110 ppm corresponding to silicon oxide were observed.
3.2 Synthesis of silicon nanoparticles presenting various surface functionalities
S0 was treated with different nucleophilic reagents to obtain a variety of surface types through formation of Si–C, Si–O or Si–N surface bonds (Eq. (3)).
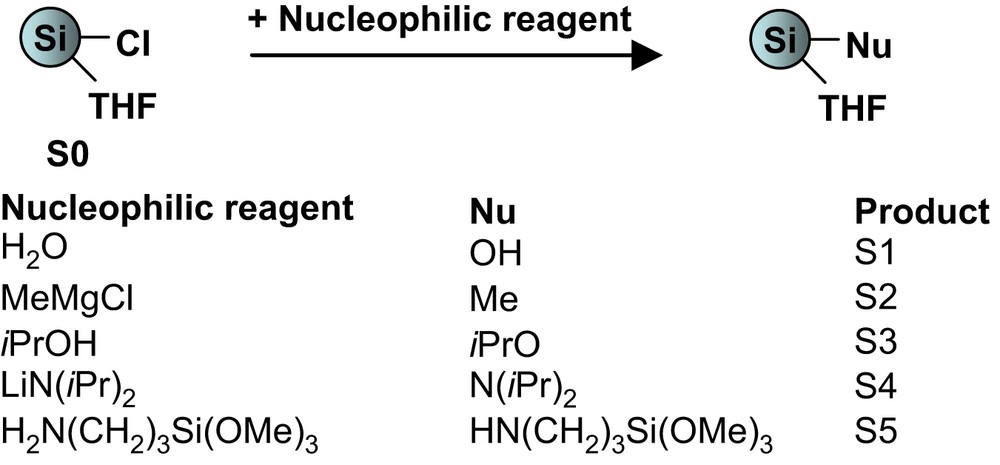
Surface functionalization of the silicon nanoparticles.
Samples S1–S5 were isolated as solids in yields ranging between 40 and 72%. They were analyzed by usual techniques. In all cases FTIR and solid-state 13C NMR exhibited peaks due to the presence of organic fragments formed by reaction with THF, as reported in the case of M0 and S0. In order to identify the characteristic bands of the organic function in each IR spectrum, we compared them to the spectrum of the hydroxy sample S1 and to the literature [36]. An example is given in Fig. 2 in the case of S2.

FTIR spectra of (a) S1 and (b) S2.
For S2, in addition to the peaks present in the spectrum of S1, peaks were observed at 1250 and 840 cm−1, corresponding to the symmetric deformation vibration of the CH3 group and to the methyl rocking vibration and the Si–C stretching vibration. Assignments of the characteristic bands for S1–S5 are provided in Table 1.
FTIR data for S1–S5
| Sample | Wave numbera | Assignment |
| S1 | 3310 | O–H |
| 1100–1000 (s) | Si–O and Si–O–Si | |
| S2 | 1250 and 840 (s) | Si–C |
| 1385 and 1370 | CH(CH3)2 | |
| S3 | 1200–1030 (s) | Si–O |
| 880 (s) | SiOCH(CH3)2 | |
| S4 | 1180 | N–C |
| 788 (w) | Si–N | |
| 3390 | N–H | |
| S5 | 1191 (s) | N–C |
| 780 (w) | Si–N |
a In cm−1, s: strong, w: weak.
The same approach was used in order to interpret the 13C and 29Si MAS NMR spectra of the samples. The 13C NMR spectra depicted in Fig. 3 illustrate this in the case of S1 and S2. For S2, characteristic peak of the methyl carbon appears at 2 ppm (Fig. 3).
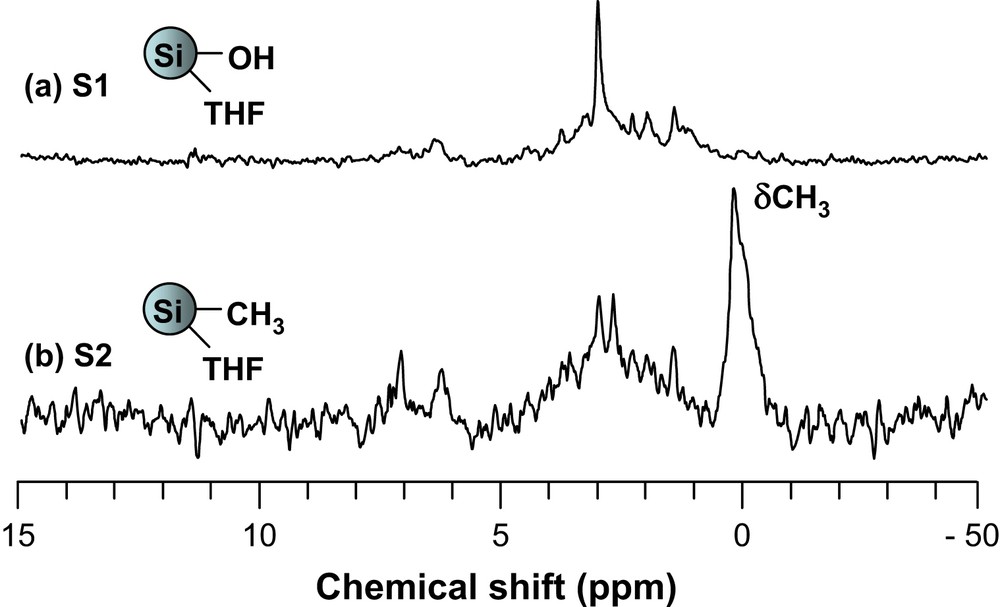
Solid-state 13C MAS NMR spectra of S1 and S2.
Thus, the peaks expected for each sample, depending on their surface functionality, could be identified in their respective spectra. Their chemical shifts and assignment are listed in Table 2.
13C Chemical shifts and their assignment for S2–S5a
| Sample | 13C NMR | |
| δ | Assignment | |
| S2 | 2.4 | SiCH3 |
| S3 | 26.2 | SiOCH(CH3)2 |
| 65.4 | SiOCH(CH3)2 | |
| S4 | 27.3 | SiN[CH(CH3)2]2 |
| 63.7 | SiN[CH(CH3)2]2 | |
| S5 | 6.1 | SiNH–(CH2)2–CH2-Si(OMe)3 |
| 21.5 | SiNH–CH2–CH2–CH2–Si(OMe)3 | |
| 43.7 | SiNH–CH2–(CH2)2–Si(OMe)3 | |
| 50.7 | SiNH–(CH2)3–Si(OCH3)3 |
a Although they are present in all cases, the signals due to the by-products formed by reaction with THF are not mentioned for reasons of clarity.
Solid-state 29Si NMR spectra showed three peaks at −64, −101 and −110 ppm in all cases. These were attributed to Si−OR, Si−OH and Si−O−Si bonds [37], respectively (Fig. 4). Again, signals corresponding to silicon atoms with their corresponding functional groups could be identified in each sample (Table 3).
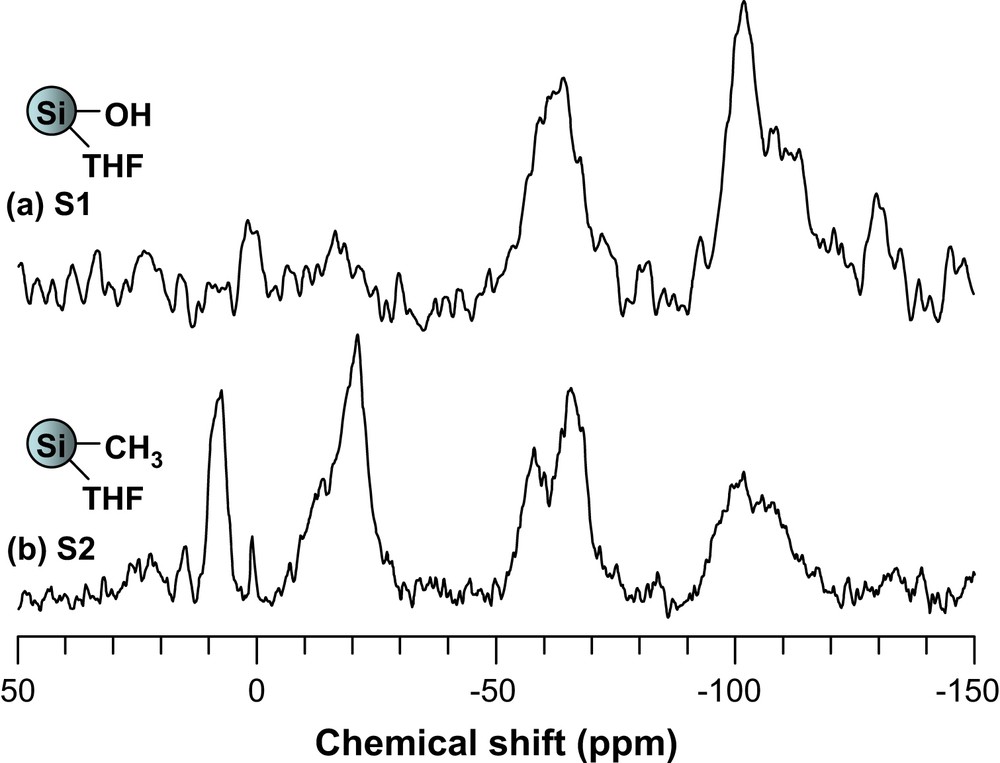
Solid-state 29Si MAS NMR spectra of S1 and S2.
29Si Chemical shifts and their assignment for S2–S5
| Sample | 29Si NMR | |
| δ | Assignment | |
| S2 | 7.6, −20.9 | SiCH3 |
| S3 | −83.7 | SiOCH(Me)2 |
| S4 | −17.7 | SiN[CH(Me)2]2 |
| S5 | −16.1 | SiNH(CH2)3Si(OMe)3 |
| −42.4 | SiNH(CH2)3Si(OMe)3 |
It is important to note that silicon nuclei linked to four other silicon nuclei present high relaxation times [37] and their contribution to the spectrum under the experimental conditions is very weak.
In summary, spectral analyses of the samples S1–S5 were in agreement with the substitution of chlorine atoms by nucleophiles. They also confirmed the presence of organic fragments coming from a side-reaction with THF.
TEM was used to investigate the Si-np structure. The Si-np were deposited on a holey carbon grid via evaporation of a pentane suspension. It is to note that particles cast on the grid from other solvents gave similar results. The number of Si-np, their shape and size were similar regardless of their surface functionality. Fig. 5 shows a picture for the methyl-terminated Si-np.

Micrographs of S2: (a) ×50, 000 times (scalebar = 100 nm) and (b) electronic diffraction.
A large number of homogeneous round-shaped particles were observed (Fig. 5a). Measurements of more than 800 particles sizes gave a mean diameter of 13 nm with a standard deviation of 3.4 nm (Fig. 6). The observed polydispersity may be viewed as satisfying, considering the fact that no templating agent and no post-synthesis treatment such as HPLC was used [23].
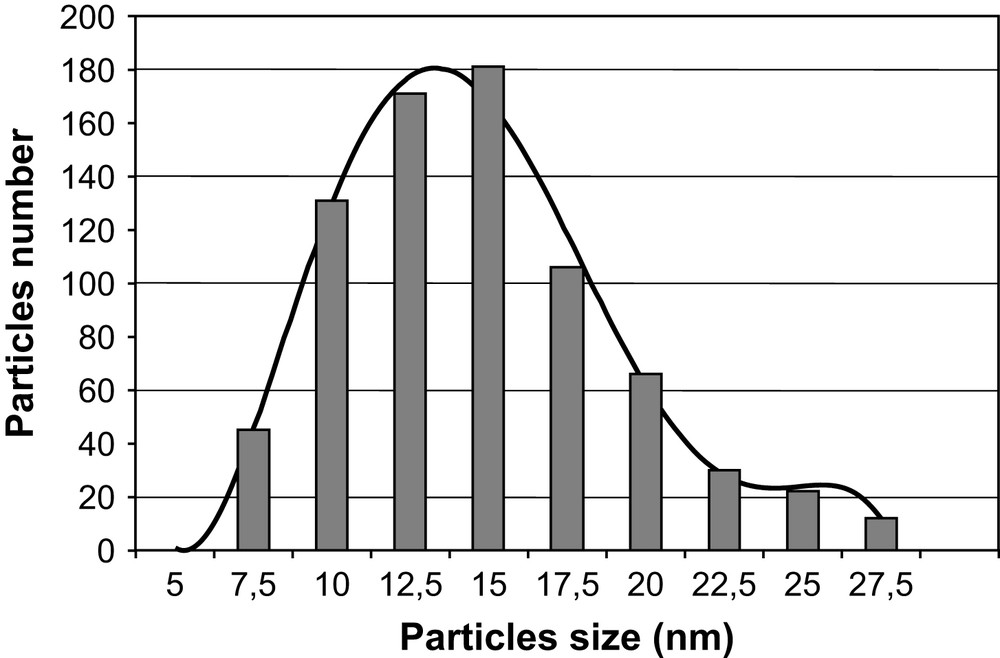
Histogram of particle sizes from a survey of 800 particles from different regions of the grid.
Selected area electron diffraction (SAED) showed a diffraction pattern characteristic of a sample presenting disordered particles, with well-defined rings presenting localized, more luminous, spots (Fig. 5b). General intensity of these patterns varied according to the observed area and the number of crystalline particles. Interreticular distances were measured on the first five rings using a gold sample as reference. Our experimental values corresponded well with the standard values of diamond cubic crystalline silicon from JCPDS (Joint Committee for Powder Diffraction Standard) (Table 4). It is to note that Si-np exhibited no diffraction pattern on powder XRD despite their electron diffraction pattern in TEM. This is consistent with particles with a small crystalline core [38] and unstructured peripheral matter. The absence of any signal confirmed that there was no trace of graphite or KCl, by-products of the reaction, in the sample. Variation of experimental parameters such as time or temperature during either the first step of the synthesis or the surface functionalization did not change the morphology, size or aggregation of the Si-np described here.
Experimental and literature interreticular distances
| dhkl measured (Å) | dhkl silicon (Å) | {hkl} |
| 3.0 ± 0.2 | 3.14 | 111 |
| 1.93 ± 0.09 | 1.92 | 220 |
| 1.58 ± 0.06 | 1.64 | 311 |
| 1.21 ± 0.04 | 1.25 | 331 |
| 1.08 ± 0.04 | 1.11 | 422 |
Finally, the optical properties of Si-np were studied by UV–vis and photoluminescence spectroscopies at room temperature. It appeared that all compounds exhibited a maximum absorption between 250 and 380 nm. They had an emission at about 410 and 430 nm when they were excited at 334 nm, that is, in the range already reported in the literature [13,23,26]. It has been suggested that the nature of the surface of Si-np has an influence on its light emission [39–41]. However, the origin of Si-np photoluminescence is still controversial. The nanostructure gives to Si particular physical and electronic properties because the ratio between surface atoms and bulk is high [39,42]. To date, many theories have been proposed from surface state to quantum confinement effects [43]. In the present case no major difference was observed between S2–S5, but the presence of organic residues linked on the surface by covalent bonds due to side reactions with THF does not allow a simple interpretation. The fact that our particles are larger on average than those described in literature give rise to similar emission spectra implies that the electronic properties of silicon nanoparticles are not related to quantum confinement effects. This observation is consistent with the fact that the silicon nanoparticles are of covalent nature. They differ from metallic nanoparticles that are dependent on quantum confinement effects.
4 Conclusion
The novel solution route reported here allowed us to obtain silicon nanoparticles at different levels of polycondensation. It resulted in a graphite/Si-np composite with ordered silicon particles at the nanoscale when the stoichiometry between the two reactants was 0.25 SiCl4 for 1C8K. In the presence of an excess of SiCl4, the resulting chloride-capped silicon nanoparticles allowed an easy access to a large range of surface functionalities by simple nucleophilic substitutions: hydroxide, methyl, isopropoxy or amino groups. However, a side reaction with the solvent (THF) was observed. It appears that while the size of our silicon nanoparticles is larger than those described in the literature, their emission properties are comparable. This implies that size is not a main parameter in orienting the optical properties of silicon nanoparticles. As opposed to metals, the silicon bond is covalent and as such obeys the rules of covalent chemistry: it is not dependent on quantum confinement effects. Hence, we believe that covalent links between silicon atoms and surface functional groups might be more inclined to directly influence the properties of the particles than their size. Work is in progress in our group to confirm this statement and will be published in due time.
Acknowledgements
The authors thank Dr. Michel Granier (CMOS – UM2) for assistance in the photoluminescence experiments.