

1 Introduction
1.1 Low-molecular-weight spies
The small-molecular world includes an extremely large collection of chemical compounds, existing or virtual, sometimes called the chemical space. The size of chemical space is a matter of speculation, but figures of the order of 1060 have been suggested [1]. This number is certainly much larger than the number of existing proteins and there is a reasonable chance that, for each protein, one could find a specific small ligand, allowing a one-to-one connection to be made between the small- molecular weight world and the macromolecular world. Since the nuclear magnetic resonance (NMR) properties of the members of the two worlds are rather different, one can envisage new approaches in which small molecules are used as ‘spies’ to detect by NMR macromolecules and their interactions (i) in complex mixtures, (ii) at low concentrations, and (iii) using non-isotopically labelled samples. These three limitations are typical of physiological conditions. Direct NMR observation of individual macromolecules under these conditions is presently impossible for sensitivity and selectivity reasons.
Small ‘spy’ molecules should display selectivity for their macromolecular targets in the order that they can be addressed individually in a complex mixture. The variety of chemical environments in the small-molecular world is much larger than that in biological macromolecules, providing an enhanced spectroscopic resolution. This makes much easier the selective NMR observation of each of the small-molecular-weight spies in a complex mixture than the direct selective observation of their high-molecular-weight correlates (Fig. 1).
Schematic depiction of the relationship between small spy molecules and their macromolecular counterparts. Small molecules can detect not only the presence of specific macromolecules but also their interactions with other macromolecular components.
The use of small ‘spy’ molecules is directly related to the study of ligand–protein interactions, which is fuelled by the search for low-molecular-weight drugs. There are, however, major differences: drug candidates are usually expected to bind strongly to the macromolecule and change its properties (e.g., inhibit its natural activity) and to be non-toxic for the whole organism. In contrast, low-molecular-weight spies are expected to bind weakly to the macromolecule, not perturbing its activity and, if used in vitro, they are subjected to far less stringent conditions of toxicity or bioavailability.
In addition, small spy molecules should be able to sense different states of their target (e.g., different conformations or the interaction with other macromolecules) and encode this information in a way that can be ‘read’ in their free state.
1.2 Ligand screening
The use of NMR to study protein–ligand interactions is a mature field and has been extensively reviewed [2–4]. The typical problem is to screen a collection of compounds to find suitable ligands for a macromolecule of interest. One classically distinguishes between methods based on protein observation and those that rely on ligand detection. The use of small-molecular-weight spies is clearly related to ligand detection methods used for screening purposes. In this case, a known ligand–macromolecular pair is observed in order to detect additional perturbations in the macromolecular component.
1.2.1 NMR experiments for lead generation in drug discovery
The most used ligand-based screening methods have the desired properties of not requiring isotopically labelled proteins, not being limited by the molecular weight of the biomolecule, and being applicable to samples of low macromolecular concentration.
Binding is detected through the changes in the NMR spectroscopic properties of the free ligand induced by the temporary interaction with the macromolecular target. This requires fast exchange between the free and the bound forms and puts a lower limit to the ligand koff, the rate constant for ligand release from the complex.
In the fast exchange limit, the value of the observed properties is the weighted average of those corresponding to the free and bound forms and the sensitivity of the method depends on the relative values of the measured property in the two forms. Ligand chemical shifts are, in general, only weakly affected by its interaction with diamagnetic proteins. In contrast, the molecular weights of small ligands and their complexes with macromolecules are widely different and so are the diffusion coefficients (translational and rotational) of free and bound ligands. Translational diffusion is readily measured by the attenuation of NMR signals in gradient echo experiments and is used in different ligand screening experiments. Rotational diffusion governs relaxation and is the basis of the most widely used NMR methods for ligand screening: Saturation Transfer Difference (STD) [5,6] and WaterLOGSY (Water–ligand observed via gradient spectroscopy) [7,8].
Both STD and WaterLOGSY are based on (i) the maintenance of a steady state away from equilibrium by the capacity to selectively saturate the macromolecular component without directly affecting the free ligand, (ii) the efficient cross-relaxation in slowly tumbling molecules that extends saturation to all spins in the complex (including those of the bound ligand) and (iii) the slow relaxation of the free ligand that allows the build up of the concentration of saturated free ligand as it exits the complex and is replaced with non-saturated ligand from an excess free ligand pool. Accumulation of free ligand wearing the ‘mark’ of its pass through a macromolecular complex depends on the balance between binding/saturation/release events and free ligand relaxation.
2 Using ligands to detect protein–protein interactions
2.1 Relaxation based detection of protein–protein complexes
Large changes in correlation time are associated with protein–protein interactions. These changes can be measured indirectly through low-molecular-weight reporters of each of the interacting partners. This approach was pioneered by the group of Konrat [9]. Their approach makes use of a ligand-binding domain fused to the protein of interest and monitors protein–protein interactions through changes in the observed relaxation time of the ligand. The approach is schematically shown in Fig. 2.
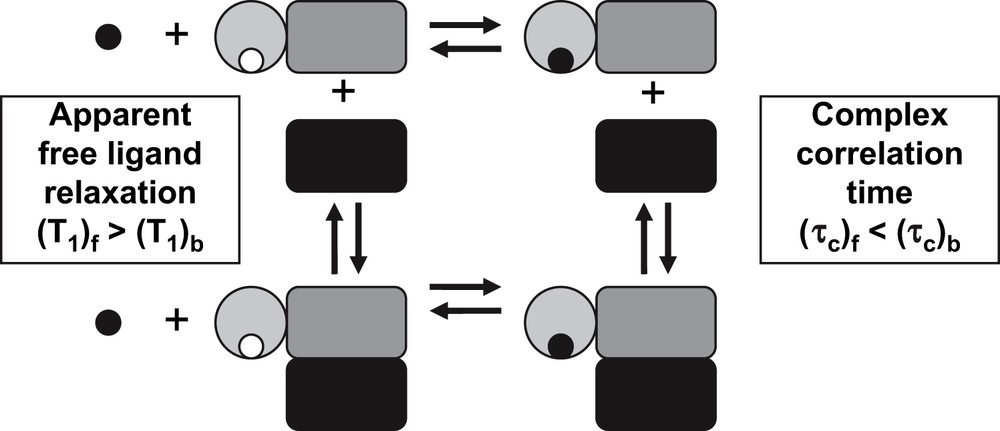
Changes in correlation time of the ligand–macromolecule complex resulting from the interaction between two macromolecules can result in changes in the relaxation properties measured in the free ligand. If the dissociation constant of the ligand–macromolecular complex is not affected by the additional interaction, ligand STD can be used to measure the formation of protein–protein complexes.
The concept was demonstrated by measuring selective T1 of the meta protons of phenyl phosphate binding to a src homology 2 domain (SH2 domain) fused to v-Myc. A decrease in T1 was observed when this protein binds to Max and that the effect could be reversed by adding un-tagged Myc, demonstrating that phenyl phosphate is indeed reporting on the formation of a protein–protein complex. Relatively large protein concentrations were required due to the direct measurement of the relaxation rates.
In order to decrease the protein requirement, one could use ligands whose relaxation rates are more strongly affected by changes in the correlation time of the complex, e.g., fluorinated ligands owing to their large chemical shift anisotropy (CSA) contribution to relaxation [10,11].
An alternative approach that has the potential of drastically reducing the protein concentration requirements is to measure relaxation rates, indirectly through an STD experiment.
The intensity of the STD signal is a complex function of the ligand on and off rates, the structure and dynamic of the complex and the ligand, and the correlation time of the complex. Thus, STD may be considered as a very sensitive method to measure the correlation time of the protein and protein–protein interactions can be detected through changes in the STD of a small molecular weight reporter.
Exploratory simulations have been carried out using the complete relaxation and conformational exchange matrix (CORCEMA) method [12]. This program calculates the fractional STD intensity {(I0 − I)/I0} assuming fast chemical exchange on the chemical shift and relaxation rate scales on the basis of the structure of the complex, the correlation times of the different species involved and the dissociation constant of the complex. We used the crystal structure (Protein Data Bank (PDB) reference identifier: 1JQ3) of a spermidine synthase complexed with S-adenosyl-1,8-diamino-3-thiootane (AdoDATO) [13] and the correlation time and dissociation constant of the complex were varied to simulate different experimental situations. Fig. 3 shows the calculated intensity of the STD signal for a ligand proton as a function of the correlation time of the macromolecule and the dissociation constant of the complex. A nearly linear dependency of the STD signal with the correlation time is observed. The maximum signal is predicted for ligand dissociation constants from 10−4 to 10−5 M, although ligands with dissociation constants in the 10−3–10−7 M range should also allow the detection of changes in correlation time induced by protein–protein interactions.

Calculated STD for the proton of the AdoDATO ligand closest to spermidine synthase in the crystal structure IJQ3 assuming different values for the dissociation constant and the correlation time of the complex. Curves in the left panel correspond to correlation times from 15 to 45 ns in 5 ns steps. In the right panel, the dependency of the STD signal with the correlation time is plotted for different values of the dissociation constant. Note that the best reporters are expected to have dissociation constants between 1 and 100 μM.
The advantages of detecting protein–protein interactions indirectly through STD are its sensitivity and selectivity. Thus, protein–protein interactions can be detected in complex mixtures of proteins at low micromolar protein concentrations without isotopic enrichment, provided that a selective ligand for the protein of interest is available.
The concept was tested experimentally using spermidine synthase 2 (SPDS2), a 39-kDa protein from Arabidopsis thaliana and its natural substrate, putrescine, which binds reversibly and selectively with a dissociation constant of 240 μM at 298 K, close to the optimum predicted by the simulations. Lowering the temperature to 278 K increases the viscosity two-fold, causing an equivalent increase in the correlation time. This induces the expected change in the STD intensity, after correcting for the change in dissociation constant of the complex that changes from 238 μM at 298 K to 454 μM at 278 K.
An artificial protein–protein interaction was introduced by incorporating an S-peptide tag (15 residues) in the C-terminal end of SPDS2. The S-peptide binds with high affinity (KD = 10−9 M) to the 104 amino acid S-protein (11.5 kDa) derived from pancreatic ribonuclease A [14]. Under the experimental conditions used, addition of S-protein to the SPDS2·S-Tag fusion protein produced a ca. 25% increase in the STD effect of putrescine, as expected for the formation of the SPDS2–S-protein complex. No STD could be observed between S-protein and putrescine. STD spectra of putrescine intreacting with SPDS2 alone or in a complex with S-protein are shown in Fig. 4.
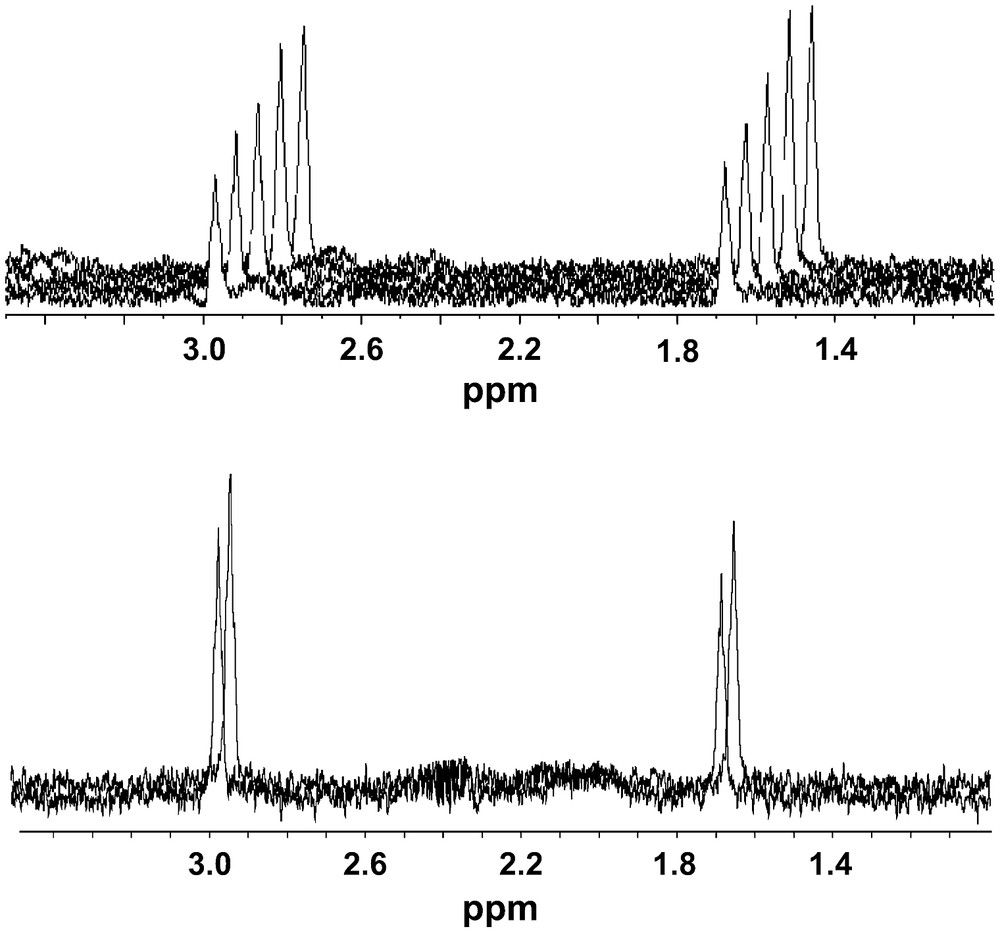
Top panel: the observed STD for the putrescine ligand of spermidine synthase increases by ca. 50% when the temperature is changed from 298 to 278 K (an approximately two-fold increase in viscosity). Bottom panel: the observed STD for the putrescine ligand increases by 25% in the presence of protein S binding to an S-tag attached to spermidine synthase. Putrescine shows no STD with protein S.
2.2 Ligand selection
The selection of the appropriate reporter for a given protein is a key step for the general application of the method. The optimal dissociation constants for small molecule reporters of protein–protein interactions are in the range of 10−3–10−5 M. Ligands of proteins with known structure in this affinity range can be readily identified with a fast protocol that involves a minimum number of experiments selected from a short list of candidates identified with a very efficient computational screening approach developed in our group [15]. The method is based on the LINGO concept that provides a very fast evaluation of chemical similarities and the prediction of relevant properties, such as water solubility, resulting in a very efficient selection of the most promising candidates to be tested experimentally [16]. In a typical application using a low-molecular-weight phosphatase of known structure, 9 ligands with dissociation constants lower than 1 mM (of which 4 were below 10−4 M) were found after only 34 experiments starting with a virtual library of 500 000 small molecules [17].
2.3 Chemical shift based detection of protein–protein complexes
While protein chemical shift perturbations are widely used, ligand chemical shift changes are usually small and can only be observed using high protein concentrations. 129Xe is an exception. The high polarizability of the electronic cloud of Xe atoms makes 129Xe chemical shifts very sensitive reporters of its environment.
The interaction of Xe atoms with proteins has been extensively studied. In protein crystals, Xe binds preferentially close to hydrophobic cavities of proteins [18]. 129Xe chemical shift effects caused by solutions of amino acids and proteins, under native and denatured conditions, have been studied systematically by Rubin et al. [19]. 129Xe deshielding resulting from weak dispersive interactions with the different amino acids shows a correlation with the Xe accessible surface. Folded proteins have a large fraction of their side chains buried in their core; however, the 129Xe NMR chemical shifts in the absence of specific interactions are roughly proportional to the molecular weight of the protein and, in proteins like bovine serum albumin (BSA), the deshielding effect of the native protein is larger than that of the denatured form. This suggests that Xe binding sites may be present in the surface of folded proteins.
The presence of well-defined hydrophobic cavities in the interior of proteins has been observed and characterized by 129Xe NMR in other cases like myoglobin [20], lipid-transfer protein 1 [21], or T4 lysozyme [22]. Concentration and composition normalised 129Xe deshielding factors of around 5 × 10−3 ppm/mmol of residue are taken as the threshold for detecting the presence of specific interactions.
Specific Xe–protein interactions are sensitive reporters of protein structure and have been used to study structural changes associated with protein function, for example in the case of maltose binding to maltose binding protein (MBP) [23], or changes in ligand binding in Escherichia coli CheY protein [24].
The X-ray study of Prange et al. located Xe-binding sites at the interface between oligomers [18]. We have very recently shown that 129Xe NMR is indeed a good technique to study protein–protein interactions, as shown in the study of the oligomerization of bovine low-molecular-weight protein tyrosine phosphatase (lmwPTP) [25]. In this system, 129Xe NMR shows a clear non-linear dependency on the protein concentration that reflects the different interaction of Xe with the different species (monomer, dimer, tetramer) that are being populated as the total concentration of the protein increases. The interpretation of 129Xe chemical shift data is complicated because the observed signal is the average of specific and non-specific interactions to the different species. A much simpler picture is obtained when 129Xe NMR spectra are measured in the presence of 50 mM arginine and 50 mM glutamic acid. This buffer system had been previously described to decrease non-specific protein–protein interactions responsible for low solubility and NMR line broadening in a variety of unrelated proteins [26]. We have observed that non-specific Xe–protein interactions are strongly attenuated in the presence of arginine and glutamic acid [27]. At low protein concentrations where monomer and dimer lmwPTP are the major species, very small 129Xe NMR shifts are observed in the presence of arginine and glutamic acid. In contrast, the interaction with lmwPTP tetramers is preserved. An estimate of the chemical shift induced by each of the species was possible because their individual concentrations were deduced independently from 15N NMR relaxation measurements. The 129Xe NMR shifts induced by monomer, dimer and tetramer lmwPTP are αM/N = 0.016 ± 0.007 × 10−2, αD/N = 0.28 ± 0.037 × 10−2, and αT/N = 2.7 ± 0.4 × 10−2 ppm/mmol of residue. These values indicate very weak non-specific binding to monomer and dimer and a strong specific binding to lmwPTP tetramers. Even ignoring the presence (binding) of monomer and dimer (to Xe), a very good correlation with an R2 = 0.993 between the overall Xe chemical shift and the concentration of tetramer is obtained (Fig. 5). Xe binding to oligomer interfaces may be a general property making 129Xe a fast and very sensitive reporter of protein–protein interactions.
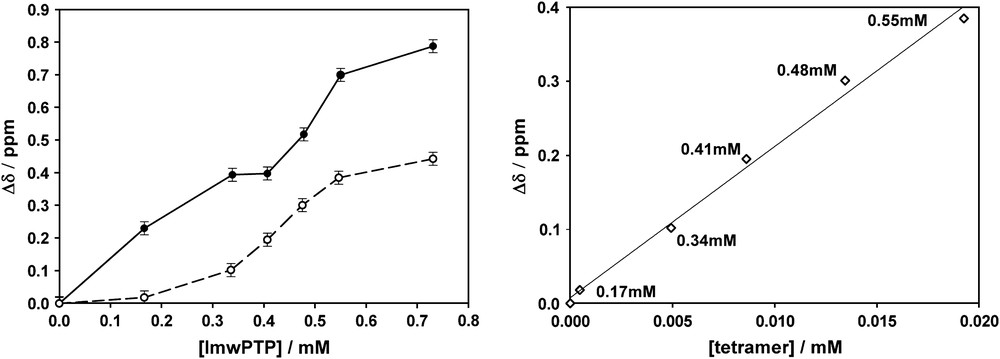
129Xe chemical shifts induced by lmwPTP. The left panel shows the effect of different total concentrations of lmwPTP in the presence (dashed curve) and in the absence (continuous curve) of 50 mM arginine and glutamic acid. The right panel shows the linearity of the effect with the concentration of lmwPTP tetramers present in the solution and independently detected by relaxation measurements (reproduced from Ref. [25] with permission from the copyright owner).
Despite of the obvious advantages of inertness and NMR sensitivity, the interpretation of 129Xe NMR shifts can be complicated by the competition between multiple binding sites in fast exchange. Xe based biosensors have been developed to alleviate this problem. Cryptophane-A binds reversibly Xe atoms with an apparent association constant of 3000 M−1 [28]. The process is slow on the 129Xe NMR time scale and bound Xe gives a separate signal shifted 160 ppm upfield in tetrachoroethane at 298 K. The chemical shift of bound Xe is very sensitive to minor changes in its environment. This sensitivity is dramatically demonstrated by the observation of measurable chemical shift changes between Xe complexes of cryptophane-A with different degrees of deuteration [29]. By attaching a targeting biotin moiety to a functionalized cryptophane, changes in 129Xe chemical shifts can be used to detect binding to the protein, avidin [30]. It is expected that additional interactions of the target protein with other macromolecules should also result in perturbations of the 129Xe chemical shift.
2.4 Increasing the sensitivity by using non-equilibrium polarized probes
The sensitivity of 129Xe as a reporter can be enhanced by several orders of magnitude by using non-equilibrium (hyperpolarized) gas obtained by collision exchange with rubidium atoms' laser polarized with circular polarized light [31]. The same method is applicable to other noble gases which can also be used as reporters or in high-sensitivity imaging applications [32,33].
In addition, relaxation times of 129Xe can be extremely long, thus perturbations induced by the interactions of Xe sensors with their targets can be recorded in a physically distant, optimized, detection chamber. This is the principle of ex situ NMR first proposed by the group of Pines [34].
The long relaxation time of 129Xe, the large non-equilibrium 129Xe polarization and the chemical exchange between free and bound forms can be combined to obtain an additional sensitivity advantage for the indirect detection of very small concentrations of target molecules. Exchange-enhanced detection makes use of the fact that exchange between two spectroscopically different environments is rapid in comparison to the long longitudinal relaxation times of xenon. The process is strongly reminiscent of the sensitivity gain obtained by accumulation of free saturated ligand in STD experiments. In contrast to STD, where saturation of the ligand is achieved by cross-relaxation with protein signals, the large chemical shift difference between free and bound xenon allows for direct selective inversion or saturation of only the bound xenon. However, the intensity of free xenon signal is attenuated by exchange and the decrease in intensity can be related to the presence of xenon bound to its target [35]. This principle has been recently applied to image a 5 μM solution of a target and foreseeable improvements could increase the sensitivity to allow detection of nanomolar to picomolar concentrations [36].
3 Prospects for future developments
The unique sensitivity of 129Xe as a molecular spy arises from its peculiar properties: the availability of hyperpolarized 129Xe and its slow relaxation rates. Could other molecular probes share similar desirable properties? There is currently a high interest in the preparation of non-Zeeman equilibrium states in small molecules to increase sensitivity and slow down relaxation. Both properties may be used to prepare more sensitive spy molecules.
Solid-state dynamic nuclear polarization (DNP) at low temperatures followed by rapid thawing and transfer to the NMR instrument can provide orders of magnitude enhancement in the polarization of small molecules [37]. The decreased concentration at which these hyperpolarized samples can be observed by NMR should allow probing their target macromolecules at much lower concentrations.
Non-equilibrium nuclear singlet states may be created in isolated pairs of coupled spins [38] and even in systems containing more than two coupled spins [39]. If singlet–triplet exchange is prevented, the lifetime of these states can be much longer than T1. Singlet states may become useful to maintain hyperpolarized spin order generated by DNP or other methods and be used as sensitive molecular spies in a way reminiscent of the use of DQ relaxation for screening [40].
The connection between the small molecular world and the macromolecular interactome through NMR is a growing interdisciplinary frontier where different branches of chemistry, molecular biology and spectroscopy can find a fruitful common ground.
Acknowledgments
We gratefully acknowledge a fruitful continuous collaboration with Prof. Eike Brunner (University of Regensburg), Dr. Pau Bernadó (Institute for Research in Biomedicine), Dr. Oscar Millet (CICBiogune), Dr. David Vidal and Dr. Michael Thormann (Origenis). This work was supported in part by funds from the Spanish Ministerio de Educación y Ciencia, FEDER (BIO 2004-5436).