

1 Introduction
The emergence of cheap methods for preparing diamond using chemical vapor deposition (CVD) technology has resulted in a growing interest in the properties and uses for polycrystalline diamond films. Consequently, electrochemistry is an area in which diamond films are generating particular enthusiasm. Because of their attractive properties, including very low background currents, wide electrochemical potential window and chemical inertness [1–4], boron-doped diamond (BDD) electrodes have been recognized as a promising electrode material in electrochemistry, principally for electroanalysis and electrolysis [5–7]. However, there are several factors which can influence the electrochemical behavior of diamond. One is the effect of oxidative treatment which changes the terminations from hydrogen to oxygen and affects the electron transfer rates to various redox species, providing in some cases enhanced electroanalytical selectivity [8,9]. Thus, the effect of surface oxidation on BDD electrochemical behavior has provided several fundamental studies in the literature [10,11]. However, the great diversity of results proves that the role of oxygen on BDD reactivity is not completely understood [12–14]. For example, the reactivity of oxidized BDD electrodes with some redox species, like Fe(CN)63−/4− shows controversial results, which exhibits the ambiguous behavior of the diamond/electrolyte interface and the charge transfer mechanisms [15,16].
As well as cyclic voltametry, capacitance–voltage measurements constitute another good probe of diamond/electrolyte interface. The general trend in the literature is to associate surface oxidation with a positive shift of the flat-band potential (VFB), deduced from Mott–Schottky plots [1,17,18]. By using oxygen plasma treatments, strong anodic treatments or boiling in strong acid, different authors have reported VFB values raised by 2 V. The generation of oxygenated terminations is usually proposed to explain this phenomenon.
However, as reported in previous works [15,20], electrochemical treatments could lead to a particular evolution of VFB, which is not in agreement with the general trend. For example, during the transition from “C–H” to “C–O” terminations, it has been established that a slight negative shift of flat-band potential can occur before the positive shift expected [19,20]. This result has been obtained by using successive mild anodic treatments coupled to systematic C–V measurements, and this particular phenomenon has been proved to take place while oxygen terminations are present on the surface. Furthermore, it has also been shown that a strong cathodic treatment applied on an anodized electrode leads to a negative displacement of VFB, which returns towards its initial value, although oxygenated terminations are still present at the surface [15]. Consequently, the role of oxygen on flat-band potential needs to be clarified.
In order to correlate VFB position to surface chemistry, the aim of the present paper is to describe the chemical modifications induced by anodic treatments, and especially to identify the type of oxygenated terminations involved during surface oxidation. For this purpose, surface analyses performed by XPS have been coupled to capacitance–voltage measurements after each anodization, in order to establish a correlation between VFB shift and chemical surface terminations.
2 Experimental
Polycrystalline boron-doped diamond (BDD) films ([B] = 2 × 1020 cm−3) of 1.6 μm thickness were deposited on high conductivity (100) silicon substrates by hot filament chemical vapor deposition.
Electrochemical experiments were performed using a classical three-electrode configuration: mercury sulfate electrode (MSE) as reference, BDD as working electrode and platinum as counter electrode.
Successive anodic treatments were performed in 0.5 M H2SO4. Each of them has been performed during 10 s in galvanostatic mode. Current density has been fixed at 500 μA cm−2. For each anodic treatment, the spent coulometric charge (Q) has been calculated. In order to “quantify” the successive anodizations, the coulometric charges involved in each process have been cumulated (QA).
Capacitance–voltage (C–V) measurements have been carried out in 0.5 M H2SO4 with a frequency set at 1107 Hz, using a HP oscillator 4204A and a lock-in amplifier (EG&G 5208). A linear potential sweep has been fixed at 20 mV/s for all experiments. The reproducibility of the capacitance–voltage measurements has been confirmed with experiments at different frequencies (from 100 Hz to 30 kHz).
XPS chemical analyses were performed with an Escalab 220i XL V.G. spectrometer equipped with a focused monochromated X-ray beam Al Kα. The constant analyzer energy mode was used with a pass energy of 8 eV.
All experiments were performed by using the same procedure: anodization and C–V measurements were performed successively in 0.5 M H2SO4. Prior to the transfer in the XPS chamber, samples were dipped in pure H2O solution and then handled under a solution drop protection that was tangentially dried under an argon stream.
3 Results and discussion
In situ capacitance–voltage measurements have been performed on as-deposited and treated electrodes. Mott–Schottky plots (C−2 vs. E, where C is the interfacial capacitance and E is the applied polarization) corresponding to as-deposited samples and anodized samples are shown in Figs. 1 and 2. All curves reveal a classical and reproducible Mott–Schottky linear behavior. This result allows considering that BDD electrodes have a classical semiconductor behavior in H2SO4, which is preserved all along the oxidation process.

Mott–Schottky plots (C−2 = f(E)) in 0.5 M H2SO4 obtained from as-deposited electrodes and anodized electrodes for QA ≤ 10 mC cm−2 (f = 1107 Hz).
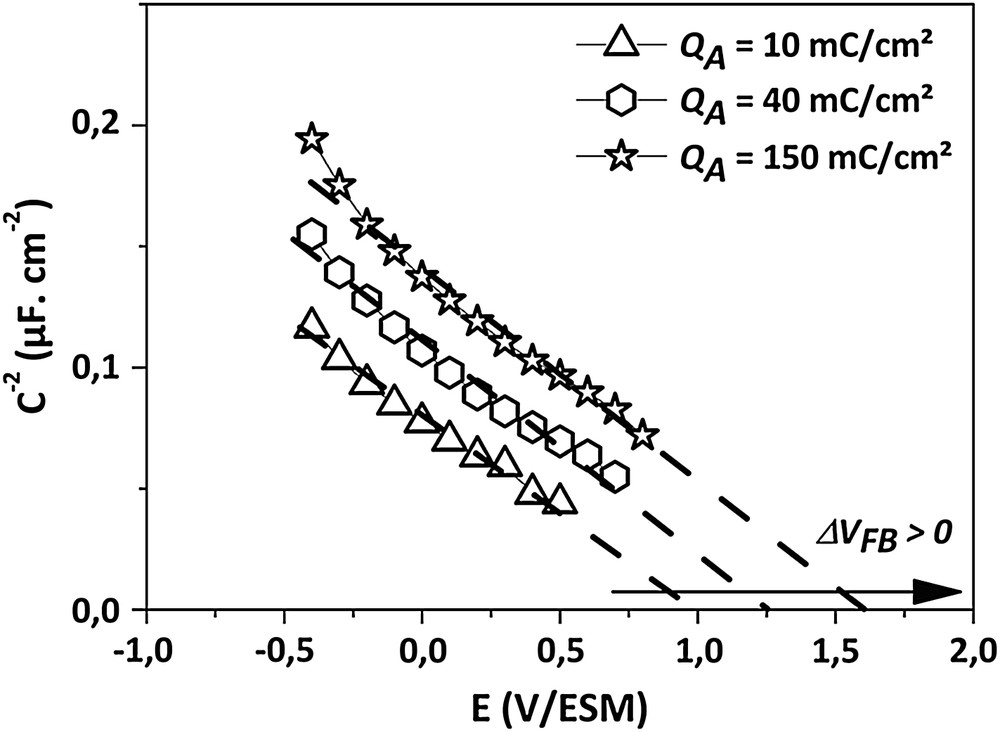
Mott–Schottky plots (C−2 = f(E)) in 0.5 M H2SO4 obtained from anodized electrodes for QA > 10 mC cm−2 (f = 1107 Hz).
From Mott–Schottky plots, space-charge density (NI) and flat-band potential (VFB) could be extrapolated. For an as-grown sample (Fig. 1), VFB = +1.2 V/MSE has been measured with a very good reproducibility. With NI = 2 × 1020 cm−3, the acceptor level is close to the doping level expected. All these values show weak frequency dependence in the range from 100 Hz to 30 kHz. The literature is exhaustive on flat-band potential values for hydrogenated electrodes [21–23]. Nevertheless, according to the specificity of each growing technology and samples preparation, the flat-band values reported by different authors scatter over a wide potential range, from 0 V/MSE to 1.5 V/MSE, which is in accordance with the present work.
The surface oxidation of the electrodes has been carried out by using successive anodic treatments on a single sample. Then, the accumulation of mild anodizations (ja = 500 μA cm−2 during 10 s, corresponding to a coulometric charge of Q = 5 mC cm−2 for each treatment) gives rise to an accumulated anodic charge (QA). Between each anodization, a capacitance–voltage measurement has been performed. The noteworthy C−2–V curves are reported in Figs. 1 and 2. When successive anodic treatments are applied to the electrode, a shift of the flat-band potential occurs without evolution of the slope. However, the shift direction depends on the accumulated coulometric charge. When the accumulated anodic charge (QA) is lower than ca. 10 mC cm−2, the anodic process gives rise to a negative displacement of VFB (Fig. 1). The minimal VFB value obtained is equal to 0.8 V/MSE. Then, for QA > 10 mC cm−2, a positive shift of VFB is observed, and for ca. 150 mC cm−2, VFB is up to 1.6 V/MSE (Fig. 2).
In order to investigate the chemical origin of VFB displacement, XPS analyses have been carried out on as-grown samples and after each anodization. From well-defined C1s and O1s core level spectra, respectively, at 284 eV and 532.5 eV, O1s/C1s atomic concentration ratios have been systematically calculated. In this paper, the study has focused on C1s spectra, since O1s peak fitting does not provide easily reliable information. Fig. 3 shows XPS spectra of the C1s spectral region of an as-grown sample. Binding energies (EB) and full widths at half maximum (FWHM) have been established according to data reported in Refs. [13,24,25]. The most intense peak (2), centered at 284.0 eV, can be attributed to the bulk diamond component. The peak at 284.7 eV (3) has been associated with the polyhydride carbon species (CHx) adsorbed on the surface. The peak centered on 285.6 eV (4) has been assigned to hydroxyl or ether groups (designated “C–O”). The peaks at higher binding energies, up to 287 eV (5), are related to carbonyl, carboxylic or ester carbon species (designated “CO”). The small peak at 283 eV corresponds to the graphitic carbon species. For an as-grown sample, the O1s/C1s atomic ratio is equal to 9%.
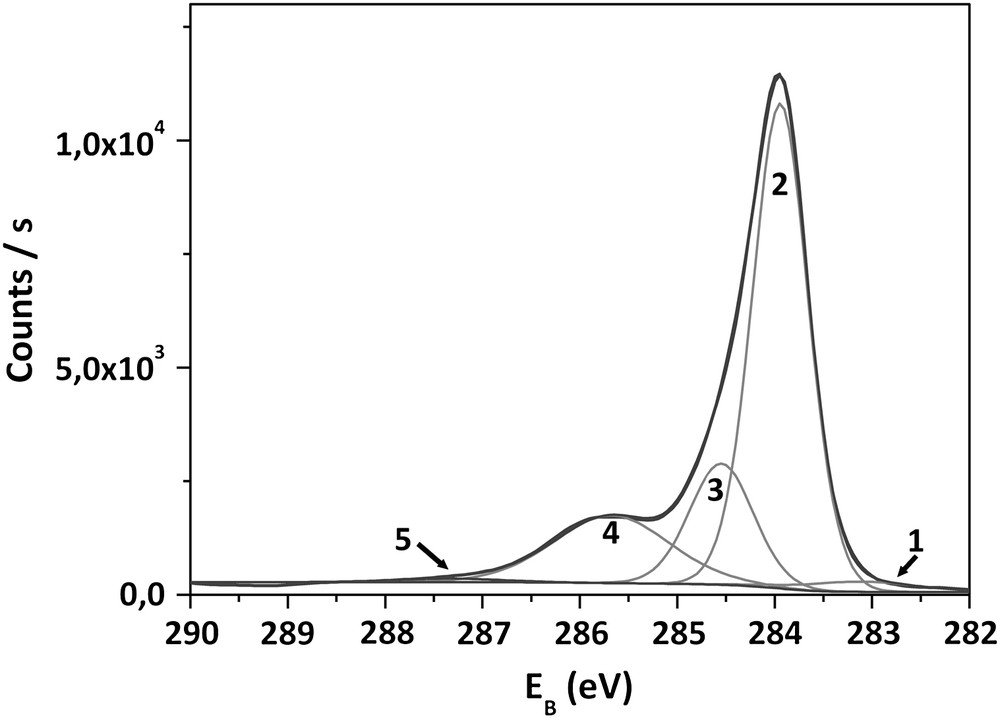
Fitted C1s XPS spectrum obtained from as-deposited diamond electrode surface.
C1s spectra obtained after anodic treatments are given in Figs. 4 and 5, respectively, for QA < 10 mC cm−2 and QA > 10 mC cm−2. In the first steps of surface oxidation (QA < 10 mC cm−2), the main evolution concerns the components related to oxygen functions, with a decrease of peak 4 centered at 285,6 eV, from 18% to 8%. In the same time, the proportion of peak 5, relative to the most oxidized functionalities, increases from 1% to 4%. During these first anodizations, the other components remain stable and the O1s/C1s atomic ratio is constant and equal to 9%.
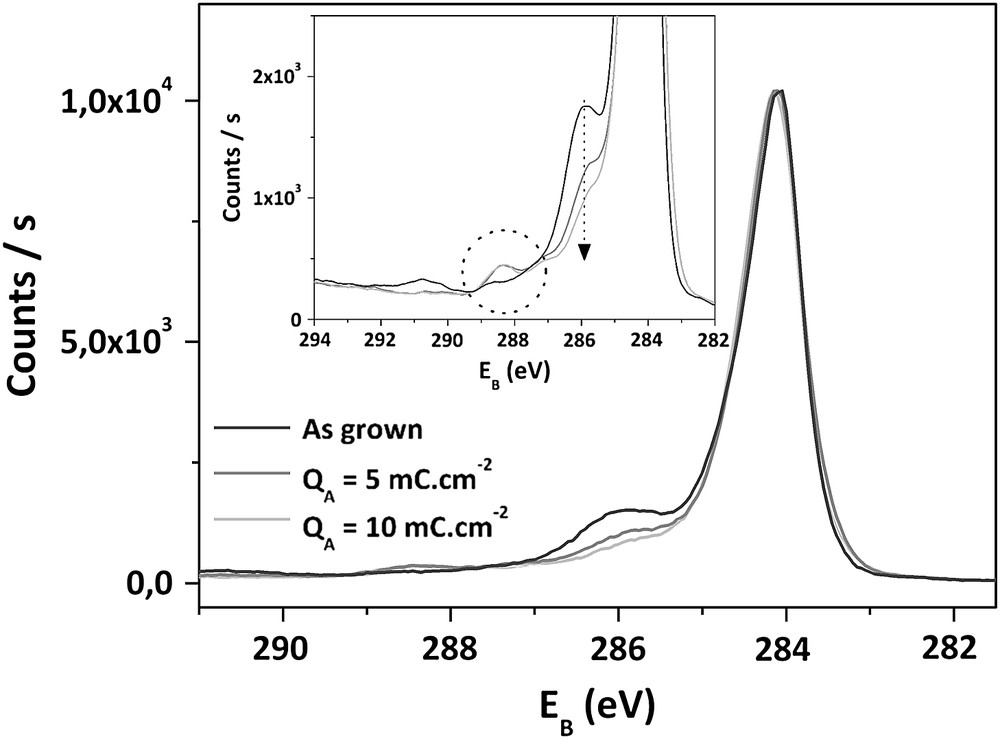
Superposition of C1s XPS spectra of as-deposited and treated electrodes for QA ≤ 10 mC cm−2.
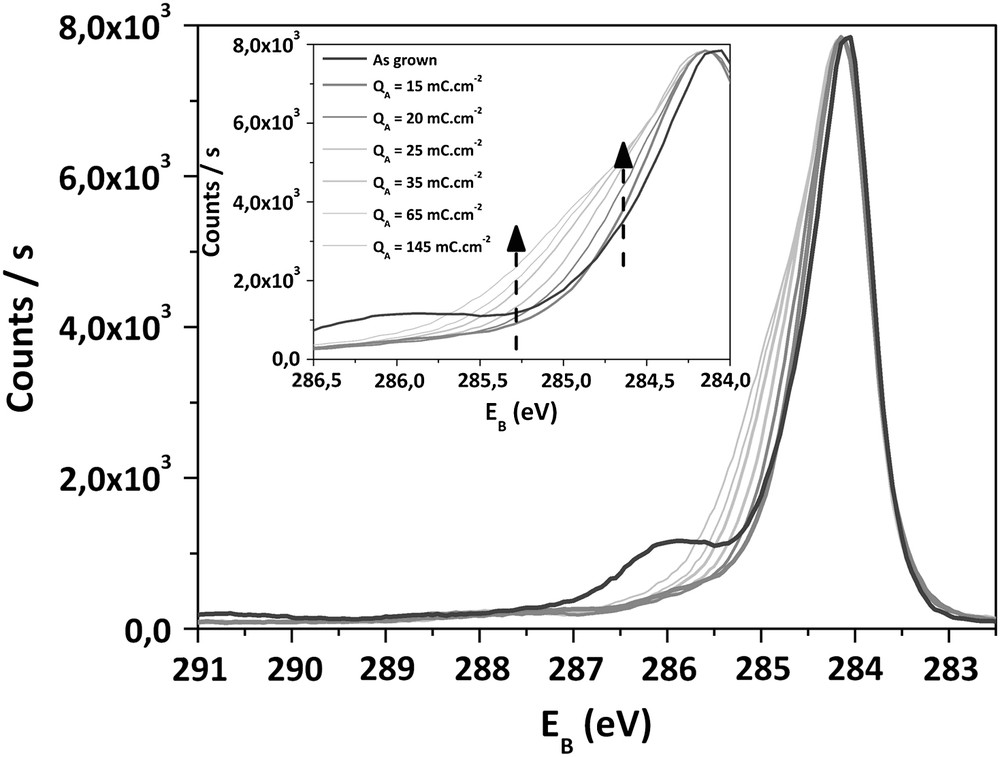
Superposition of C1s XPS spectra of as-deposited and treated electrodes for QA > 10 mC cm−2.
When QA > 10 mC cm−2, the evolution of the C1s spectra is different. A broadening of the main peak occurs due to the raise of components 3 and 4. From 8%, the peak 4 relative to “C–O” groups increases up to 15%. In the same time, the position of peak 4 is shifted to lower binding energies (−0.3 eV), which suggests a slight evolution of the chemical nature of these “C–O” groups. This modification of the surface chemistry occurs while an increase of peak 3 (relative to CHx) takes place. From 17% on as-grown sample, this component reaches 28% after an anodic process involving 200 mC cm−2. The proportion of “CO” groups remains stable at 3%. When QA exceeds 10 mC cm−2, a constant augmentation of the oxygen ratio at the surface is observed, from 9% to 12% for QA = 150 mC cm−2.
From XPS analyses and capacitance–voltage measurements, an evolution during the surface oxidation process occurs when accumulated coulometric charge is equal to ca. 10 mC cm−2. Below this threshold value, the proportion of surface oxygen remains constant, while a slight negative shift of VFB is observed. In the literature, a modification of VFB is generally related to an evolution of the oxygen proportion at the surface [1,17]. This apparent contradiction can be enlightened by taking into account the chemical type of oxygenated terminations at the surface. Even though the proportion of oxygen at the surface remains constant, first anodizations lead to an evolution of oxygen functionalities. The concomitant decrease and increase of, respectively, “C–O” and “CO” functionalities suggest that an oxidation of the “C–O” initially existing on as-grown samples takes place, leading to the formation of “CO” groups. This first stage in the oxidation process goes on up to the threshold value of ca. 10 mC cm−2. Then, anodization of the BDD surface leads to the formation of fresh “C–O” groups, which slightly differ from the original ones (see the moderate shift of binding energies in Fig. 5). Therefore, new oxygenated terminations are created at the surface, which explains the enhancement of the surface oxygen ratio (from 9% to 12%).
This particular evolution for oxygen functionalities during anodic treatments is also responsible for VFB displacement. Fig. 6 shows VFB variations and surface oxygen groups evolution according to the accumulated coulometric charge. This figure reveals a correlation between the proportion of “C–O” bounds at the surface and the flat-band potential position. Although dependence of flat-band potential on oxygen at the surface has already been mentioned by several authors, it is generally considered that all kinds of oxygen induce a positive shift of VFB. Our original approach of successive anodic treatments systematically coupled to XPS analyses strongly suggests that the flat-band potential position of BDD electrodes is mainly influenced by the presence of specific “C–O” groups, like alcohol and ester. This assumption is confirmed by an observation reported in a previous work, in which a negative shift of VFB was observed after a strong cathodic treatment inducing a constant “CO” proportion and a decrease of specific “C–O” groups [15]. Therefore, this work allows specifying the general trend in literature which links the flat-band potential shift to presence of all kinds of oxygenated terminations.

Flat-band potential variation and surface oxygen functionalities evolution according to the accumulated coulometric charge.
4 Conclusion
By using successive anodic treatments coupled with systematic XPS analyses and C–V measurements, a specific study of surface chemistry and BDD energetic diagram during surface oxidation has been performed. Firstly, it has been established that global oxygen ratio at the surface is not sufficient to explain the VFB shift. The chemical type of oxygenated terminations has to be considered, and a tight relation has been evidenced between the proportions of “C–O” functionalities, like hydroxyl or ether groups, and the flat-band potential's position. Furthermore, thanks to the control of the coulometric charge, the progress of BDD surface oxidation has been detailed. The transition of surface terminations from “C–H” to “C–O” undergoes two stages. In the first stage, modifications seem to be limited to the oxidation of preexistent oxygenated terminations. In the second stage, anodizations induce the formation of new oxygenated terminations.