

1 Introduction
Benzodifurane based compounds are very useful for a large variety of applications. They are used in bio-organic and medical fields [1,2] or, in the chemo-physical one, as dye-based molecular switches of luminescence, as building blocks for opto-electronic devices, as efficient organic dyes for solar cells and as a hole-transporting material in multilayered organic light-emitting diodes (OLEDs) [3–6]. Moreover, the benzodifurane core can be variously functionalised in order to modulate the absorption–emission spectral properties and can be employed in polymerization reactions [7]. Interest towards diamino benzodifuranes started a long time ago. The reaction of 1,4-benzoquinone with ethyl cyanoacetate in ethanol in the presence of a high concentration of aqueous ammonia was first reported by Craven [8] in 1931 and successively examined by various authors [9–13].1 Some arylsulphenyl derivatives were also reported [14]. Different hypotheses have been proposed over the years to explain the nature of the product of the Craven reaction, however, a complete and systematic study of the synthetic route and characterization is still missing. Compound 1 (Scheme 1) was first proposed by Wood [11] on the basis of the hydrolytic behavior of the Craven product. A tricyclic structure 2 (Scheme 1) was proposed by Jeffreys [12], and the same tricyclic core, containing two furane rings, was assumed by Derkosch [15] on the basis of IR spectroscopic data concerning the tetraacetyl derivative of the diethyl ester of 2,6-diamino-benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 3 (Scheme 1).

Subsequent evidence (by 1H NMR spectroscopy) of the basic benzofurane structure was given for compounds obtained by the reaction of 2,4-dimethyl-o-quinol acetate with ethyl cyanoacetate [16]. Evidence in favour of structure 3 for the Craven and related products was given by King and Newall [17] who also described some acetyl- and benzoyl-derivatives. They also pointed out the need for a strong excess of acetylating reagent to obtain the N,N′-tetraacetylated product and the feasibility of hydrolysis of N,N′-tetrabenzoylated compounds to the corresponding N,N′-dibenzoylated derivatives. Finally, recall that Obushak et al. [18] investigated the interaction of 2-aryl-1,4-benzoquinones with cyanoacetic esters in basic media, proposing the synthetic Scheme 2, and obtained a large number of arylbenzodifurane sparingly soluble compounds.

In the search for new materials potentially useful in the field of opto-electronics, we focused our attention on type 3 (Scheme 1) diamino benzodifurane based compounds to test the feasibility of functionalised moieties that are employable in the synthesis of polymers. To this purpose, a good solubility in common organic solvents and a good reactivity of amino groups are required. Moreover the synthetic route should be simple, not expensive and afford pure products in high yields.
A series of 2,6-diamino-benzo[1,2-b:4,5-b′]difurane based compounds were synthesized; the free diamine derivatives (1a–1c, Scheme 3), the N,N′-tetrabenzoylated (2a–2c, Scheme 3) and N,N′-tetraacetylated derivatives (3a–3c, Scheme 3), the N,N′-diacetylated (4a–4c, Scheme 3) and N,N′-dibenzoylated (5a–5c, Scheme 3) derivatives, and the mixed N,N′-diacetyl-N,N′-dibenzoyl derivative (6, Scheme 3).
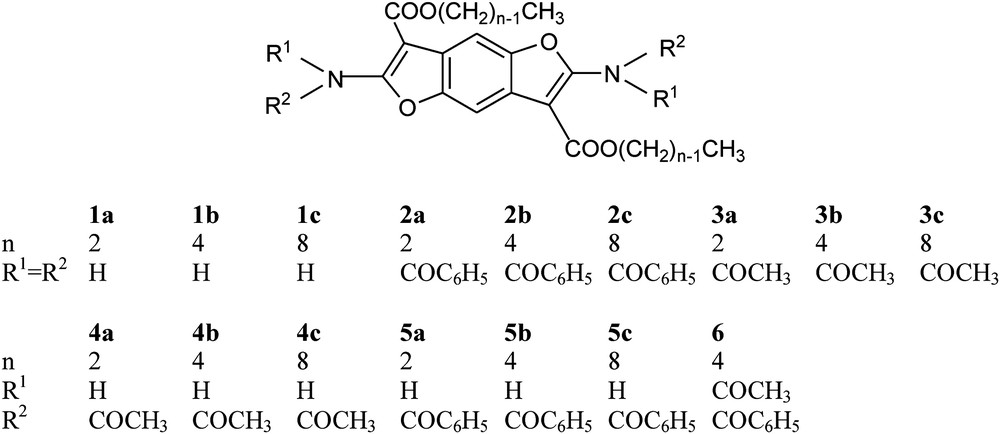
Compound 1a is already described in the literature [17], but its synthetic procedure is scarcely clear, laborious and produces low yield. Moreover, a systematic study of the reactivity of the amino groups has never been performed, being known only for 2a, 3a and 5a [17] and their direct structural characterization has not yet been reported. A better synthetic procedure for the systematic preparation of a class of 2,6-diamino-benzo[1,2-b:4,5-b′]difurane and their derivatives is described here. In order to improve the solubility in common organic solvents, and assuming that reaction scheme is analogous to Ref. [18] (see also Scheme 2), we reacted unsubstituted 1,4-benzoquinone and cyanoacetate alkyl esters having long n-alkyl chains. It is reason to expect, in fact, that the presence of such chains in the molecule lowers the melting point and improves the solubility in organic solvents, while the presence of substituents on the arylic group seems to be not effective [18].
The reactivity of amino groups towards acetylating and benzoylating reagents was investigated. Moreover, the ability of the free 1a–1c diamines and the N,N′-disubstituted derivatives to be employed as difunctional monomers in the obtainment of polymeric materials was tested through the synthesis of two new polyamides.
The synthesized compounds were characterized by NMR spectroscopy and elemental analyses. Single crystal X-ray diffraction was performed for 1a, 3a and 6.
2 Results and discussion
2.1 Preparation and characterization of diamino benzodifurane derivatives
The prepared benzodifurane compounds and their derivatives are labeled in Scheme 3 as 1a–1c (free diamine derivatives), 2a–2c (N,N′-tetrabenzoylated derivatives), 3a–3c (N,N′-tetraacetylated derivatives), 4a–4c (N,N′-diacetylated derivatives), 5a–5c (N,N′-dibenzoylated derivatives) and 6 (mixed N,N′-diacetyl-N,N′-dibenzoyl derivative).
For the known compounds [17] synthetic conditions were improved, in fact we were able to reduce considerably the reaction time and obtain pure products in high yields. Starting from 1a, the corresponding N,N′-diacetylated or N,N′-dibenzoylated derivatives were obtained following two different synthetic pathways. Firstly, a strong excess of acetylating and benzoylating agents was used to obtain the tetraderivatives (Scheme 4, path b) in agreement with the literature [17], but operating at the boiling temperature of the solvent. In this way the time of the reaction reduces from two days to less than 1 h. The tetra-substituted compounds have then been partially hydrolyzed [17] (Scheme 4, path c) to obtain the corresponding N,N′-diacetamides.
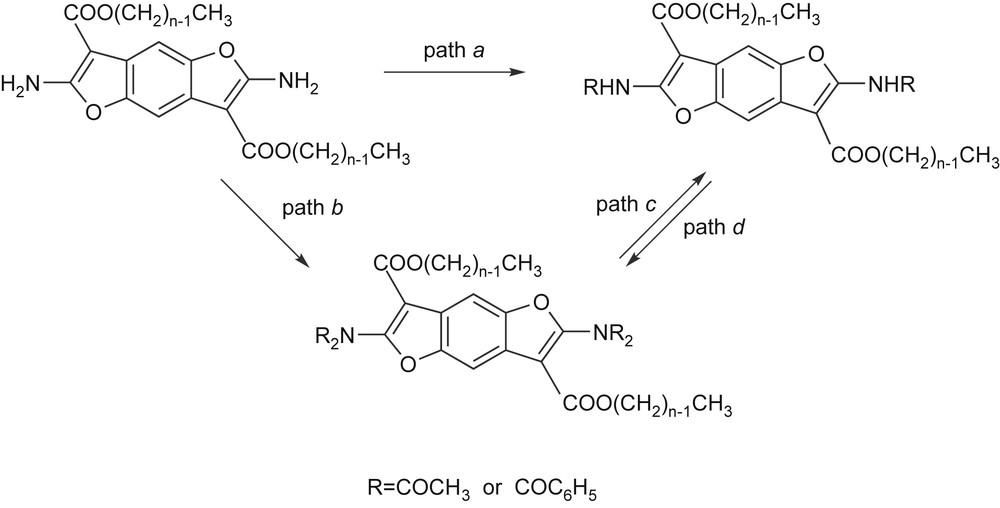
In this way, 4a was obtained with high purity and good yield, while the yield of 5a was unsatisfactory. In order to obtain a higher yield of 5a, a different synthetic approach was used (Scheme 4, path a). A stoichiometric amount of benzoylating agent was used in pyridine solution, operating at gentle boiling for 1 h. These conditions guarantee the isolation of pure N,N′-dibenzoylated products in good yields. In this case the reactivity of –NH2 groups seems comparable to that of aniline. The reaction following path b in Scheme 3 is also known for aniline derivatives [19], but it is more easy and complete in the case of the benzodifurane derivatives described here.
When the synthesis of N,N′-dibenzoylated derivatives was carried out according to the literature conditions [17] (Scheme 4, paths b + c), a considerable amount of 2,8-diphenyl-4,10-dihydro[1,3]oxazino[5‴,4‴,4″,5″]furo[3″,2″:4′,5′]benzo[4,5]furo[2,3-d][1,3]oxazine-4,10-dione (7, Fig. 1) was obtained unexpectedly.
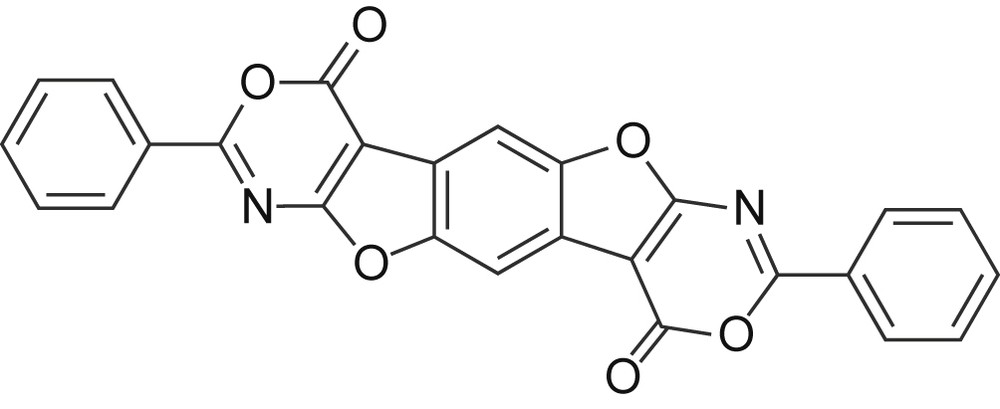
Oxazine derivative 7.
This compound was fully characterized and the UV–vis absorption–emission spectrum is reported in Fig. 2.
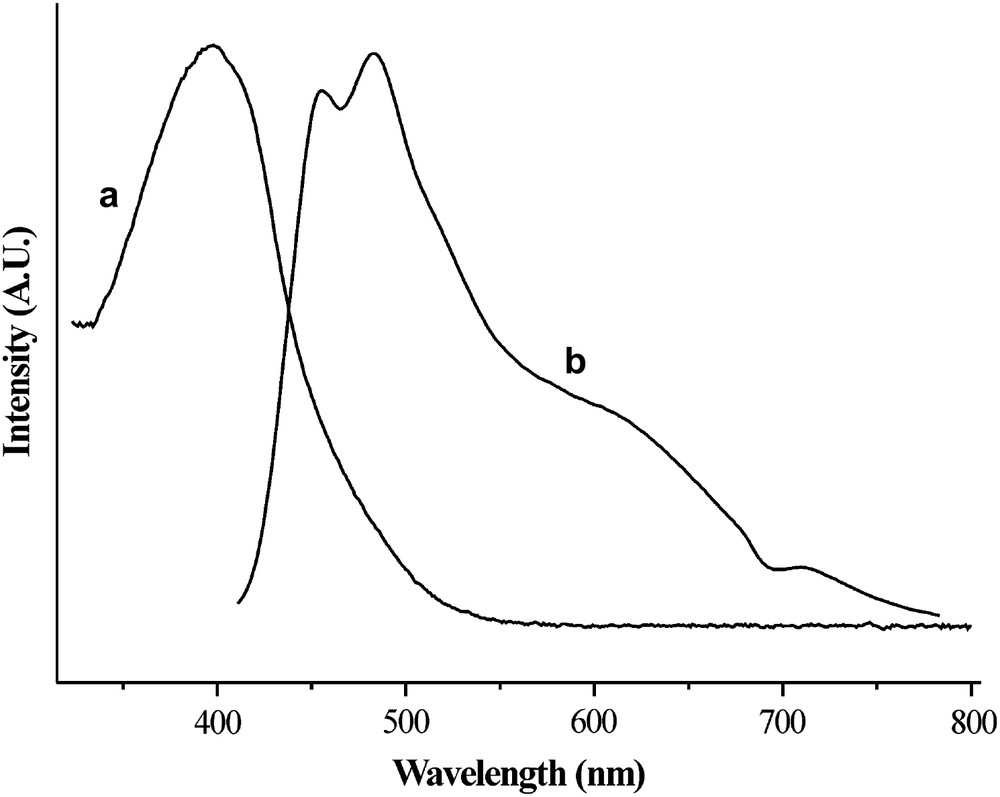
UV–vis absorbance (a) and emission (b) spectra of 7 in NMP solution (λexcit = 400 nm).
Compound 7 results from the cyclization reaction involving the alkylester and the benzoamide groups contiguous to the furane ring, proceeding with the elimination of two alcohol molecules. This kind of cyclization reaction is known [20,21] but, while benzoxazinone derivatives are frequent in the literature, only one case of furo–oxazinone has been reported up to our knowledge [22]. It is worth noting that in the field of heterocyclic compound chemistry the oxazino derivatives are frequently quoted as intermediates in mechanistic studies.
Of remarkable interest was the thermal stability of oxazinone, which can be sublimated at 340 °C, without decomposition. This feature makes this highly heterocondensed and extensively conjugated compound very interesting for the potential electric properties.
Compound 1a and its derivatives are poorly soluble and low handling materials. In fact, they can be dissolved in significant concentration only in hot DMF or pyridine. For this reason we tried to improve the solubility properties of 1a (m.p. 278 °C) and its analogous by adding long alkyl chains to the ester functions. As expected, the new derivatives 1b and 1c melt at temperatures lower than 1a (228 and 162 °C, respectively) without decomposition, and are fairly soluble at room temperature in common organic solvents like chloroform, acetone, THF or dioxane. These stable and soluble diamino benzodifuranes were further acylated on both aminic groups to afford new compounds that are shown in Scheme 3.
The reactivity of –NHR hydrogen atom was studied by performing further acetylation or benzoylation of the diamides. It was found that the N,N′-diacetamides undergo the reaction (Scheme 4, path d) by using a moderate excess of acylating agent. Moreover, it was also found that it is easy to effect a successive acetylation of the dibenzoylated compound 5b. In this way the mixed N,N′-diacetyl-N,N′-dibenzoyl 6 derivative was obtained, the mixed nature of which was unequivocally proved by single crystal X-ray structural analysis. In this way it was demonstrated that the N,N′-disubstituted amides can be employed as monomers in polycondensation reactions since the –NHR hydrogen atom can act like an amine hydrogen.
In Table 1, UV–vis data in N-methyl-2-pyrrolidone (NMP) solution (0.500 g/dL) are reported for all the synthesized compounds. It can be seen that, as expected, the UV–vis absorption pattern depends on the type and the number of N-substituents and not on the extension of alkyl chain.
UV–vis absorbance and fluorescence emission maxima in NMP solution (0.500 g/dL) for compounds 1a–5c, 6 and 7
| λa | ɛb | λc | |
| 1a | 326 | 26 900 | 360 |
| 1b | 326 | 26 200 | 369 |
| 1c | 325 | 23 200 | 366 |
| 2a | 323 | 25 000 | 480 |
| 2b | 323 | 27 600 | 474 |
| 2c | 323 | 29 600 | 475 |
| 3a | 305 | 24 600 | 411 |
| 3b | 304 | 24 300 | 415 |
| 3c | 304 | 25 400 | 410 |
| 4a | 324 | 34 400 | 377 |
| 4b | 324 | 31 100 | 378 |
| 4c | 324 | 32 300 | 378 |
| 5a | 393 | 41 200 | 491 |
| 5b | 392 | 42 500 | 497 |
| 5c | 392 | 44 200 | 492 |
| 6 | 394 | 20 800 | 488 |
| 7 | 400 | 26 900 | 457, 485 |
a Wavelength of UV–vis absorbance maximum (excitation wavelength for the fluorescence emission spectrum in nm).
b Molar UV–vis absorbance coefficient (L mol−1 cm−1).
c Wavelength of fluorescence emission maximum (nm).
Fluorescence emission was also observed and some spectra are reported as examples in Fig. 3.
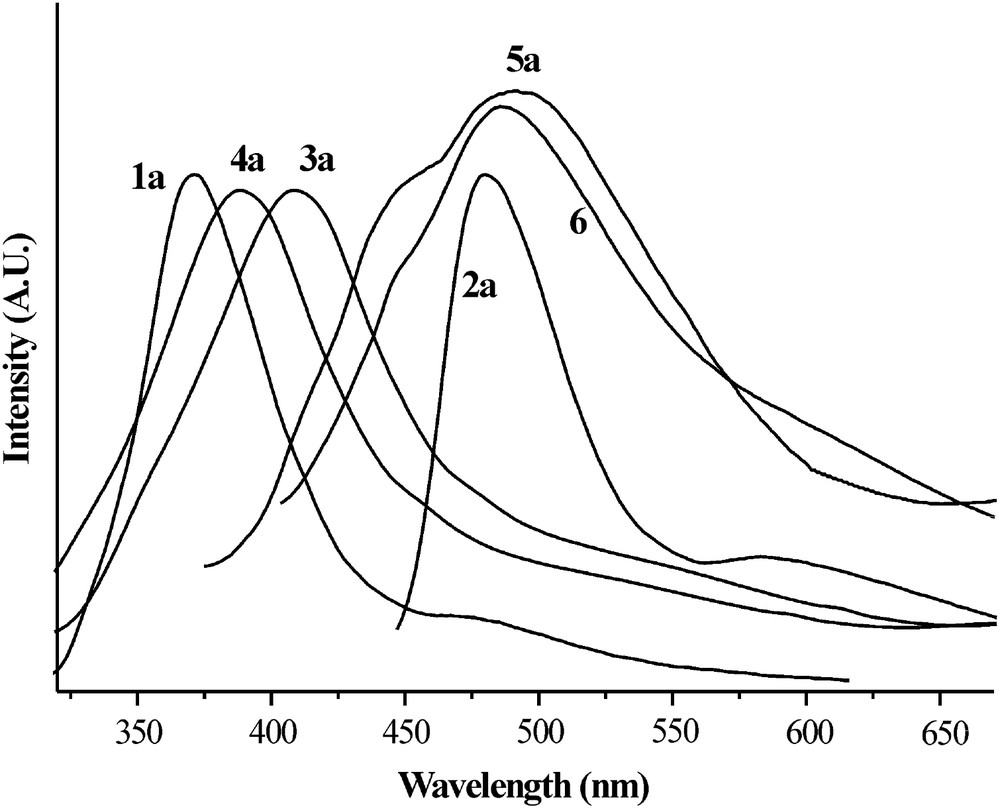
Fluorescence emission spectra of 1a (λexcit = 326 nm), 2a (λexcit = 323 nm), 3a (λexcit = 305 nm), 4a (λexcit = 324 nm), 5a (λexcit = 393 nm) and 6 (λexcit = 394 nm) in NMP solution.
The free amine and the acetylated derivatives emit blue light when hit by UV radiation while the benzoylated compounds and 6 give a green/yellow emission.
The high reactivity of the –NH2 and the –NHCOCH3 groups towards acylic agents that were previously experienced allows the easy preparation of polymers P1 and P2 (Scheme 5) by the reaction of 1b and 4b monomers with sebacoyl chloride.
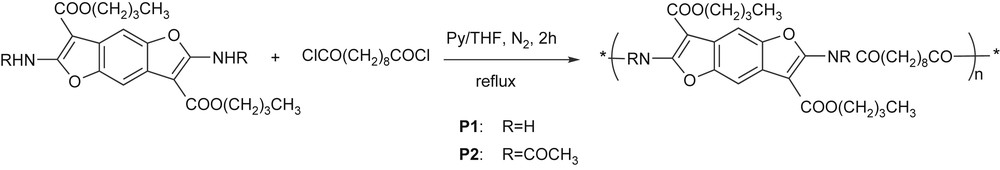
A stoichiometric amount of acylic dichloride dissolved in THF was simply added, at room temperature, to the pyridine solution of 1b or 4b and the mixture was gently refluxed for 2 h. Standard work-up allowed to us recover P1 and P2 as a gray and a brown solid, respectively.
For P1, the 1H NMR spectrum (in DMF-d6) shows the broad signals expected for a polymeric material. In particular, the isolated resonance at 10.50 ppm relative to the amidic hydrogen atoms, as found for the model compound 4b, and another isolated signal at 2.73 ppm due to the CH2 protons nearest to the amidic group (in the expected 1:2 integration ratio with respect to the –NHCO signal) can be observed. The inherent viscosity is 0.26 dL/g (measured at 80 °C, 0.500 g/dL concentration in NMP solution). The DSC analysis pattern shows two endothermic broad signals (at 148 and 212 °C), and thermal decomposition begins just after the second peak. X-ray diffraction pattern recorded at room temperature on a sample extruded from the melt shows the semicrystalline nature of the sample.
For P2, the resonance related to the two aromatic hydrogen protons (8.22 ppm) in the 1H NMR spectrum (in CDCl3) is broad and shifted with respect to the monomer 4b (7.83 ppm), due to the de-shielding effect of the additional acylic group on N atom. In the same way as for P1, the signal at 2.72 ppm due to CH2 protons nearest to the amidic group in the expected 1:1 integration ratio with respect to the aromatic signal is detected. The inherent viscosity is 0.18 dL/g (measured at 25 °C, 0.500 g/dL concentration in CHC3). The DSC pattern shows only the glass transition temperature signal (Tg = 40 °C), thus suggesting an amorphous nature of the product. Thermal stability is maintained up to 250 °C.
It was also observed that polymer P2 is much more soluble than P1 in common organic solvents, possibly due to the absence of hydrogen bond interactions. Transparent and homogeneous films of P2 can be very easily obtained by casting or from the melt.
Polymers P1 and P2 were also characterized by UV–vis spectrum measurement (P1: λmax = 325 nm, ɛ = 21 000 L mol−1 cm−1; P2: λmax = 309 nm, ɛ = 25 000 L mol−1 cm−1). It was noticed that when the polymer solutions were hit by UV radiation (at 325 and 309 nm, for P1 and P2, respectively), a faint fluorescent blue emission (λemiss = 380 and 412 nm, for P1 and P2, respectively) is produced, with a broad maximum comparable with their model compounds 4b and 3b. As an example, the fluorescence spectra in NMP solution of P2, its monomer 4b and its model compound 3b are reported in Fig. 4. A close similarity between the spectra of polymer P2 and its model compound 3b can be noted.
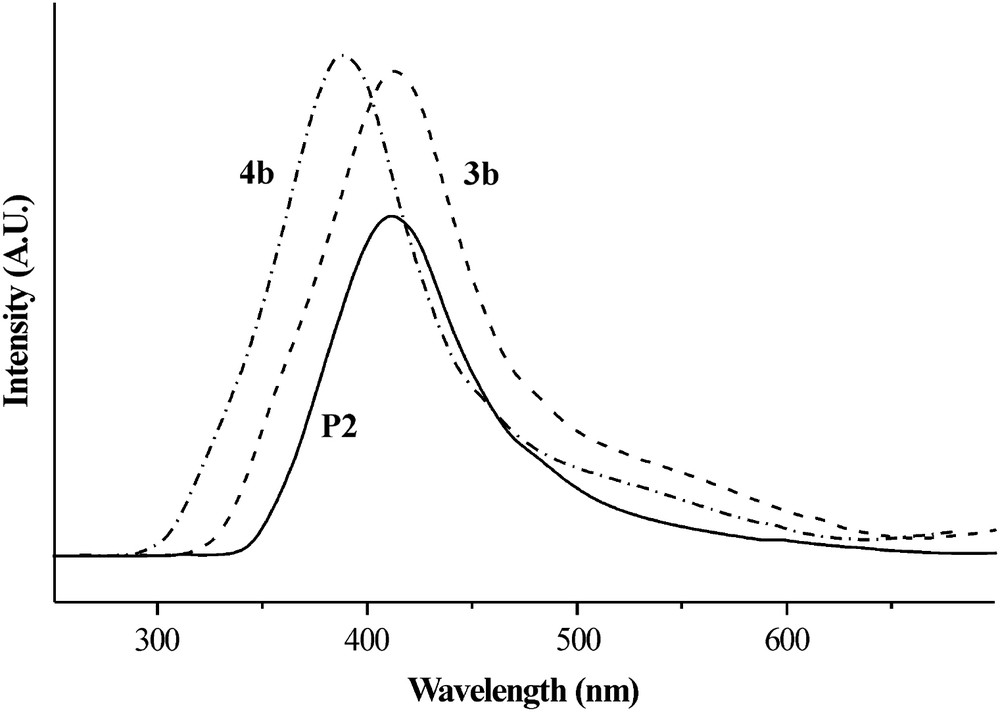
Fluorescence emission spectra of P2 (λexcit = 309 nm), 4b (λexcit = 324 nm) and 3b (λexcit = 304 nm) in NMP solution.
Broad fluorescence emission bands were also observed on thin (about 400 nm) films obtained by casting from NMP (P1) or chloroform (P2) solutions. This emission covers a range of about 180 nm (from 300 to 480 nm), which may account for the observed whitish-pale blue emission of the films.
2.2 X-ray crystal structure of 1a, 3a and 6
The molecular structures of 1a, 3a and 6 are shown in Figs. 5–7. Selected bond lengths and angles are reported in Table 2.

ORTEP view of one independent 1a molecule. Thermal ellipsoids are shown at 50% probability level.
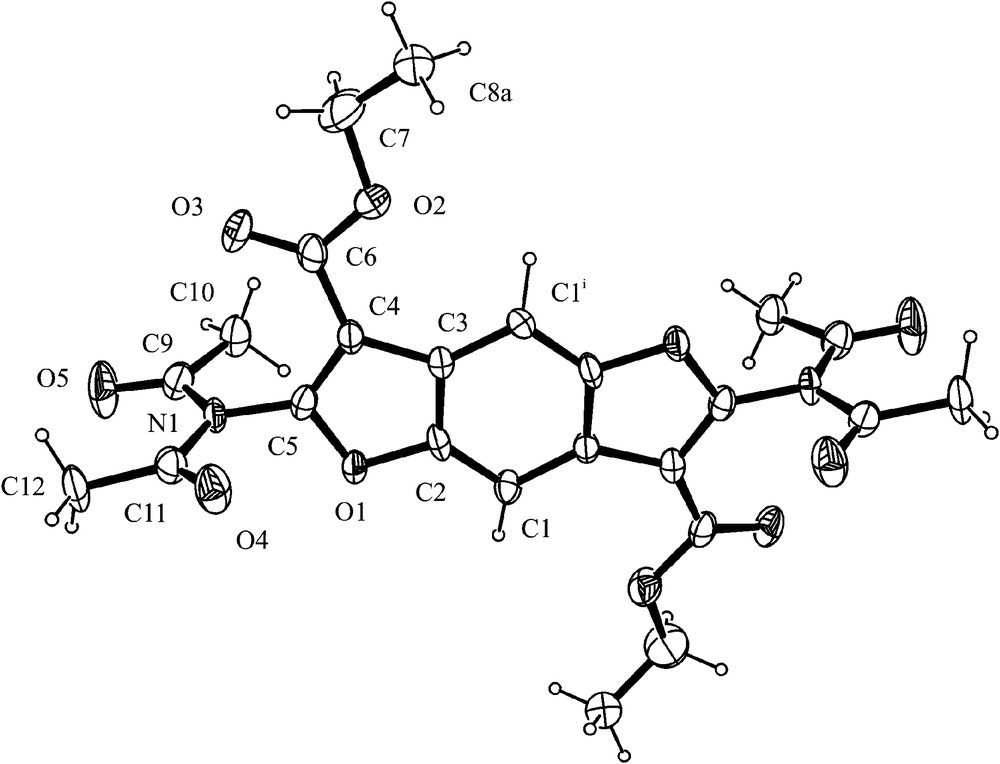
ORTEP view of 3a. Thermal ellipsoids are shown at 50% probability level, only one of the two statistical terminal methyls of ester group is reported for clarity. Symmetry operation: i = −y + 1, −x + 1, 1/2 − z.
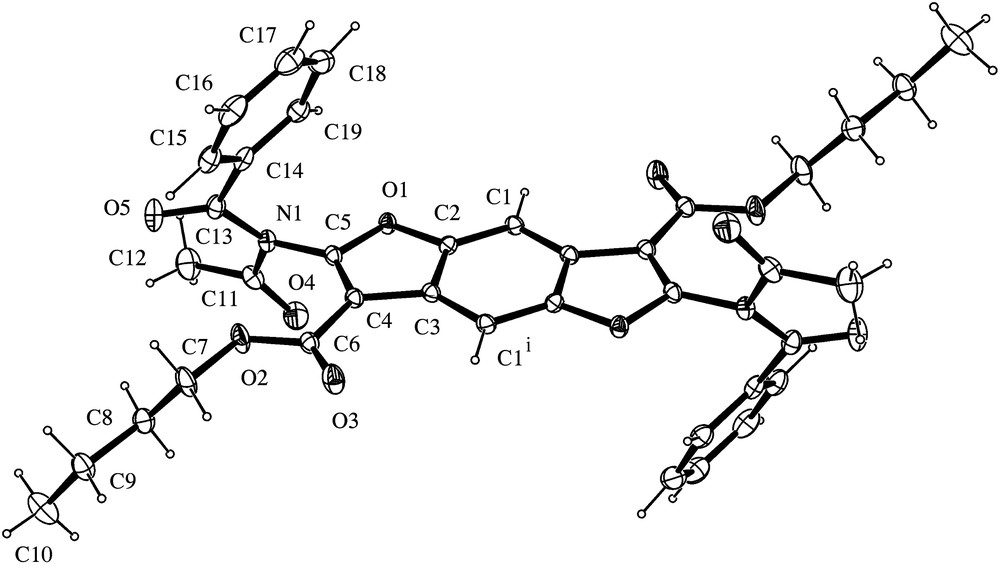
ORTEP view of 6. Thermal ellipsoids are shown at 30% probability level. Symmetry operation i = −x, −y, −z.
Selected bond lengths (Å) and angles (°) for 1a, 3a and 6 with esd's in parentheses
| 1a | |||
| N–C(ring) | 1.330(3)–1.342(2) | ||
| N–C(ring)–C(ring) | 132.2(2)–133.4(3) | ||
| N–C(ring)–O(ring) | 115.0(2)–115.6(2) | ||
| 3a | |||
| N1–C5 | 1.406(4) | C2–O1 | 1.391(4) |
| N1–C9 | 1.421(5) | C5–O1 | 1.373(4) |
| N1–C11 | 1.425(5) | ||
| N1–C5–C4 | 132.8(3) | N1–C5–O1 | 114.4(3) |
| C5–N1–C9 | 118.8(3) | C9–N1–C11 | 126.5(3) |
| C5–N1–C11 | 114.7(3) | ||
| C5–N1–C9–O5 | −178.7(4) | C5–C4–C6–O2 | 172.5(4) |
| C5–N1–C11–O4 | 2.9(5) | C4–C5–N1–C9 | 86.2(6) |
| 6 | |||
| N1–C5 | 1.408(2) | C2–O1 | 1.364(2) |
| N1–C11 | 1.424(2) | C5–O1 | 1.385(2) |
| N1–C13 | 1.426(2) | ||
| N1–C5–C4 | 131.8(1) | N1–C5–O1 | 115.3(1) |
| C5–N1–C11 | 114.2(1) | C11–N1–C13 | 125.2(1) |
| C5–N1–C13 | 118.3(1) | ||
| C4–C5–N1–C11 | −100.6(1) | C5–C4–C6–O2 | 3.3(2) |
| C4–C6–O2–C7 | 178.0(1) | C6–O2–C7–C8 | −129.5(1) |
| C5–N1–C13–O5 | −152.2(1) | O1–C5–N1–C11 | 78.1(2) |
| C5–N1–C11–O4 | 0.0(2) | N1–C13–C14–C15 | −138.6(1) |
In 1a the crystallographic independent unit contains one molecule and two halves. One molecule exhibits a local non-crystallographic inversion centre, the other two sit on crystallographic inversion centres. They differ from each other only to a minimum extent so that only one molecule is discussed and is shown in Fig. 5.
The geometry around N atom is substantially planar (sum of the bond angles around N is in the range 350.2(3)°–356.3(3)°) and the molecule is characterized by an overall planarity. The substantial coplanarity of –NH2 group with the benzodifurane and the value of N–C bond length (mean value is 1.333(3) Å) suggest a high degree of conjugation of N atom with the ring system. In the crystal packing a complex N–H⋯O intermolecular hydrogen bonding pattern affords a three-dimensional network of parallel molecules, thus indicating an easy rationale for the observed high melting point and the poor solubility.
In 3a (Fig. 6), the molecule is located on a crystallographic binary axis and a substantial coplanarity of the aromatic core with the –COOC2H5 groups is observed. A clear planar geometry is observed at N atom (sum of angles around N is 359.5(3)°). The –N(COCH3)2 group is all planar and shows the usual syn–anti conformation of carbonyls [19b,23]. Its mean plane is almost perpendicular to the benzodifurane plane (angle is 87.75(8)°) and a lengthening of the amidic C–N bond distance (1.406(4) Å) is observed with respect to 1a. These observations are in keeping with a loss of conjugation of the N atom with the benzodifurane ring system in the tetra-substituted compounds.
In 6 (Fig. 7), the molecule sits on a crystallographic inversion centre and the aromatic core is coplanar with the carbonyl group of the ester moiety. The alkyl chain is in all-trans conformation but, at variance with 1a and 3a, it is not coplanar with the aromatic core as a consequence of the value of C6–O2–C7–C8 torsion angle (129.5(1)°). The geometry at N atom is planar (the sum of bond angles around N atom is 357.7(1) Å) and a lengthening of the amidic C–N bond distance (1.408(2) Å) is also observed in analogy with 3a. N-Acetyl-N-benzoyl-amido group is planar (except for a torsion of phenyl group around C13–C14 bond) and is almost perpendicular to benzodifurane plane (torsion angle O1–C5–N1–C1178.1(2)°). Carbonyl groups adopt a syn–anti conformation (syn- for acetyl and anti- for benzoyl).
3 Conclusions
A series of diamino benzodifurane type compounds and their N,N′-di- and -tetraacetylated or -benzoylated compounds have been prepared following two different synthetic paths in high yields. Solubility in common organic solvents was improved by the introduction of long chains into the ester groups. Single crystal X-ray analysis was employed to confirm the molecular structure of the three derivatives, i.e. the unsubstituted 1a, the tetra-substituted 3a and the mixed 6. The results give unequivocal information on the extent of electronic conjugation. Moreover, by performing the benzoylation reaction according to the literature conditions, the remarkable cyclized product 7 was isolated and characterized.
Finally, the feasibility of the polymerization reaction between the –NH2 or –NHR groups and a carboxylic dichloride to give linear polyamides (P1 and P2) was demonstrated. In particular P2, which contains a disubstituted amidic function can be considered a promising material for application in the field of organic electronics, as it is amorphous, soluble and stable up to high temperature.
4 Experimental
4.1 General
All solvents and reagents were of AnalR grade and used without further purification, as provided by Sigma–Aldrich, Lancaster Across or Carlo Erba. The characterization of the final compounds and the intermediates was performed by 1H NMR and 13C NMR using Gemini Spectrometers operating at 200 MHz. Optical observations were performed by using a Zeiss Axioscop polarizing microscope equipped with FP90 Mettler temperature heating stage. Phase transition temperatures and enthalpies were measured using a DSC scanning calorimeter from Perkin Elmer Pyris with a scanning rate of 10 °C/min and under a nitrogen flow. UV–vis absorption spectra were recorded at room temperature by use of a Jasco V-560 spectrophotometer (range 200–800 nm, scanning rate 120 nm/min). Fluorescence spectra were recorded at room temperature by a Jasco FP 750 apparatus (bandwidth at 5 nm, scan rate at 125 nm/min, integration time of 0.5 s). X-ray spectra were obtained on a BAS-MS imaging plate (FUJIFILM) with a cylindrical camera (radius 57.3 mm, monochromatized Cu Kα radiation) and processed with a digital scanner (FUJI-BAS 1800). Inherent viscosities of NMP solution (0.500 g/dL) for P1 and CHCl3 solution (0.500 g/dL) for P2 were measured using an Ubbelohde viscosimeter.
4.2 Synthesis of 1a–1c
The compounds were obtained starting from the suitable alkyl-cyanoacetate by using the experimental conditions detailed for compound 1a.
4.2.1 2,6-Diamino-benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, diethyl ester [17]: 1a
To a solution of 1,4-benzoquinone (2.0 g, 18.5 mmol) in absolute ethanol (40 mL), ethyl cyanoacetate (3.0 mL, 26.5 mmol) was added with stirring at room temperature. The addition of 30% w/w aqueous solution of ammonia (7.0 mL) caused fast exothermic reaction with temperature rising up to 40 °C and precipitation of a gray solid. After 30 min of stirring the solid was recovered by filtration and washed three times with 20 mL portions of ethanol. After drying in vacuo 1.35 g (44% yield) of product, sufficiently pure for further reactions, was obtained. A more pure product (light gray) displaying melting point at 278 °C with decomposition, in good agreement with the literature [17], was obtained by recrystallization from DMF/water (9:1). Single crystals suitable for X-ray analysis were obtained by slow evaporation of an acetic acid solution of 1a.
C16H16N2O6 (332.310): calcd: C, 57.83; H, 4.85; N, 8.43; found: C, 57.80; H, 4.88; N, 8.47.
1H NMR (200 MHz, DMSO-d6, 25 °C): δ = 7.61 (s, 4H), 7.32 (s, 2H), 4.24 (q, J = 6.7 Hz, 4H), 1.32 (t, J = 6.7 Hz, 6H) ppm. 13C NMR (200 MHz, DMSO-d6, 25 °C): δ = 195.0, 158.3, 153.0, 126.6, 123.2, 123.1, 34.9, 8.2 ppm.
4.2.2 2,6-Diamino-benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, dibutyl ester: 1b
Compound 1b was recrystallized from ethanol/water or dioxane/water obtaining large greenish needles. Yield: 38%. A solid–solid transition is observed at 140 °C (ΔH = 10 J/g) with melting point at 228 °C (ΔH = 108 J/g). C20H24N2O6 (388.417): calcd: C, 61.85; H, 6.23; N, 7.21; found: C, 61.72; H, 6.35; N, 7.24.
1H NMR (200 MHz, DMSO-d6, 25 °C): δ = 7.61 (s, 4H), 7.29 (s, 2H), 4.20 (t, J = 6.7 Hz, 4H), 1.65 (m, 4H), 1.41 (m, 4H), 0.94 (t, J = 6.7 Hz, 6H) ppm. 13C NMR (200 MHz, DMSO-d6, 25 °C): δ = 195.0, 158.3, 153.0, 126.6, 123.2, 123.1, 37.9, 26.1, 22.3, 13.8 ppm.
4.2.3 2,6-Diamino-benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, dioctyl ester: 1c
Compound 1c was recrystallized from ethanol/water obtaining gray flat crystals. Yield: 33%. Two solid–solid transitions are observed at 139 °C (ΔH = 32 J/g) and 162 °C (ΔH = 19 J/g) with melting point at 170 °C (ΔH = 57 J/g). C28H40N2O6 (500.631): calcd: C, 67.18; H, 8.05; N, 5.60; found: C, 67.05; H, 7.98; N, 5.60.
1H NMR (200 MHz, DMSO-d6, 25 °C): δ = 7.61 (s, 4H), 7.29 (s, 2H), 4.19 (t, J = 6.6 Hz, 4H), 1.70 (m, 4H), 1.41 (m, 4H), 1.26 (m, 16H), 0.81 (t, J = 6.6 Hz, 6H) ppm. 13C NMR (200 MHz, DMSO-d6, 25 °C): δ = 195.0, 158.3, 153.0, 126.6, 123.3, 123.2, 38.2, 29.3, 23.8, 22.8, 14.0 ppm.
4.3 Synthesis of 2a–2c and 3a–3c
The compounds were prepared following the same general procedure of 2a, reported in detail, by using benzoyl chloride (2-type compounds) or acetic anhydride (3-type compounds). They were recrystallized from chloroform/heptane (1:5).
All the compounds can also be obtained from the corresponding disubstituted derivatives, by acetylation or benzoylation with an excess (100%) of benzoyl chloride or acetic anhydride.
4.3.1 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(dibenzoylamino)-, diethyl ester [17]: 2a
To a solution of 1a (1.0 g, 3.11 mmol) in pyridine (30 mL), benzoyl chloride (8.5 mL, 73.23 mmol) was added with stirring at room temperature. The solution was gently boiled for 1 h, then cooled at room temperature and poured on crushed ice to afford precipitation of a solid. This was recovered by filtration, washed with water, then dissolved in chloroform (80 mL). The solution was dried with sodium sulphate and filtered. After the addition of heptane (80 mL), the solution was concentrated to a small volume affording a pale yellow product (yield: 46%). The warm suspension was filtered and the product recrystallized from CHCl3/heptane, by slow evaporation. A crystalline compound, melting at 263 °C (ΔH = 100 J/g), was obtained. C44H32N2O10 (748.740): calcd: C, 70.58; H, 4.31; N, 3.74; found: C, 70.55; H, 4.35; N, 3.69.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 8.00 (s, 2H), 7.84 (m, 8H), 7.40 (m, 12H), 4.34 (q, J = 7.0 Hz, 4H), 1.32 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 172.9, 159.4, 151.1, 140.3, 134.0, 132.7, 129.0, 127.5, 123, 118.7, 117.2, 64.0, 14.1 ppm.
4.3.2 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(dibenzoylamino)-, dibutyl ester: 2b
Yellow crystals; yield: 50%; melting point: 243 °C (ΔH = 59 J/g). C48H40N2O10 (804.847): calcd: C, 71.63; H, 5.01; N, 3.48; found: C, 71.60; H, 5.02; N, 3.42.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 7.97 (s, 2H), 7.84 (m, 8H), 7.40 (m, 12H), 4.29 (t, J = 6.4 Hz, 4H), 1.66 (m, 4H), 1.42 (m, 4H), 0.92 (t, J = 6.8 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 173.0, 159.0, 151.1, 140.3, 134.0, 132.1, 129.0, 127.2, 123.9, 118.7, 117.0, 64.0, 31.2, 19.5, 13.9 ppm.
4.3.3 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(dibenzoylamino)-, dioctyl ester: 2c
Yellow crystals; yield: 40%; melting point: 161 °C (ΔH = 73 J/g). C56H56N2O10 (917.062): calcd: C, 73.34; H, 6.15; N, 3.05; found: C, 73.29; H, 6.14; N, 3.02.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 8.00 (s, 2H), 7.90 (s, 8H), 7.56 (m, 12H), 4.45 (t, J = 6.8 Hz, 4H), 1.86 (m, 4H), 1.59 (m, 4H), 1.05 (t, J = 6.5 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 172.3, 159.0, 150.1, 140.2, 134.0, 132.2, 128.9, 127.2, 123, 118.7, 117.1, 65.0, 32.1, 29.5, 29.2, 26.1, 23.1, 14.0 ppm.
4.3.4 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(diacetylamino)-, diethyl ester [17]: 3a
Whitish crystals; yield: 55%; melting point: 241 °C (ΔH = 127 J/g), in agreement with literature [17]. Single crystals suitable for X-ray analysis were obtained by slow evaporation of a dioxane/water solution. C24H24N2O10 (500.457): calcd: C, 57.60; H, 4.83; N, 5.60; found: C, 57.52; H, 4.85; N, 5.62.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 8.21 (s, 2H), 4.41 (q, J = 7.1 Hz, 4H), 2.39 (s, 12H), 1.41 (t, J = 7.1 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 151.0, 150.0, 140.5, 124.3, 119.0, 116.0, 60.8, 48.7, 13.8 ppm.
4.3.5 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(diacetylamino)-, dibutyl ester: 3b
White crystals; yield: 58%; melting point: 171 °C (ΔH = 72 J/g). C28H32N2O10 (556.564): calcd: C, 60.43; H, 5.79; N, 5.03; found: C, 60.41; H, 5.80; N, 5.07.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 8.20 (s, 2H), 4.36 (t, J = 6.8 Hz, 4H), 2.38 (s, 12H), 1.76 (m, 4H), 1.48 (m, 4H), 0.99 (t, J = 6.8 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 151.0, 150.0, 140.5, 124.3, 119.0, 116.0, 60.8, 48.7, 31.0, 19.0, 14.0 ppm.
4.3.6 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-bis(diacetylamino)-, dioctyl ester: 3c
Whitish crystals; yield: 69%; melting point: 141 °C (ΔH = 69 J/g). C36H48N2O10 (668.779): calcd: C, 64.65; H, 7.23; N, 4.19; found: C, 64.57; H, 7.18; N, 4.17.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 8.20 (s, 2H), 4.35 (t, J = 6.8 Hz, 4H), 2.38 (s, 12H), 1.75 (m, 4H), 1.33 (m, 20H), 0.87 (t, J = 6.5 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 151.0, 150.0, 140.5, 124.3, 119.0, 116.0, 64.8, 48.7, 31.0, 29.4, 29.1, 25.0, 23.0, 14.0 ppm.
4.4 Synthesis of 4a–4c and 5a–5c
The compounds were prepared following two synthetic pathways: (i) by partial hydrolysis from the tetra-substituted compounds, as described in the literature [17] (Scheme 4, paths b + c); (ii) by employing benzoyl chloride (5-type compounds) or acetic anhydride (4-type compounds) in stoichiometric amount (Scheme 4, path a). The latter procedure allowed to obtain a more pure product for N,N′-dibenzoylated. N,N′-diacetylated derivatives were prepared by partial hydrolysis.
4.4.1 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-diacetamido-, dibutyl ester: 4b
This synthesis is described as an example of paths b + c synthetic way in Scheme 4.
To a solution of 1b (0.500 g, 1.29 mmol) in dry pyridine (15 mL), acetic anhydride (5.00 g, 48.9 mmol) was added under stirring at room temperature. The solution was gently boiled for 1 h, then water (20 mL) was added. The solution was boiled again for 30 min. From the warm solution, a compound in the form of thin needles, precipitated. This compound was recovered, washed with a mixture water/ethanol, and dried. Yield: 85%; melting point: 231 °C (ΔH = 115 J/g). C24H28N2O8 (472.491): calcd: C, 61.01; H, 5.97; N, 5.93; found: C, 61.04; H, 5.96; N, 5.96.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 10.0 (s, 2H), 7.83 (s, 2H), 4.38 (t, J = 6.3 Hz, 4H), 2.37 (s, 6H), 1.83 (m, 4H), 1.53 (m, 4H), 1.01 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 154.0, 150.0, 140.6, 124.2, 118.4, 116.0, 64.8, 48.7, 31.9, 19.4, 13.8 ppm.
4.4.2 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-diacetamido-, diethyl ester: 4a
It was obtained as whitish prismatic crystals, which melt with decomposition at 313 °C. Yield: 84%. C20H20N2O8 (416.383): calcd: C, 57.69; H, 4.84; N, 6.73; found: C, 57.60; H, 4.85; N, 6.80.
1H NMR (200 MHz, DMSO-d6, 25 °C): δ = 8.56 (s, 2H), 7.37(s, 2H), 4.34 (q, J = 6.8 Hz, 4H), 2.17 (s, 6H), 1.35 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 154.0, 150.0, 140.6, 124.2, 118.4, 116.0, 64.8, 48.6, 14.3 ppm.
4.4.3 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-diacetamido-, dioctyl ester: 4c
Whitish crystals; yield: 89%; melting point: 173 °C (ΔH = 106 J/g). C32H44N2O8 (584.705): calcd: C, 65.73; H, 7.58; N, 4.79; found: C, 65.65; H, 7.60; N, 4.72.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 10.00 (s, 2H), 7.85 (s, 2H), 4.37 (t, J = 6.8 Hz, 4H), 2.37 (s, 6H), 1.75 (m, 4H), 1.33 (m, 20H), 0.87 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 159.5, 154.2, 150.0, 140.6, 124.0, 118.4, 116.0, 64.8, 48.6, 31.9, 29.4, 29.0, 25.9, 22.7, 14.0 ppm.
4.4.4 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-dibenzamido-, diethyl ester [17]: 5a
This synthesis is described as an example of path a synthetic way in Scheme 4.
To a solution of 1a (0.500 g, 1.55 mmol) in pyridine (20 mL), benzoyl chloride (0.360 mL, 3.10 mmol) was added with stirring at room temperature. The solution was gently boiled under stirring for 1 h, then cooled at room temperature, thus prompting the precipitation of a gold yellow solid. This was recovered by filtration, recrystallized first from pyridine and then from chloroform/hexane (as for the other 5-type derivatives). Yield: 67%; melting point: 297–302 °C (ΔH = 40 J/g) in agreement with the literature, with a solid–solid transition at 293 °C (ΔH = 70 J/g). C30H24N2O8 (540.525): calcd: C, 66.66; H, 4.48; N, 5.18; found: C, 66.71; H, 4.46; N, 5.22.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 11.04 (s, 2H), 8.36 (m, 2H), 8.07 (m, 4H), 7.90 (s, 2H), 7.59 (m, 4H), 4.49 (q, J = 7.2 Hz, 4H), 2.59 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 164.9, 159.4, 150.3, 140.6, 134.2, 132.7, 129.0, 127.4, 124.1, 118.5, 116.0, 61.0, 14.1 ppm.
4.4.5 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-dibenzamido-, dibutyl ester: 5b
Yellow crystals; yield: 93%; melting point: 249 °C (ΔH = 16 J/g). C34H32N2O8 (596.632): calcd: C, 68.45; H, 5.41; N, 4.70; found: C, 68.44; H, 5.39; N, 4.65.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 11.0 (s, 2H), 8.32 (m, 2H), 8.00 (m, 4H), 7.90 (s, 2H), 7.56 (m, 4H), 4.45 (t, J = 6.4 Hz, 4H), 1.86 (m, 4H), 1.59 (m, 4H), 1.05 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 164.9, 159.4, 150.3, 140.6, 134.2, 132.7, 129.0, 127.4, 124.1, 118.5, 116.0, 64.6, 31.1, 19.1, 13.8 ppm.
4.4.6 Benzo[1,2-b:4,5-b′]difuran-3,7-dicarboxylic acid, 2,6-dibenzamido-, dioctyl ester: 5c
Yellow crystals; yield: 70%; melting point: 191 °C (ΔH = 29 J/g). C42H48N2O8 (708.847): calcd: C, 71.17; H, 6.83; N, 3.95; found: C, 71.09; H, 6.85; N, 3.88.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 11.0 (s, 2H), 8.33 (m, 2H), 8.05 (m, 4H), 7.90 (s, 2H), 7.56 (m, 4H), 4.44 (t, J = 6.8 Hz, 4H), 1.90 (m, 4H), 1.34 (m, 2H), 0.90 (t, J = 6.5 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 164.9, 159.4, 150.3, 140.6, 134.2, 132.7, 129.0, 127.4, 124.1, 118.5, 116.0, 64.8, 30.0, 25.9, 29.2, 22.7, 14.0 ppm.
4.5 2,8-Diphenyl-4,10-dihydro[1,3]oxazino[5‴,4‴,4″,5″]furo[3″,2″:4′,5′]benzo[4,5]furo[2,3-d][1,3]oxazine-4,10-dione: 7
The product 1b (0.500 g, 1.29 mmol) was dissolved in gently boiling dry pyridine (15 mL) with an excess of benzoyl chloride (3.3 mL, 28.4 mmol), then hydrolyzed by adding water (2 mL) and ethanol (2 mL), following paths b + c according to the literature [17].
The so-obtained boiling suspension was filtered. Two different products were separated. In the solution, 5b was recovered by further addition of water. The yellow solid, recovered by filtration, was washed twice with chloroform, then crystallized as bright yellow crystals (from boiling 1,1,1,2-tetrachloroethane). Yield: 24%. C26H12N2O6 (448.388): calcd: C, 69.65; H, 2.70; N, 6.25; found: C, 69.70; H, 2.64; N, 6.26.
1H NMR (200 MHz, TCE-d2, 25 °C): δ = 8.41 (d, 2H), 8.22 (s, 2H), 7.61 (m, 3H) ppm. 13C NMR (200 MHz, TCE-d2, 25 °C): δ = 156.1, 153.1, 152.1, 141.0, 131.0, 130.0, 129.2, 128.9, 124.5, 118.1, 118.4 ppm.
ESI-MS m/z: 449.41 (found), 449.41 (expected for [M + 1H]+); 920.33 (found), 919.79 (expected for [2 M + Na]+).
4.6 Synthesis of 6
To 5b (0.200 g, 0.48 mmol) dissolved in dry pyridine (12 mL), acetic anhydride (0.196 g, 1.92 mmol) was added under stirring at room temperature. The solution was gently boiled for 30 min, then stirred at room temperature for 2 h. The solution was poured into water to precipitate as a gray solid, which was washed with water and recrystallized from chloroform/heptane. Yield: 80%; melting point: 192 °C (ΔH = 72 J/g). C38H36N2O10 (680.706): calcd: C, 67.05; H, 5.33; N, 4.12; found: C, 67.14; H, 5.29; N, 4.18.
1H NMR (200 MHz, CDCl3, 25 °C): δ = 7.85 (s, 2H), 7.52 (d, 4H), 7.27 (m, 2H), 7.14 (m, 4H), 4.20 (t, J = 6.9 Hz, 4H), 2.50 (s, 6H), 1.63 (m, 4H), 1.31 (m, 4H), 0.86 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (200 MHz, CDCl3, 25 °C): δ = 172.5, 172.1, 159.4, 150.0, 140.6, 134.2, 132.2, 129.0, 127.4, 124.1, 118.5, 116.0, 64.6, 31.1, 19.1, 18.0, 13.8 ppm.
4.7 Synthesis of polymers P1 and P2
The polymer P1 was prepared by mixing under stirring a solution of 1b (0.500 g, 1.29 mmol) in dry pyridine (10 mL) with sebacoyl chloride (0.308 g, 1.29 mmol) previously dissolved in a minimum amount (2 mL) of dry THF. The mixture was gently refluxed for 2 h. The solid product was recovered by pouring the polymerization mixture into hot (about 80 °C) water. The solid was recovered by filtration and washed several times with hot water. The purification was effected by dissolving the polymer in N,N-dimethylformamide, precipitating it in ethanol and drying at 80 °C. Yield: 60%. P1: C30H38N2O8 (554.63): calcd: C, 64.97; H, 6.91; N, 5.05; found: C, 64.89; H, 6.93; N, 5.01.
The same synthetic procedure (using 4b) was adopted for P2. In this case the polymer was recovered by pouring on ice and washing with cold (5–10 °C) water. It was purified by dissolution in CHCl3 and precipitation in heptane, then dried at 80 °C under vacuum. Yield: 55%. P2: C34H42N2O10 (638.70): calcd: C, 63.94; H, 6.63; N, 4.39; found: C, 63.87; H, 6.60; N, 4.35.
The characterization of polymers was performed by elemental analysis, DSC and TGA analyses, 1H NMR, X-ray, UV–vis spectra and inherent viscosity measurements that were discussed above. 1H NMR spectrum is not reported in detail because the broad signals do not allow punctual assignments for all the resonances. Fluorescence spectra were registered in solution and in films (about 400 nm thickness).
4.8 X-ray analysis
Single crystals of 1a, 3a and 6 were obtained by slow evaporation of acetic acid, dioxane/water and chloroform/heptane solutions, respectively. Crystal data are reported in Table 3. Crystals were mounted at room temperature (1a) and under flowing N2 at 173 K (3a and 6) on a Bruker-Nonius KappaCCD diffractometer with Mo Kα (λ = 0.71073 Å) radiation and graphite monochromator (CCD rotation images, thick slices, φ scans + ω scans to fill asymmetric unit). Semiempirical absorption correction (SADABS) was applied. All structures were solved by direct methods (SIR97 package [24]) and anisotropically refined by the full matrix least-squares method on F2 against all independent measured reflections (SHELXL program of SHELX-97 package [25]). In all the structures hydrogen atoms were introduced in calculated positions except for the aromatic and amino hydrogen atoms that were located in difference Fourier maps. They were all refined as riding on their carrier atoms. In 3a two statistical positions for the terminal methyl of –COOC2H5 group were found and refined with an occupancy factor of 0.5.
Crystal data and structure refinement details for 1a, 3a and 6
| 1a | 3a | 6 | |
| Formula | C16H16N2O6 | C24H24N2O10 | C38H36N2O10 |
| Molecular weight | 332.31 | 500.45 | 680.69 |
| Temperature (K) | 296 | 173 | 173 |
| Crystal system | Monoclinic | Tetragonal | Monoclinic |
| Space group | P21/c | P41212 | P21/c |
| a (Å) | 9.198(1) | 9.683(1) | 13.174(3) |
| b (Å) | 30.267(6) | 9.683(1) | 8.891(2) |
| c (Å) | 14.047(2) | 24.696(6) | 17.246(1) |
| α (°) | 90 | 90 | 90 |
| β (°) | 127.66(1) | 90 | 122.01(2) |
| γ (°) | 90 | 90 | 90 |
| Volume (Å3) | 3095.9(9) | 2315.5(7) | 1712.9(7) |
| Z, Dcalcd (g cm−3) | 8, 1.426 | 4, 1.436 | 2, 1.320 |
| μ (mm−1) | 0.111 | 0.113 | 0.096 |
| θmax (°) | 27.5 | 25.0 | 27.5 |
| Reflections collected/unique | 14 992/6771 | 10 996/1248 | 12 653/3907 |
| R (int) | 0.0529 | 0.0907 | 0.0276 |
| Data/restraints/parameters | 6771/0/437 | 1248/0/166 | 3907/0/226 |
| R1, wR2 [I > 2σ(I)]a | 0.0657, 0.1476 | 0.0453, 0.1098 | 0.0398, 0.0941 |
| R1, wR2 [all data]a | 0.1522, 0.1815 | 0.0893, 0.1251 | 0.0628, 0.1050 |
| Largest diff. peak and hole (e Å−3) | 0.205, −0.220 | 0.196, −0.240 | 0.254, −0.214 |
a R1 = ∑‖Fo| − |Fc‖/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
All crystallographic data have been deposited with the Cambridge Crystallography Data Centre as supplementary publication nos. CCDC-629337 (1a), CCDC-629338 (3a) and CCDC-664339 (6). These data can be obtained free of charge via www.ccdc.cam.au.uk/data_request/cif.
Acknowledgments
The work was supported by Regione Campania, Italy. Thanks are also due to “Centro Regionale di Competenza – Nuove Tecnologie per le Attività Produttive” (CRdC – NTAP) of Regione Campania, and to “Centro Interdipartimentale di Metodologie Chimico Fisiche” (CIMCF) of the University of Naples “Federico II” for NMR and X-ray facilities.
1 Ogata et al. [13] have used the Craven procedure aiming to obtain a type 1 (Scheme 1) product, however, they remarked the absence of –CN signals in IR spectra.