

1 Introduction
As our fossil raw materials are irrevocably decreasing and as the pressure on our environment is building up, the progressive changeover of chemical industry to renewable feedstocks for their raw materials emerges as an inevitable necessity [1–4], i.e. it will have to proceed increasingly to the raw materials basis that prevailed before natural gas and oil outpaced all other sources.
Historically, the raw materials basis was substantially renewable, as depicted in Fig. 1, with the utilization of biomass and coal being in equal about 100 years ago [5]. In the 1920s, coal tar-based materials had taken the lead, reaching a maximum around 1930; thereafter, fossil gas and oil irresistibly took over, eliminating coal nearly completely and reducing renewable feedstocks to very modest levels.

Raw materials basis of chemical industry in historical perspective.
This over-reliance of chemical industry on fossil raw materials has its foreseeable limits as they are depleting and are irreplaceable, the only question being: when will affordable fossil fuels be exhausted? Or, stated more appropriately: when will fossil raw materials have become so expensive that biofeedstocks are an economically competitive alternative? Experts realistically predict the end of cheap oil for 2040 at the latest [6,7], a development that we can witness by now already, as chemical industry combats the rising costs of natural oil and gas [8]. Thus, taking the prognostication for the end of cheap oil [7], the curve for the utilization of biofeedstocks in Fig. 1 will have to rise such that it meets that of fossil raw materials somewhere around 2030–2040.
The transition to a more bio-based production system is pressing, yet hampered by a variety of obstacles: fossil raw materials are more economic at present, and the process technology for their into organic chemicals is exceedingly well developed and basically different from that required for transforming carbohydrates into products with industrial application profiles. This situation originates from the inherently different chemical structures of the two types of raw materials, as terrestrial biomass is considerably more complex, constituting a multifaceted array of low and high molecular weight products, exemplified by sugars, hydroxy and amino acids, lipids, and biopolymers such as cellulose, hemicelluloses, chitin, starch, lignin, and proteins. By far, the most important class of organic compounds in terms of volume produced are carbohydrates, as they represent roughly 75% of the annually renewable biomass of about 200 billion tons. Of these, only a minor fraction (ca. 4%) is used by man, the rest decays and recycles along natural pathways.
The bulk of the annually renewable carbohydrate biomass are polysaccharides, yet their non-food utilization is confined to textile, paper, and coating industries, either as such or in the form of simple esters and ethers. Organic commodity chemicals, however, are usually of low molecular weight, so they are more expediently obtained from low-molecular-weight carbohydrates than from polysaccharides. Accordingly, the constituent repeating units of these polysaccharides − glucose (cellulose, starch), fructose (inulin), xylose (xylan), etc., inexpensive and available on multi-ton scale − are the actual carbohydrate raw materials for basic organic chemicals.
Intense efforts within the last decade [9–17] to advance the use of inexpensive, large-scale-accessible mono- and di-saccharides as raw materials for chemical industry have so far not been able to basically bridge the conceptual, technological and economical gap between fossil hydrocarbons and renewable carbohydrates; so, this account reviews the present utilization of low-molecular-weight carbohydrates as feedstock for organic chemicals and − that being rather modest − accentuates existing methodologies that appear practical enough to be developed into industrially viable processes and products of presumed industrial relevance, i.e. bulk, intermediate, and fine chemicals, pharmaceuticals, agrochemicals, high-value-added speciality chemicals, or simply enantiopure building blocks for organic synthesis.
2 Sugars as biofeedstocks
The following overview concentrates on transformations of large-scale accessible mono- and disaccharides that either can be performed in one-pot procedures or in a few simple practicability-oriented steps, in which the carbon chain of the sugar is fully retained. As will become clear along the way, only a few of the products described have reached industrial status, yet this setting that is going to change with the rising costs for petrochemical raw materials.
2.1 Non-food valorisation of sucrose
2.1.1 Structure and conformation
Sucrose is a non-reducing disaccharide, because its component sugars, D-glucose and D-fructose, are glycosidically linked through their anomeric carbon atoms. Hence, it constitutes a β-D-fructofuranosyl α-D-glucopyranoside (Fig. 2). It is widely distributed throughout the plant kingdom, is the main carbohydrate reserve and energy source and an indispensable dietary material for humans. For centuries, sucrose has been the world’s most plentiful produced organic compound of low molecular mass, the present annual production from sugar-cane and sugar-beet being an impressive 130 × 106 t. Due to the usual overproduction, and the potential to be producible on an even higher scale if required, it is, together with starch-derived glucose, the major carbohydrate feedstock of low molecular weight from which to elaborate chemicals.
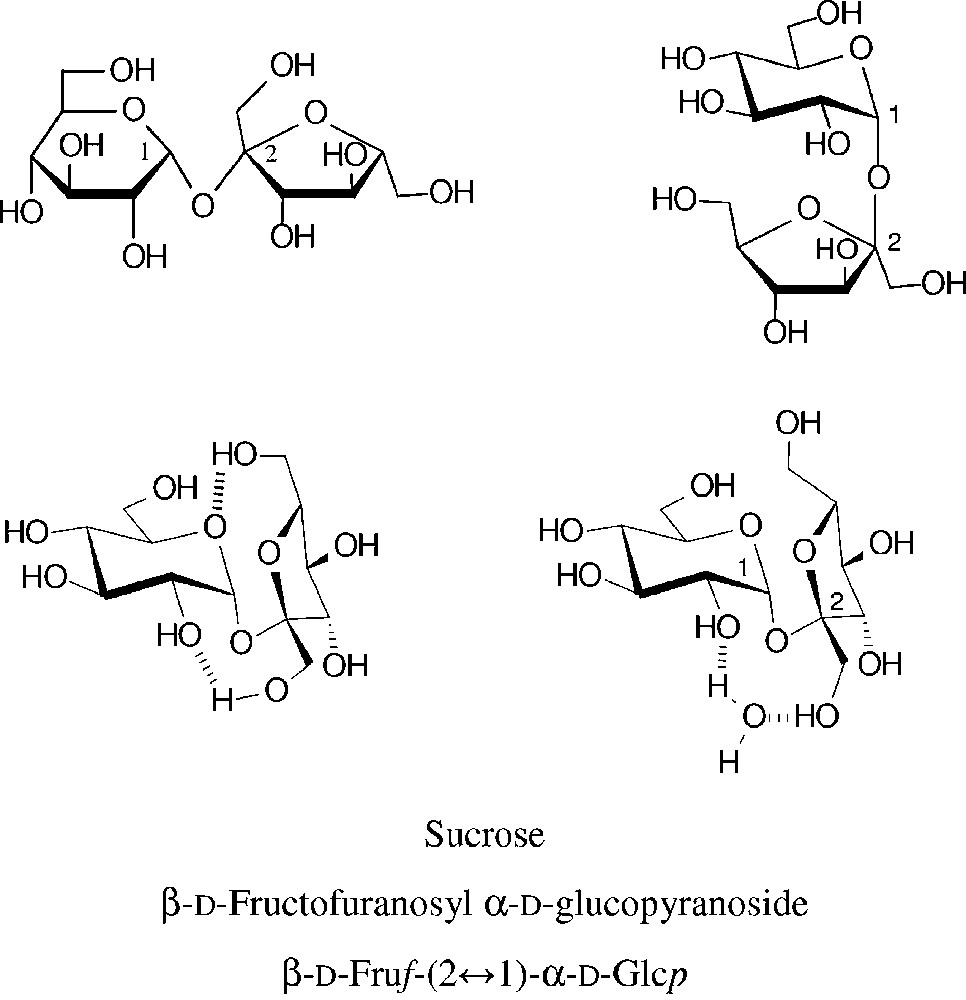
Common structural representations of sucrose (top entries). The molecular geometry realized in the crystal is characterized by two intramolecular hydrogen bonds between the glucose and fructose portion [18–20] (centre left). In aqueous solution, the two sugar units are similarly disposed towards each other, caused by insertion of a water molecule between the glucosyl-2-OH and fructosyl-1-OH [21,22], this ‘water-bridge’ being fixed by hydrogen bonding (centre right).
Due to its eight hydroxyl groups, chemical reactions of unprotected sucrose at a single hydroxyl group are difficult to achieve, i.e. getting useful regioselectivities is a basic issue [23–25]. The subtle reactivity differences between primary and secondary hydroxyl groups have been generalized such that the three primary ones are preferentially alkylated, acylated, oxidized and displaced by halogen in the order 6g-OH ≈ 6f-OH >> 1f-OH [23] − an over-generalization as this order of reactivity mainly covers comparatively bulky reagents which necessarily favour reaction at the 6g- and 6f-OH groups. (For ready differentiation of the oxygens in the fructose (primed numbers usually) and the glucose portions, they are denoted with ‘f’ and ‘g’ superscripts, respectively.) Molecular modelling of the electrostatic potential at the solvent accessible surface of sucrose clearly revealed the highest electropositivity being at the glucosyl-2-OH [20–22], entailing that it is the one most readily deprotonated. Accordingly, under basic conditions in ensuing reactions the 2g-OH is preferentially alkylated or acylated if the moiety to be attached to the oxygen is not too bulky (cf. § 2.1.2 and 2.1.3). In addition, the regioselectivity attainable is also depending on the nature of the electrophilic reagent, on the catalyst used for promoting the reaction and, not the least, on the solvent or solvent mixtures used. Sucrose is only soluble in DMF and DMSO aside water, and differently solvated in each one [10,22].
In the sequel, only those ‘entry reactions’ into sucrose derivatives are covered, which either are of industrial relevance or have the potential to attain significance as products with useful non-food application profiles.
2.1.2 Oxidation of sucrose
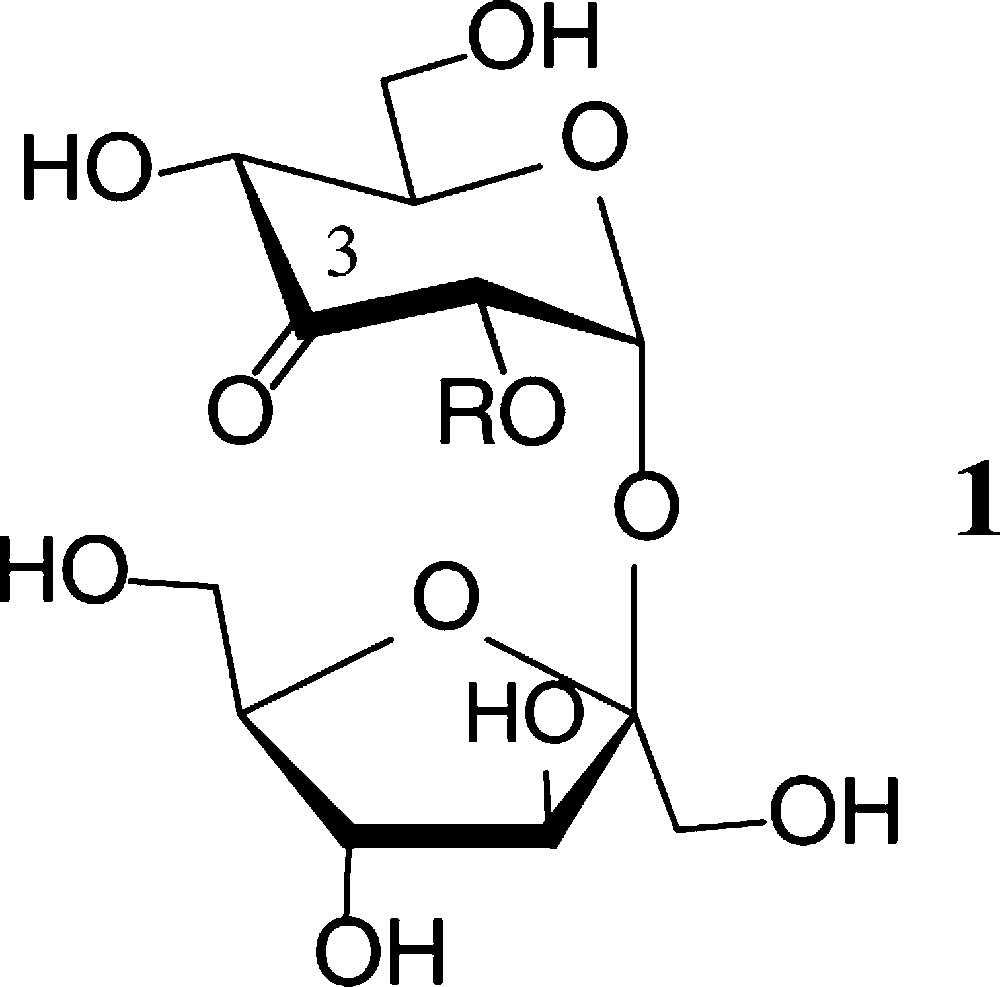
Prototype of an ‘entry reaction’ into regioselectively modified sucroses is the essentially regiospecific oxidation by Agrobacterium tumefaciens, whose dehydrogenase exclusively generates 3g-ketosucrose (1) [26]. This ready access opened the way to manifold modifications of 1 at the 3g-carbonyl function [27,28] (Scheme 1).
Chemical oxidation (Fig. 3) proceeds less uniformly, e.g. agitation of an aqueous solution of pH 6.5–7.0 at 35 °C with air in the presence of 0.5% Pt/C gave a 9:9:1 ratio of the 6g-, 6f- and 1f-saccharonic acids [29]. On further oxidation, particularly when using large amounts of the Pt catalyst and higher temperatures (80–100 °C), the preferred formation of the 6g,6f-dicarboxylic acid 2 has been observed [30], yet a preparatively useful procedure for its acquisition was developed only recently [31] through combining Pt/air-oxidation with continuous electrodialytic removal of 2, thereby protecting it from further oxidation. Otherwise, on letting the reaction proceed, the sucrose-tricarboxylate 3 is obtained [32]. An alternate useful oxidant to the tricarboxylate 3 is the NaOCl/ TEMPO system [33], particularly when applying high-frequency ultrasound, allowing yields in the 70% range [34].
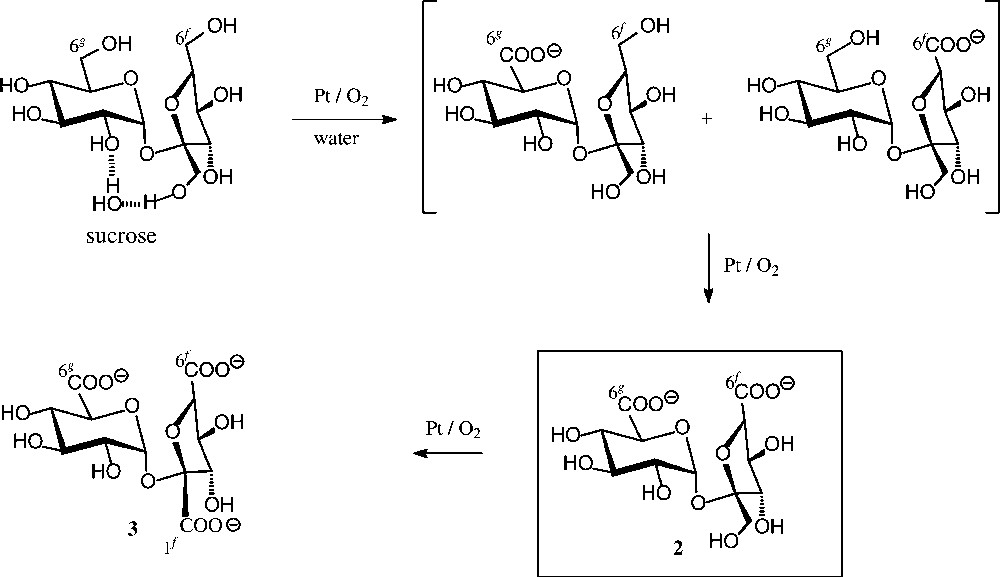
Catalytic oxidation of sucrose [29–32].
2.1.3 Sucrose esters
synthetically prepared [35] octa-fatty acid esters of sucrose have been approved by the US Food and Drug Administration in 1996 as a dietary fat substitute [36], and were marketed under the name Olestra® or Olean® [37] – a development contributing to further food utilization of sucrose.
Less highly esterified sucroses, usually mixtures of with high proportion of either mono-, di- or triesters of variable regioisomeric distribution over the 2g, 6g-, 6f- as well as other hydroxyls, are food and cosmetic emulsifiers and have favourable surfactant properties, their precise application profiles depending on the length of the fatty acid chains as well as on average degree of esterification. A concise overview on the preparation and the potential uses of sucrose esters is contained in this issue [25].
2.1.4 Sucrose ethers
Due to the persistence of an intramolecular hydrogen bond of the 2g-O...HO-1f type in aprotic solvents [38], the 2g-OH is the hydroxyl group most readily deprotonated, the resulting sucrose 2g-alkoxide then being preferentially alkylated at the glucosyl-2-oxygen. Thus, benzylation with NaH/benzylbromide in DMF is highly regioselective, resulting in an 11:2:1 mixture of 2g-O-benzyl-sucrose (5), its 1f-O- and 3f-O-isomers [38]. Being thus readily accessible, 5 proved to be a versatile intermediate for the generation of 2g-modified sucroses, e.g. the 2g-keto and 2g-deoxy derivatives as well as sucrosamine (2g-amino-2g-deoxy-sucrose) [38] (Scheme 2).
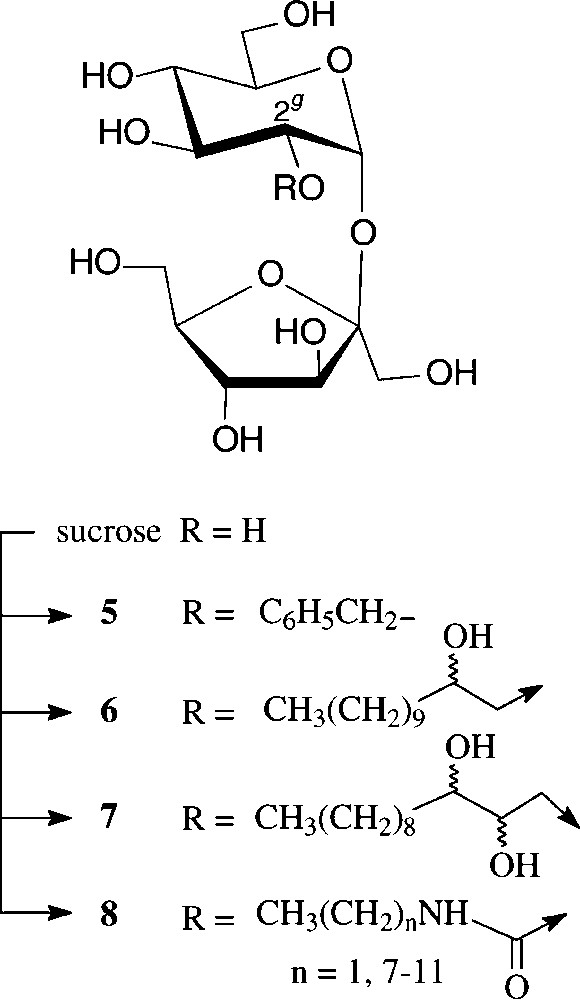
Of higher interest industrially are amphiphilic hydroxyalkyl ethers of type 6 and 7, preparable etherification of sucrose with long-chain epoxides such as 1,2-epoxydodecane [39,40] or 1,2-epoxydodecan-3-ol [41], performable as one-pot reactions in DMSO and the presence of a base (N-methylmorpholine or ground NaOH), the regioselectivities being in preference of the 2g-O- and 1f-O-positions. Unlike sucrose esters, its ethers are resistant to alkaline conditions, which considerably extends their potential application as non-ionic surfactants. They also have liquid crystal properties with mesophases depending on the point of attachment of the fatty chain to the sucrose [39].
2.1.5 Sucrose-derived urethanes
The non-reducing and polyhydroxy functionalities of sucrose have prompted various efforts to use it as a polyol for the manufacture of polyurethanes [42–44], replacing di- and polyols derived from fossil resources. Mostly though, sucrose is not directly used because it gives brittle foams on reaction with isocyanates, but is first etherified with propylene oxide to its poly(hydroxypropyl) derivative, which yields a strong polyurethane foam [43]. However, finely powdered sucrose may be used to the extent of 40% as a direct replacement of a polyol component, thereby improving mechanical properties and flame retardancy of the polyurethanes [45].
Whilst these sucrose-derived polyurethanes are industrially used, their structures, i.e. the extent with which sucrose is carbamoylated and the type(s) of crosslinking involved, are not well defined. More detailed investigations have shown that crosslinking can be avoided under certain conditions, e.g. when equimolar quantities of sucrose and aliphatic diisocyanates are subjected to polycondensation in dimethylacetamide, providing polyurethanes soluble not only in polar organic solvents, but also in water [46].
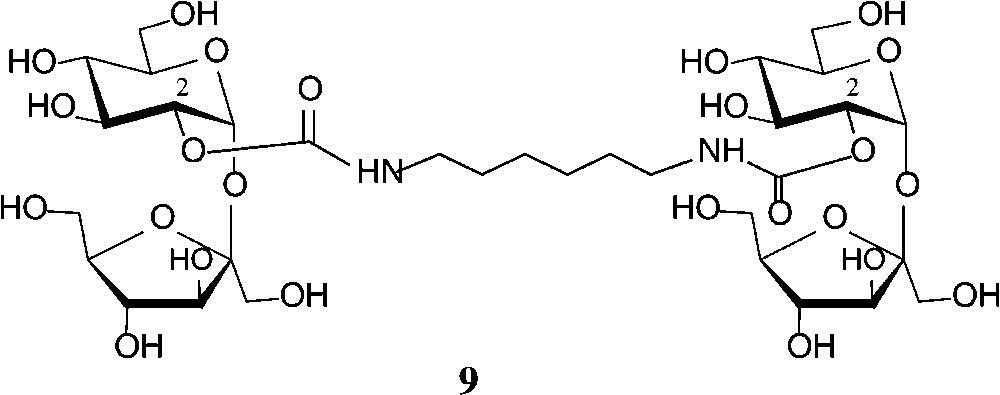
Some recent studies towards the generation of defined sucrose mono-carbamates [47-49] have again revealed the glucosyl-2-OH to be preferentially carbamoylated. Reaction of sucrose in anhydrous DMF with heptyl or undecyl isocyanate in the presence of triethylamine gave the respective 2g-O-(N-alkylcarbamoyl)-sucroses 8 (n = 7, 11) in 50% yield [47]. The analogous exposure of sucrose to ethyl isocyanate in DMF in the presence of Ca(OH)2 allowed the isolation of 72% of the 2g-O-carbamoylated product (8, n = 1), whereas use of Ba(OH)2, interestingly, afforded a 3:2 mixture of the 2g-O- and 1f-O-carbamates [48,49]. The more reactive phenyl isocyanate, however, under the same conditions, preferentially reacted with the 6g-OH followed by 2g-OH, giving a 3:2 mixture of the respective sucrose monocarbamates [49]. On application of these conditions (DMF, Ca(OH)2, −40 → +25 °C) to hexamethylene diisocyanate, the di-2-O-(N,N"-hexamethylene-1,6-di-carbamoyl)-sucrose 9 could be isolated in 31% yield [48,49] (Scheme 3).
2.1.6 Polymerisable sucrose derivatives
A large series of mono-O- and di-O-substituted sucrose derivatives – with polymerisable C=C double bonds in ester or ether moieties attached − have been prepared [50–54], usually as mixtures with average degrees of substitution: esters of acrylic or methacrylic acid, or vinylbenzyl ethers mostly. Their polymerisation as such, or in copolymerisation with styrene has led to a variety of interesting linear and crosslinked polymers with ‘sucrose anchors’ attached to the polymeric carbon chain(s) [50–53]. Despite favourable properties, as gels and chelating resins, these polymers are still only a chemical curiosity and their commercial potential has yet to be established (Fig. 4).
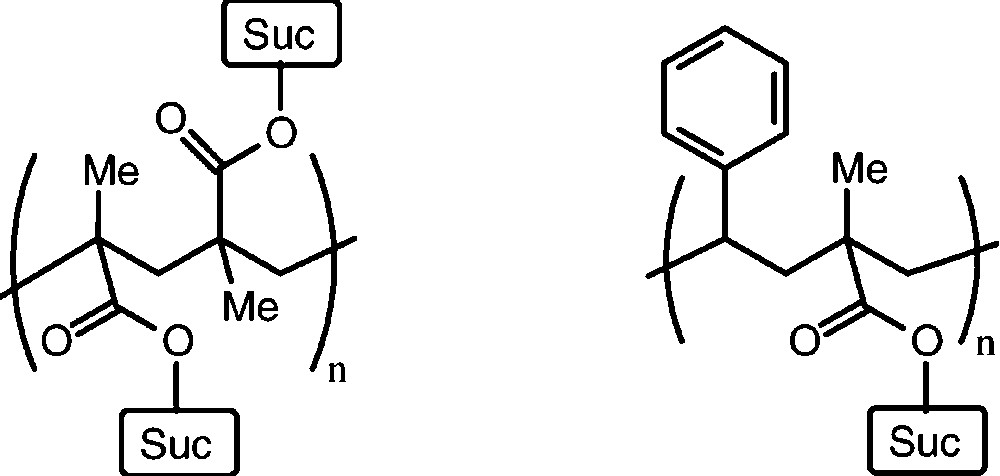
Idealized structure of a linear polymer resulting from radical polymerisation of a mono-O-methacroyl-sucrose (left) and a 1:1 copolymerization product with styrene.
2.1.7 Replacement of hydroxyl-groups in sucrose
The many studies towards the exchange of one or more of the sucrose hydroxyls by hydrogen, halogen or amino groups, or inversion of the configuration at any of the five secondary hydroxyls, have all been encumbered by regioselectivity problems when using unprotected sucrose; otherwise extensive blocking/deblocking procedures required to single out one free OH for then specific transformations [23,24,55,56]. Two ‘entry reactions’ though, both involving fairly regioselective chlorinations, may be mentioned in this context, despite the fact that the reagents required − triphenylphosphine/CCl4 and sulfuryl chloride − do not necessarily satisfy the basic principles of green/sustainable chemistry [57] (Scheme 4).
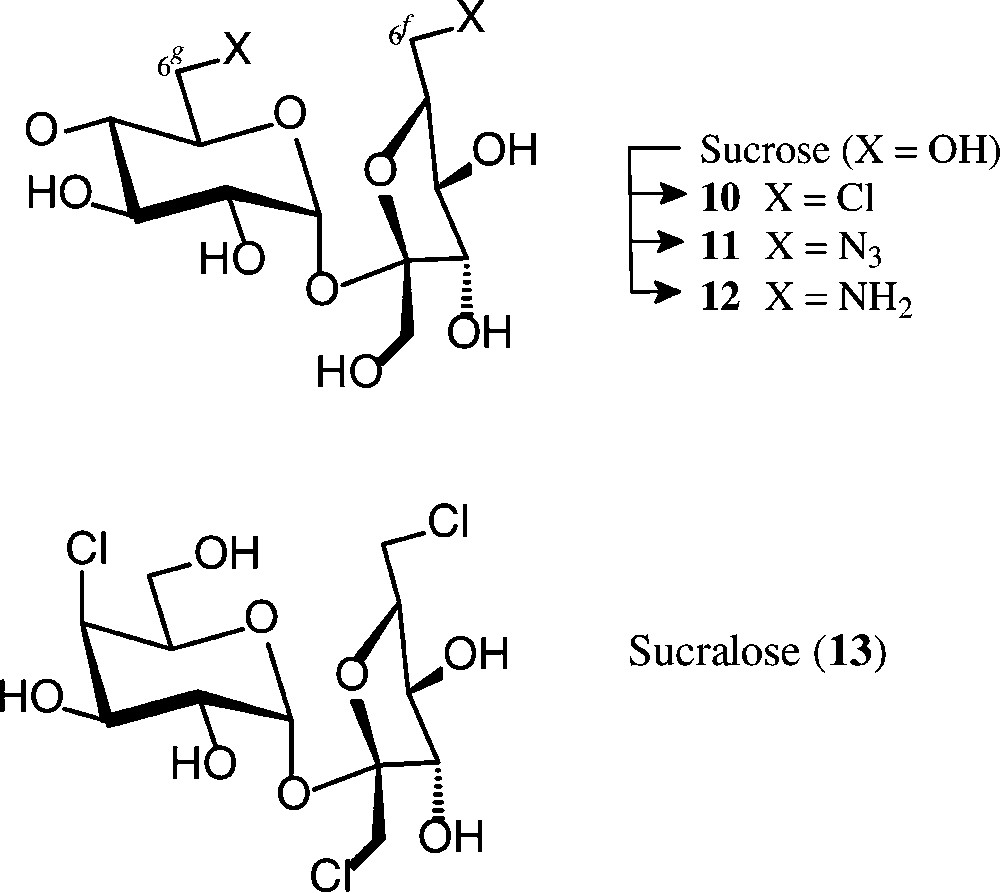
When exposing sucrose in pyridine solution to reaction with triphenylphosphine/carbon tetrachloride (3 h, 25 → 70 °C), about a 70% yield of the crystalline 6g,6f-dichlorosucrose 10 is obtained [58,59]. It can be further modified at the chlorinated positions, as, for example, to the diamine 12 via azidolysis (10 → 11) and hydrogenation [59,60]. Although this 6g,6f-diaminosucrose has potential interest as a diamine component towards hydrophilic polyamides, its unsustainable mode of preparation precludes its industrial exploitation.
Sucralose (13), a trichloro-trideoxy-galactosucrose, is 650 times sweeter than sucrose [61] and has been marketed as a non-caloric sweetener under the brand name Splenda® [62]. Its preparation involves treatment of a partially acetylated sucrose with sulfuryl chloride, whereby the initially formed chlorosulfate esters, as effective leaving groups, undergo nucleophilic displacement by chlorine, comprising inversion of configuration when esters at secondary hydroxyls are involved [63,64].
2.1.8 Conversion of sucrose to isomaltulose and ensuing products
Isomaltulose (14), a 6-O-α-D-glucosyl-D-fructose, is isomeric with sucrose, from which it is produced at an approximate 60 000-t/yr scale by Südzucker. The industrial process involves a glucosyl shift from the 2f-O of sucrose to the 6f-OH, effected by action of an immobilized Protaminobacter rubrum-derived α(1→6)-glucosyltransferase [65,66] − a transformation that most likely proceeds through a closed-shell intermediate, i.e. without full separation of the glucose and fructose portions [10]. The isomaltulose produced is subsequently hydrogenated to isomalt, an approximate 1:1 mixture of the terminally α-glucosylated glucitol and mannitol (15), which is on the market as a low-caloric sweetener [67].
Due to its the large scale availability, isomaltulose is also an attractive target for generating disaccharide intermediates of industrial utility. For example, hydrogenation of isomaltulose oxime over platinum [68] or Raney-nickel-promoted reductive amination with hydrazine [69] smoothly generates a 1:1-mixture of glucosyl-α(1→6)-2-amino-2-deoxy-D-glucitol and -mannitol, appropriately termed isomaltamine in analogy to isomalt. As a pronouncedly hydrophilic amine of a disaccharide alcohol, it is a versatile intermediate for further derivatization, e.g. with fatty acid halides to non-ionic, biologically degradable detergents of type 17, or with methacrylic acid derivatives to provide polymerisable acrylamido-disaccharides of type 18 [69].
Air oxidation in strongly alkaline solution (KOH) converts isomaltulose into the potassium salt of the next lower aldonic acid, i.e. D-glucosyl-α(1→5)-D-arabinonic acid (GPA), isolable as such, or upon neutralization, as the GPA-lactone 19 in high yields each [70]. Oxidation with hydrogen peroxide proceeds further, shortening the fructose portion by four carbon atoms to yield O-(α-D-glucosyl)glycolic acid, isolable after acetylation as its crystalline tri-O-acetyl-lactone [71] (Fig. 5).
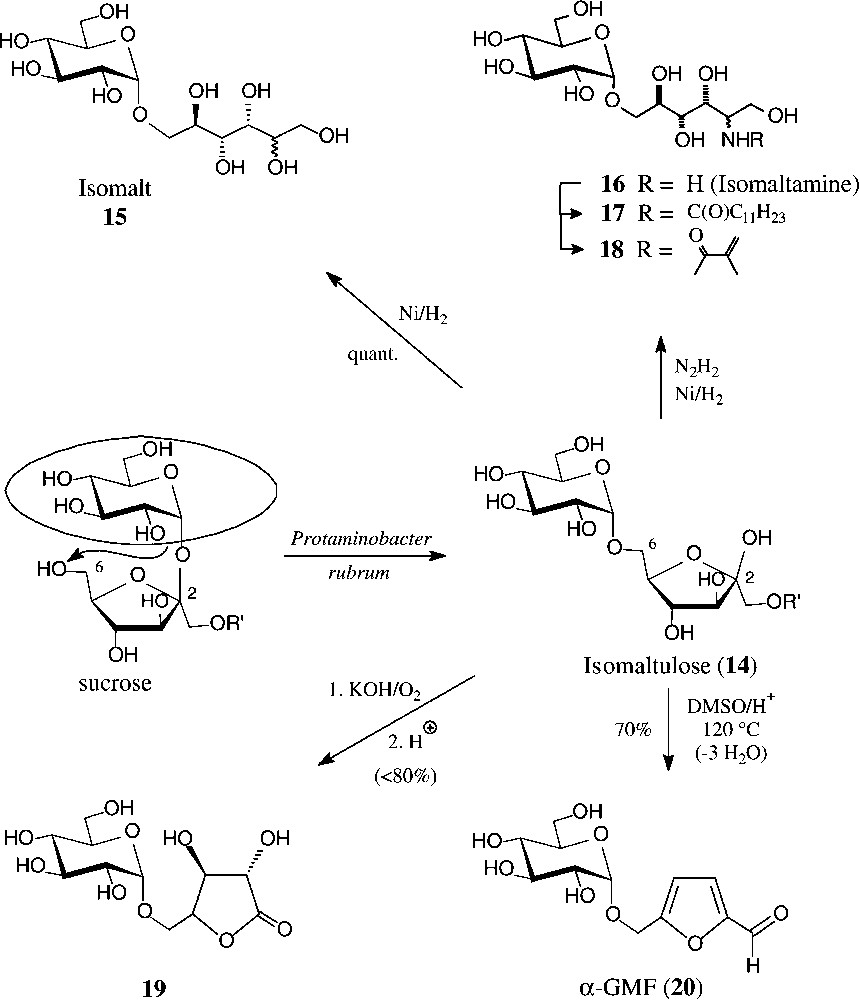
Generation of isomaltulose from sucrose, and ensuing products of industrial potential.
Another industrially relevant ensuing reaction of isomaltulose comprises its ready conversion into 5-(α-D-glucosyloxymethyl)-furfural (α-GMF, 20) by acidic dehydration of its fructose portion under conditions (acidic resin in DMSO, 120 °C) that retain the intersaccharidic linkage [72]. This process can also be performed in a continuous flow reactor [73]. Thus, a most versatile building block is available in two steps from sucrose, of which the first is already industrially realized, and the second simple enough to be performed on a large scale. A variety of industrially relevant products have already been prepared: aldol-type condensations provide derivatives with polymerisable double bonds that are expected to yield novel, hydrophilic polymers [72]. Oxidation and reductive amination generate the α-GMF-carboxylate and α-GMF-amine, respectively, which on esterification with long-chain alcohols or N-acylation with fatty acids afford compounds of type 21 and 22 − a novel type of non-ionic surfactants, in which the hydrophilic glucose and the hydrophobic fat-alkyl residue are separated by an quasi-aromatic spacer [72]; they also have favourable liquid crystalline properties [74] (Fig. 6).
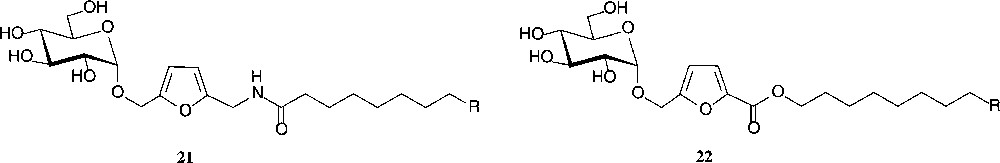
GMF-derived products with surfactant and liquid crystalline properties R = Et, Bu, Hex.
2.2 Non-food valorisation of glucose
Although D-glucose is the component sugar of cellulose and starch, only the latter is the raw material for its commercial production [75]. Glucose-containing syrups and pure, crystalline glucose are used as a nutrient in a vast variety of food applications, yet also serve as the raw material for the manufacture of sorbitol (= D-glucitol, by hydrogenation), and, by microbial processing, food-grade carboxylic acids (e.g. citric, lactic, or acetic acid), amino acids (L-lysine, L-glutamic acid), vitamins and antibiotics.
The largest non-food use of glucose is as a raw material for the production of ethanol by yeast fermentation, the annual worldwide production being in the 3 × 109 l range as of 1988 [75]. Another fairly large use of glucose goes into the alkyl polyglucosides (‘APGs’), biodegradable surfactants presently produced at an approximate 50 000-t/yr scale (cf. below), and into polylactic acid (‘PLA’), a benign packaging material (vide infra). Some minor applications comprise its use as additive in adhesives and in leather tanning, and of methyl α-D-glucoside in insulating foams. With these non-food applications of glucose, however, the huge chemical potential lying in this inexpensive and bulk-scale accessible commodity is largely untapped and waits for systematic exploitation. The most promising veins along which viable industrial products may conceivably be developed are outlined in the sequel.
The principal, preparatively useful derivatizations of D-glucose − entry reactions with which the tautomeric forms are fixed − have already been elaborated before the turn of the 19th century: mercaptalisation to the acyclic dithio acetals, isopropylidenation to furanoid systems, or the generation of pyranoid structures, such as glucosides, glucals, and hydroxyglucalesters [76] (Fig. 7).
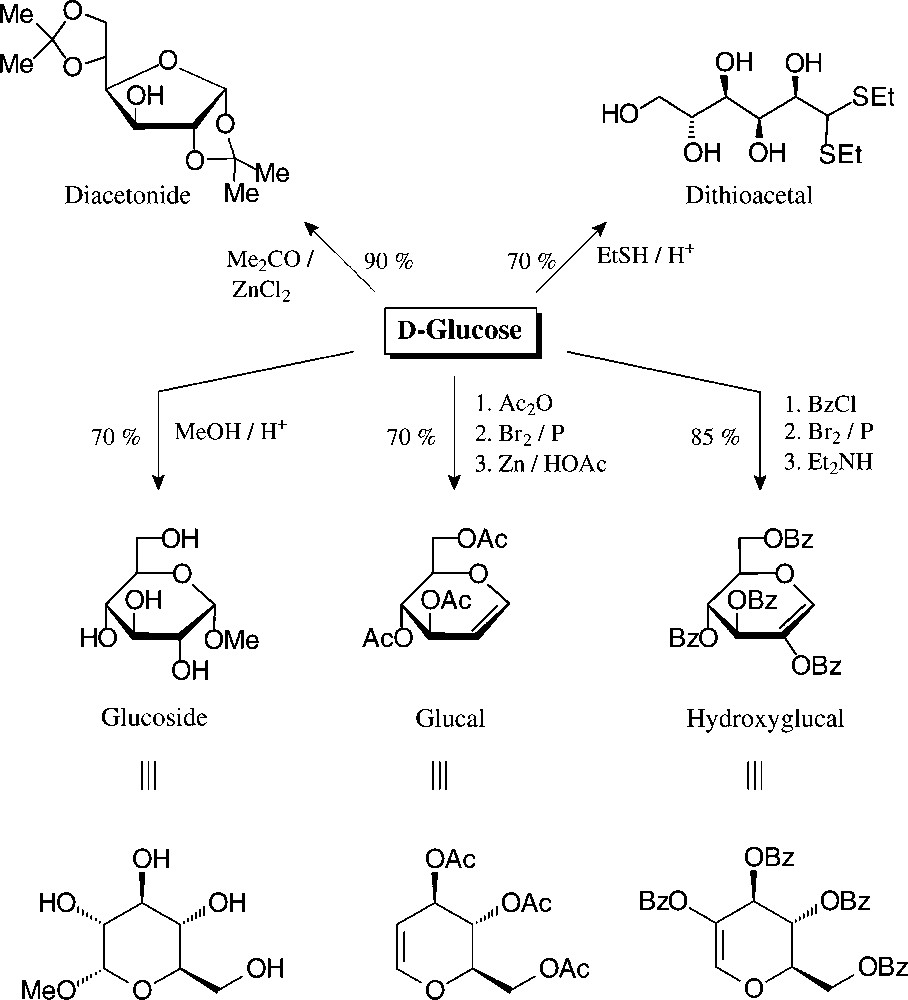
Well accessible, tautomerically fixed D-glucose derivatives with which to embark towards versatile building blocks [76].
Another simple, one-step entry from D-glucose to highly substituted furans involves their ZnCl2-mediated reaction with acetylacetone. As only the first two sugar carbons of D-glucose contribute to the formation of the furan, a distinctly hydrophilic tetrahydroxybutyl side chain is elaborated (→ 23 [77]) which can be shortened oxidatively to the dicarboxylic acid 25 or a variety of other furanic building blocks (Fig. 8). By contrast, under mildly basic conditions (aqueous bicarbonate at 85 °C), D-glucose reacts with pentane-2,4-dione in an entirely different way, elaborating via C-addition and subsequent retroaldol-type elimination of OAc–, the 2-C-glucosyl-propanone 24 [78,79]. As this conversion can be performed with the unprotected sugar and in aqueous solution with simple reagents, it may legitimately be referred to as green and/or sustainable in accord with approved principles [57]. As the procedure is equally well feasible with other monosaccharides, it is, thus, one of the cleanest and most efficient preparative entry into the area of C-glycosides, which as stable ‘mimics’ to the usual O-glycosides command major interest as glycosidase inhibitors [80].
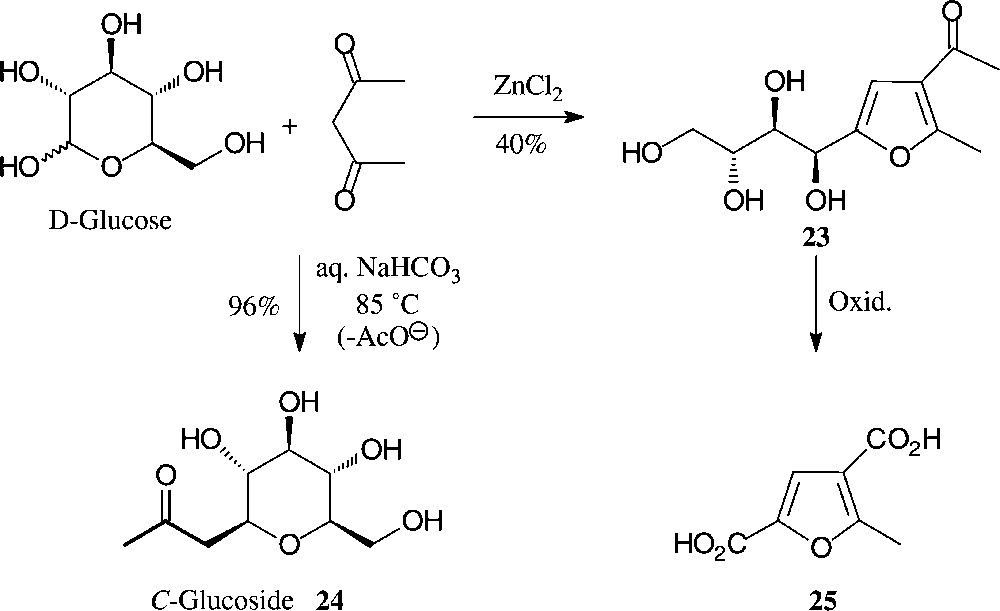
One-pot conversions of D-glucose into hydrophilic furans [77] or, alternatively, into C-glucosides by reaction with acetylacetone [78,79].
Despite the ready accessibility of these ‘entry products’, and their fairly well-developed ensuing chemistry, their exploitation towards industrial intermediates is exceedingly modest. To nevertheless emphasize their potential towards industrial intermediates, be it as enantiopure building blocks for the synthesis of non-carbohydrate natural products [81–83] or for agrochemicals and/or high-value-added pharmaceuticals, a particular versatile array of six-carbon dihydropyrans is listed in Fig. 9, all being accessible from D-glucose (via the glucal and hydroxyglucal esters) in no more than three to five straightforward steps.
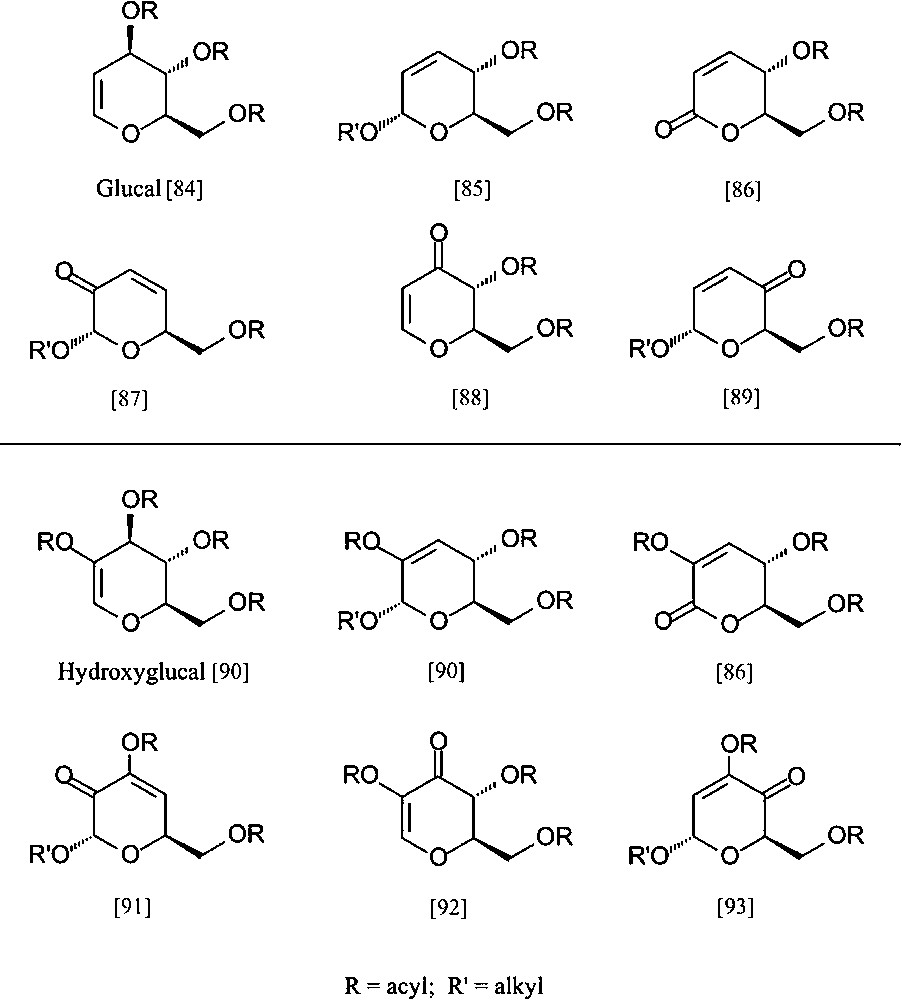
Pyranoid six-carbon building blocks accessible from D-glucose via glucal (upper half) or hydroxyglucal esters (lower entries) as key intermediates. All products require no more than 3 to 5 straightforward steps from D-glucose [84–93].
A bicyclic dihydropyranone, namely levoglucosenone, is accessible even more directly by vacuum pyrolysis of waste paper [94]. Although the yield attainable is relatively low − levoglucosan is also formed, their proportions depending on the exact conditions (Fig. 10) − relatively large amounts can be amassed quickly; levoglucosenone has been used for the synthesis of a diverse variety of natural products in enantiopure form [95].
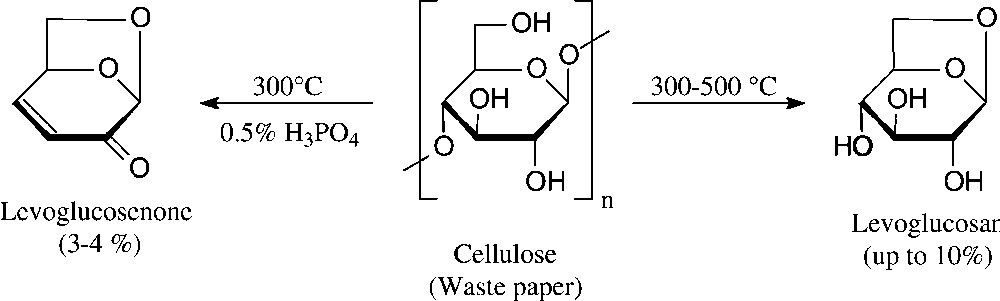
High-vacuum pyrolysis [94].
Similarly convenient are the acquisition of the two dihydropyranones 26 and 27 requiring two and three steps from maltose or sucrose respectively, their ‘left-over’ glycosyl and fructosyl residues serving as acid labile blocking groups as contrasted by the alkali-sensitive ester functions (Scheme 5).
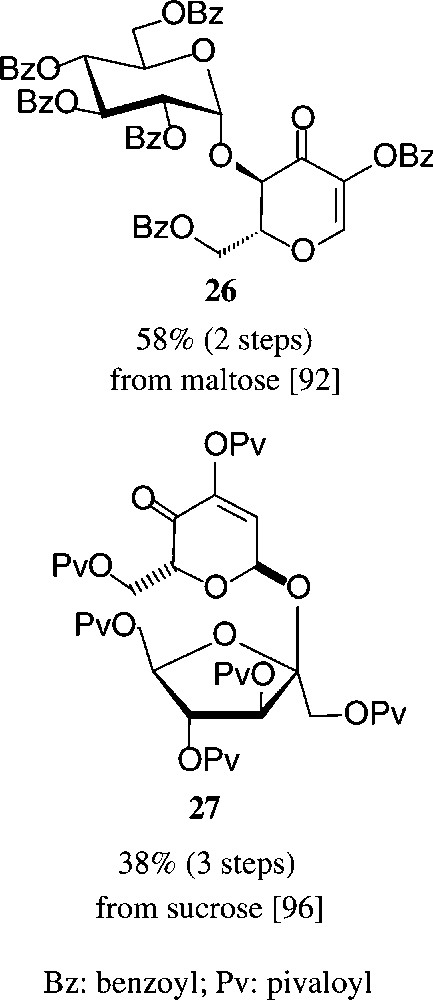
See [92,96].
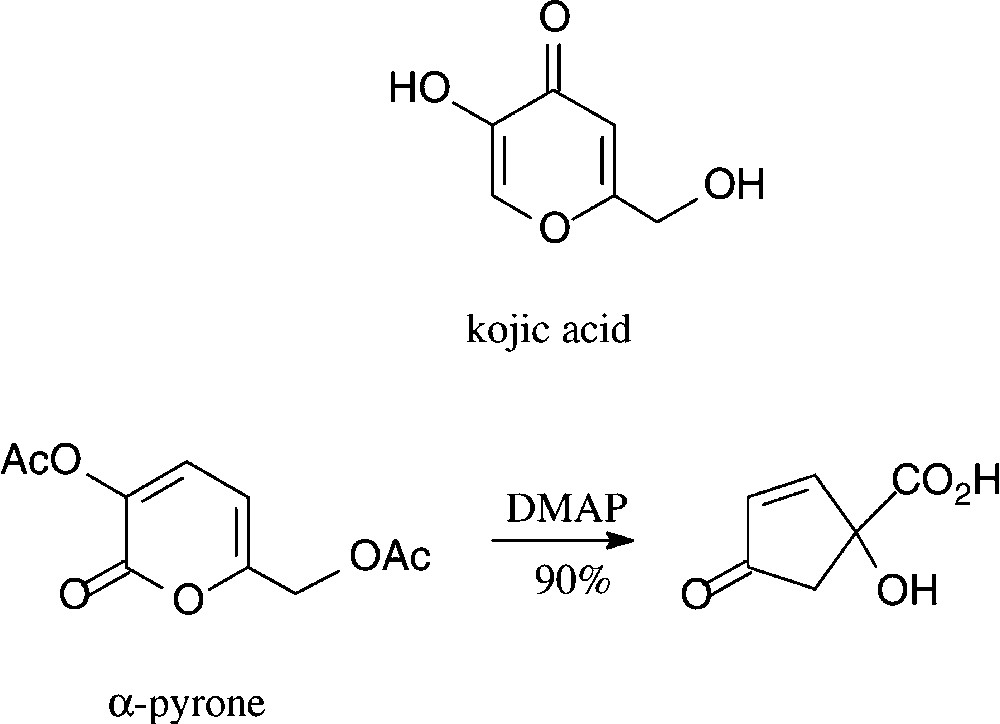
Kojic acid, a γ-pyrone, is readily obtained from D-glucose either enzymatically by Aspergillus oxyzae growing on steamed rice [97] or chemically via pyranoid 3,2-enolones [98,99]. A structurally corresponding α-pyrone can be effectively generated by oxidation of glucose to D-gluconic acid and acetylation [100]. Both, at present, are of little significance as six-carbon building blocks, despite a surprisingly effective route to cyclo-pentanoid products [101] which is surmised to have industrial potential (Scheme 6).
An established, non-food application of D-glucose are the alkyl polyglucosides (‘APGs’), which combine high performance as non-ionic surfactants with non-toxicity, low-skin irritation, and biodegradability [102]. They are technically produced − on an estimated 50 000-t scale worldwide presently − either through acid-induced glycosidation of glucose with a long chain fat alcohol or by transglycosylation of a short-chain alkyl glucoside with the appropriate long-chain alkanol. The resulting mixtures contain the α-D-glucosides majorly, as designated by the formula in Fig. 11, and are marketed as such. They are widely used in manual dishwashing detergents and in formulations of shampoos, hair conditioners, and other personal care products.
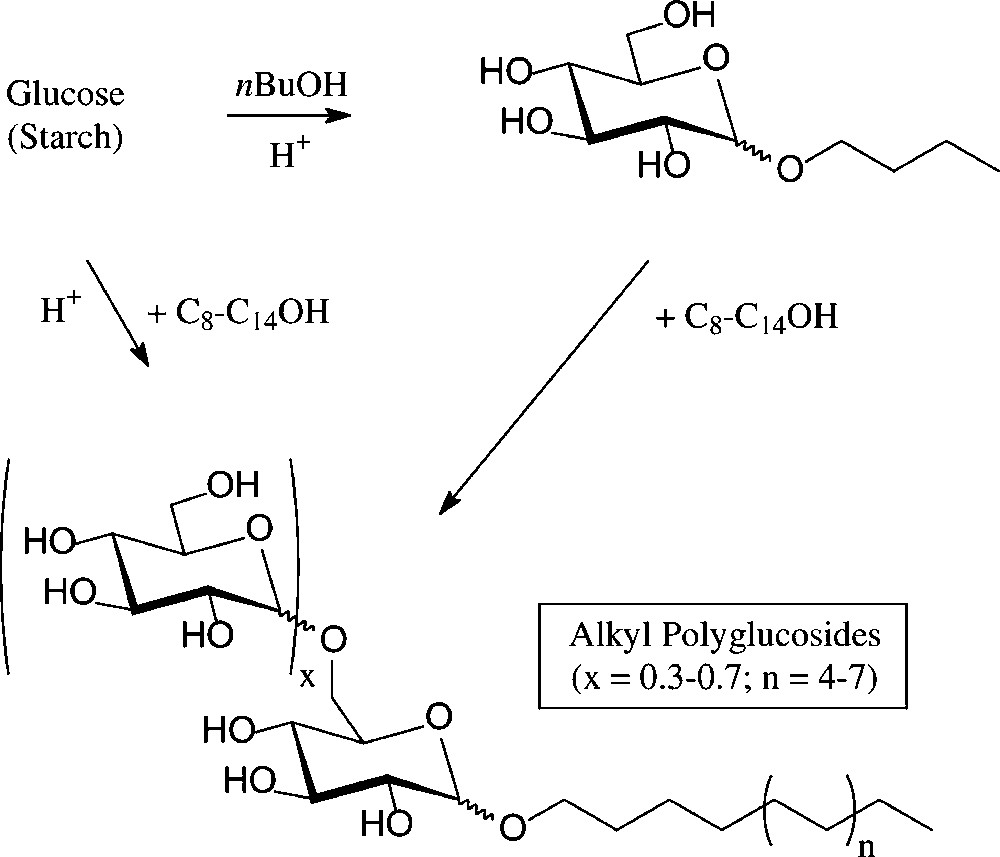
Henkel's alkyl polyglucosides (‘APGs’).
Other applications of D-glucose on an industrial scale comprise its oxidation product, D-gluconic acid, widely used as a chelating agent and textile printing additive [103], as well as sorbitol (= D-glucitol), obtained by catalytic hydrogenation [104]. Although the main consumer of its sizable annual production is the food industry, primarily as a non-caloric sweetening agent, a sugar substitute for diabetics, and as a key intermediate for the production of ascorbic acid (vitamin C) [105]. It is also applied though as a favourable moisture conditioner in printing, writing ink, and cosmetics and pharmaceutical formulations.
On exposure to mineral acid at fairly high temperatures, sorbitol readily undergoes dehydration to ‘sorbitan’, which constitutes a mixture of sorbitol, its 1,4-anhydro and 1,4:3,6-dianhydro derivatives, the exact composition depending on the conditions employed [104]. Esterification of this mixture with fatty acid chlorides/base or with their methyl esters at 200–250 °C with 0.1 N NaOH generates a sorbitan monoester (‘SMS’, R = C16/C18 acyl), di- or triester (‘SMT’), depending on the stoichiometries employed. Sorbitan monoesters (‘SMS’) are commercially available [106]; due to their low HLB values, they find use as non-ionic surfactants and as solubilizer and emulsifier in cosmetics and various other formulations [104] (Fig. 12).
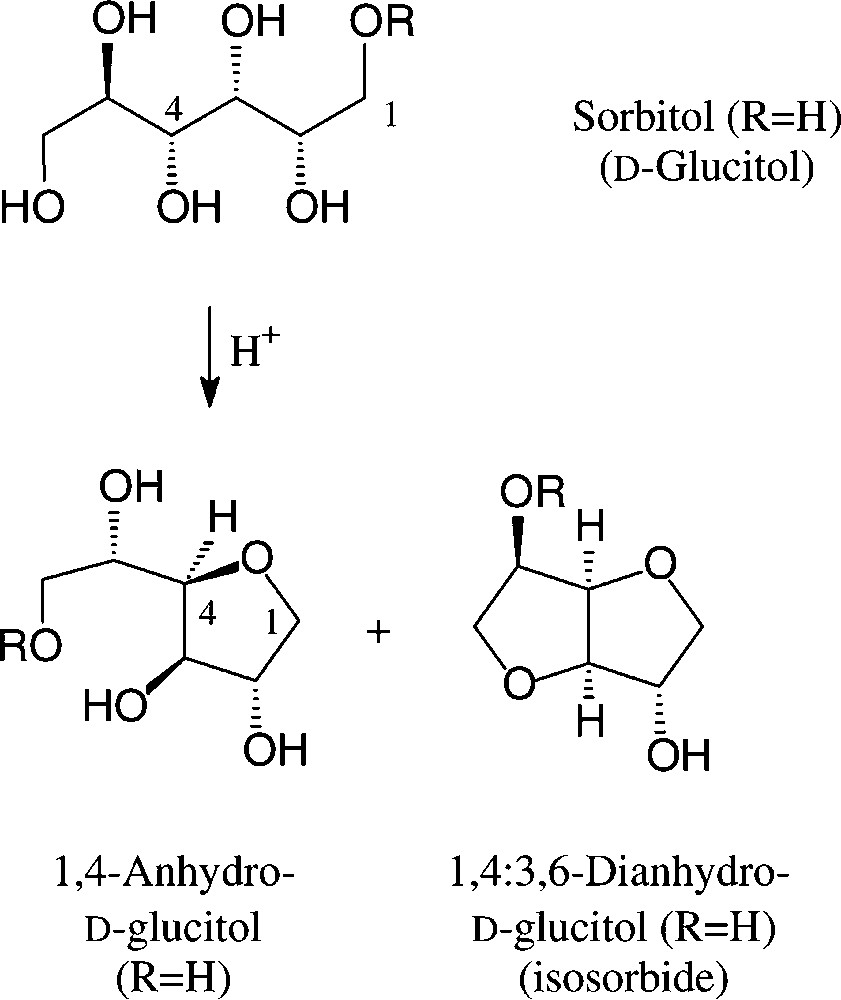
‘Sorbitan Monoester’ (SMS): R = C16-C18 acyl.
Large amounts of D-glucose – in crude form as obtainable from corn, potatoes or molasses by acid hydrolysis – enter into industrial fermentation processes towards the production of lactic acid, citric acid and various amino acids, such as L-lysine or L-glutamic acid. Whilst the major use of these products is in food and related industries, recent non-food exploitations of lactic acid have made it a large-scale, organic commodity chemical. Most of it is subsequently polymerised via its cyclic dimer (lactide) to a high molecular weight polyester: polylactic acid (‘PLA’) [107]. Due to its high strength, it can be fabricated into fibres, films, and rods that are fully biodegradable (→ lactic acid, CO2) and compostable, having degraded within 45–60 days. Accordingly, PLA and copolymers of lactic and glycolic acid are of particular significance for food packaging and in agricultural or gardening applications, but are highly suitable materials for surgical implants and sutures, as they are bioresorbable (Fig. 13).
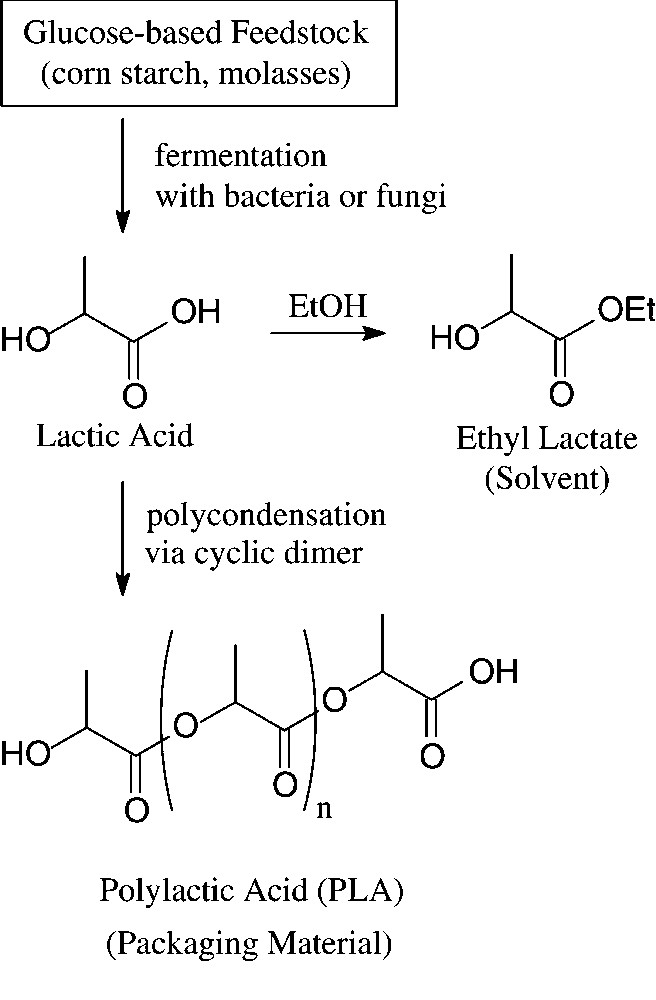
Generation and use of lactic acid.
Cargill Dow, since 1989, has invested some $ 750 million to develop and commercialise polylactic acid [108], its Nebraska plant opened in 2001 having an annual capacity of 140 000 metric tons [109,110]. Thus, polylactides, due to combining favourable economics with green sustainability are poised to compete in large volume markets that are now the domain of thermoplastic polymers derived from petrochemical sources.
Another encouraging green development based on lactic acid is its ethyl ester (‘VertecTM’) that has recently been marketed for applications in specialty coatings, inks, and straight use cleaning because of their high performance and versatility [111]. As a most benign solvent – green, readily biodegradable, and excellent toxicology records – it has the potential to displace various petrochemically based solvents such as acetone, DMF, toluene or N-methylpyrrolidone in industrial processes.
2.3 D-Fructose: potentials for non-food uses
The substantial amounts of this ketohexose are mainly prepared by base-catalysed isomerisation of starch-derived glucose [112], yet may also be generated by hydrolysis of inulin, a fructooligosaccharide [113]. As fructose – de facto only the β-D-fructopyranose tautomer – is sweeter than sucrose (up to 1.5 times), it is widely used as a sweetener for beverages (‘high fructose syrup’). Its non-food utilization is modest − not surprising since its basic chemistry is more capricious and considerably less developed than that of glucose. The ‘entry reactions’ compiled in Figs. 14 and 15, nevertheless, provide a multifaceted array of well-accessible, pyranoid (28–35) and furanoid derivatives (40–46) with which to exploit their industrial application potential.
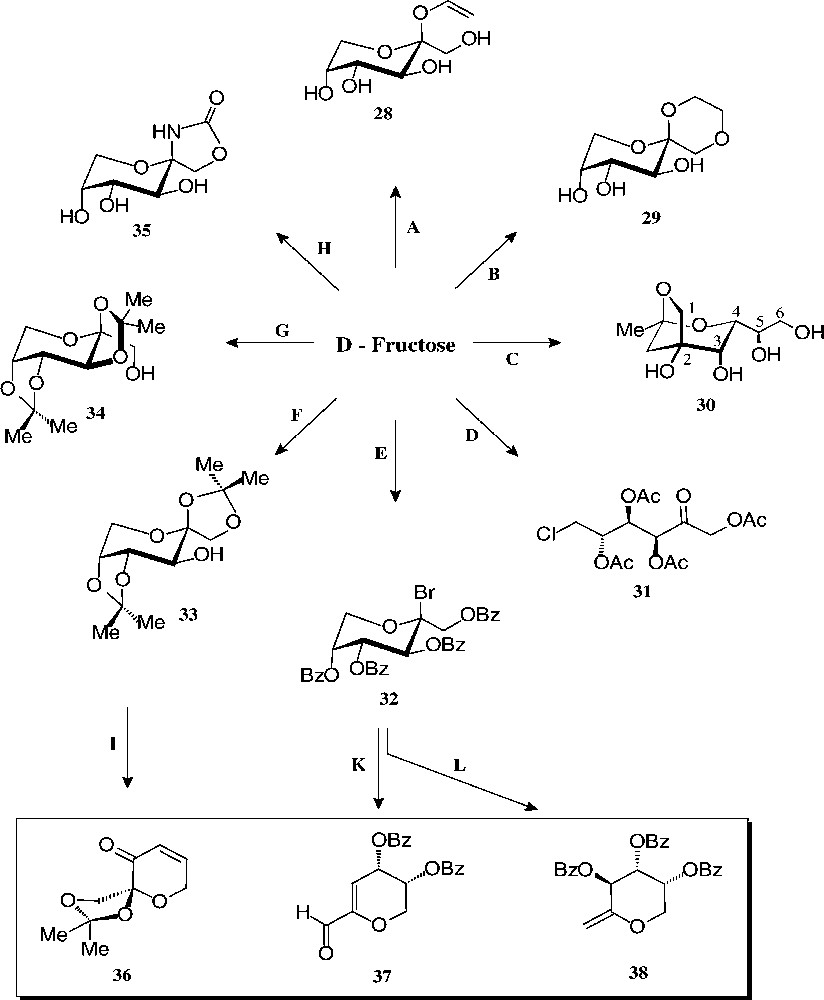
Readily accessible pyranoid derivatives of D-fructose. Key: (A) allyl alcohol/AcCl, 49% [114]; (B) glycol/H+, 74% [115]; (C) pentane-2,4-dione, aq. NaHCO3, 85 °C, 27% [116]; (D) AcCl, –10 °C → PCl5, 61% [117]; (E) BzCl, -10 °C → HBr, 63% [117]; (F) Me2CO/cat. H2SO4, 58% [118]; (G) Me2CO/>5% H2SO4, 80%[119]; (H) KOCN, 31% [117]; (I) 5 steps, 22% [120]; (K) DBU → NH2OH → MeCHO/H+, 45% [121]; (L) Zn/MIM, 90% [122].
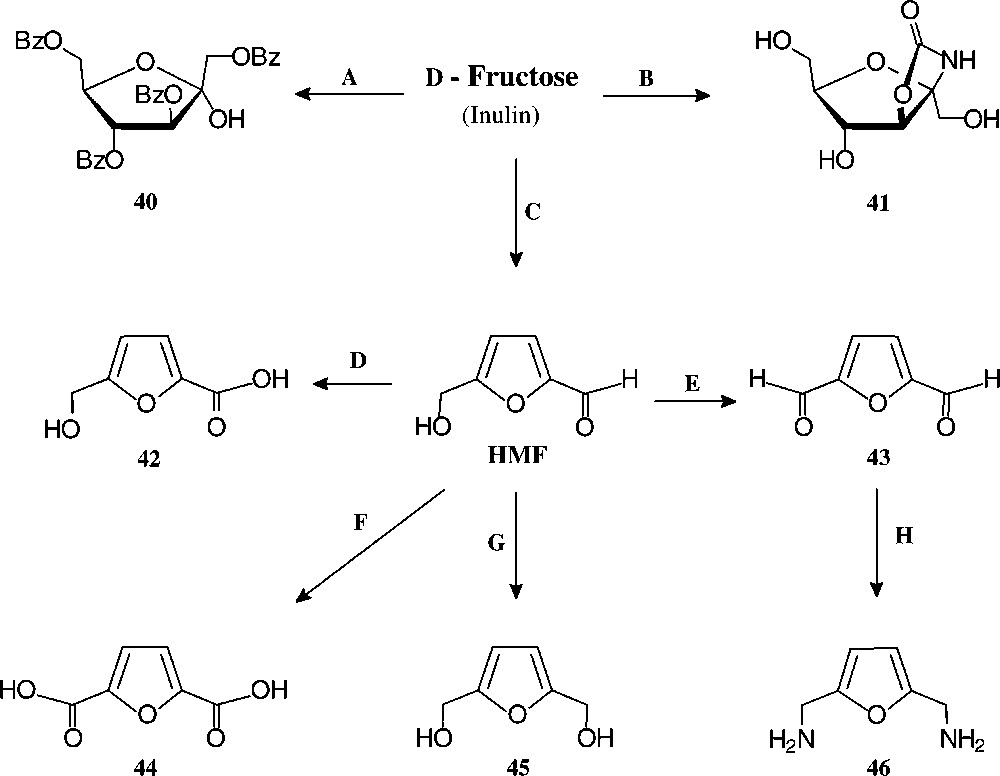
D-Fructose-derived furanoid products. Reagents and conditions: (A) BzCl, 60 °C, 60% [128]; (B) KOCN/KH2PO4, 32% [117]; (C) H+, 90% [126]; (D) Ag2O, 100 °C, 75% [129]; (E) BaMnO4/1,1,2-trichloroethane, 93% [129]; (F) Pb,Pt/O2/pH 7, quant. [130] ; (G) Pt/H2, quant. [131]; (H) NH2OH/HCl → Raney-Ni/H2, 33% [129].
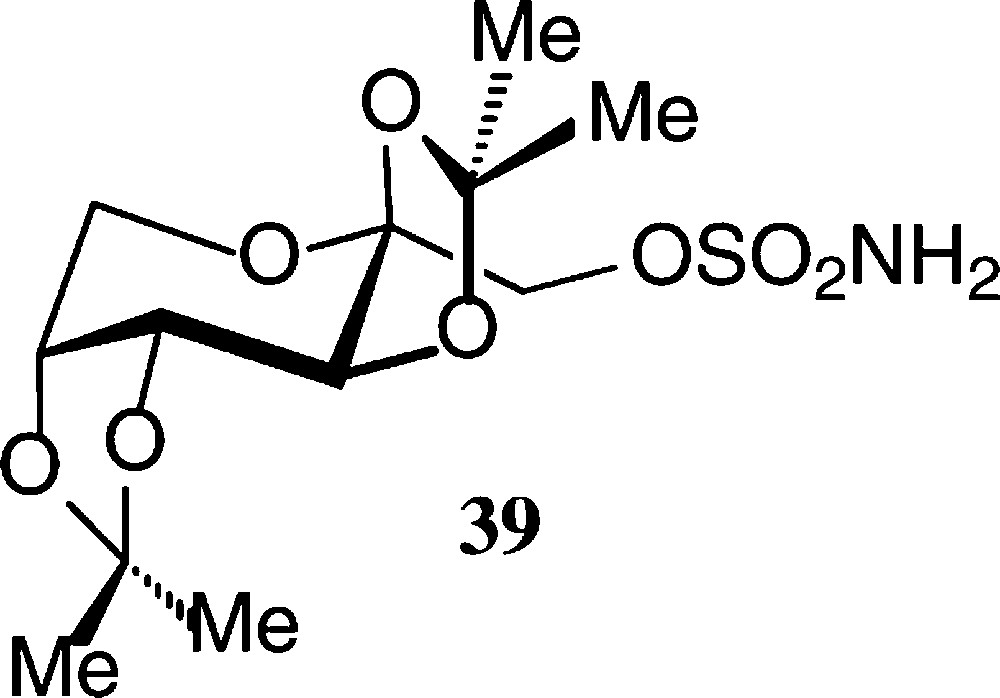
The pyranoid building blocks 36 and 37 have versatile functionalities for a rich ensuing chemistry [122] as does have the exo-D-fructal 38 [121]. The diacetone-fructose 34 similarly is the starting material for drugs, e.g. the anticonvulsant Topiramate® 39 [123].
The pyranoid building blocks 36 and 37 have versatile functionalities for a rich ensuing chemistry [122] as does have the exo-D-fructal 38 [121]. The diacetone-fructose 34 similarly is the starting material for drugs, e.g. the anticonvulsant Topiramate® 39 [123] (Scheme 7).
Of the simple furanoid products derived from fructose, the tetrabenzoate 40 and the cyclocarbamate 41 are well accessible, but by far the highest industrial potential demands 5-hydroxymethyl-furfural (HMF), which has been termed one of the few ‘petrochemicals readily accessible from regrowing resources’ [124] and ‘a key substance between carbohydrate chemistry and mineral oil-based industrial organic chemistry’ [125]. It is readily accessible from fructose or inulin hydrolysates by acid-induced elimination of 3 mol of water [126], and even a pilot plant size process has been elaborated [127].
Of the products readily generated from HMF in simple, large-scale adaptable transformations (Fig. 15), the 5-hydroxymethyl-furoic acid 42, the 2,5-dicarboxylic acid (44), and the respective 1,6-diol (45) and 1,6-diamine (46) are most versatile intermediate chemicals of high industrial potential, as they represent six-carbon monomers that could replace adipic acid, or alkyldiols hexamethylenediamine in the production of polyamides and polyesters. HMF as such has been used for the manufacture of special phenolic resins, as acid catalysis induces its aldehyde and hydroxymethyl group to react with phenol [132], and an impressive series of such furanic polymers with highly promising properties have been prepared [133], yet none has proved competitive to existing products. Thus, HMF is, as of now, not produced on an industrial scale. A recent assessment of its economics as compared to petrochemical raw materials [134] clearly unfolds the reasons underlying: ton prices of naphtha and ethylene are in the 150–400 € range and that of inulin (500 €/t) or fructose (~1000 €/t), entailing an HMF-marketing price of at least 2500 €/t, which is too expensive at present for a bulk-scale industrial product. Accordingly, as long as the economic situation favours fossil raw materials, applications of HMF lie in high value-added products, such as pharmaceuticals or special niche materials.
2.4 Furfural from pentoses (pentosans)
With an annual production of about 250 000 tons, furfural (2-furfuraldehyde) appears to be the only unsaturated large-volume organic chemical prepared from carbohydrate sources. Indeed, its price of about 250 €/t lies in the range of the basic petrochemicals mentioned, benzene and toluene being in the € 225–250 region. The technical process involves exposure of the lignocellulosic material in agricultural or forestry wastes (pentose–polysaccharides in straw, oat hulls, corn cobs, etc.) to aqueous acid and fairly high temperatures, the pentosans first being hydrolysed to pentoses and then undergoing cyclodehydration [135].
The chemistry of furfural is well developed, providing a host of versatile industrial chemicals by simple straightforward operations (Fig. 16): furfuryl alcohol and its tetrahydro derivative (hydrogenation), furfurylamine (reductive amination), furoic acid (oxidation) and furanacrylic acid (Perkin reaction), or furylidene ketones (aldol condensations). Furfural is also the key chemical for the commercial production of furan (through catalytic decarbonylation) and tetrahydrofuran (hydrogenation), thereby providing a biomass-based alternative to its petrochemical production via dehydration of 1,4-butanediol [135].
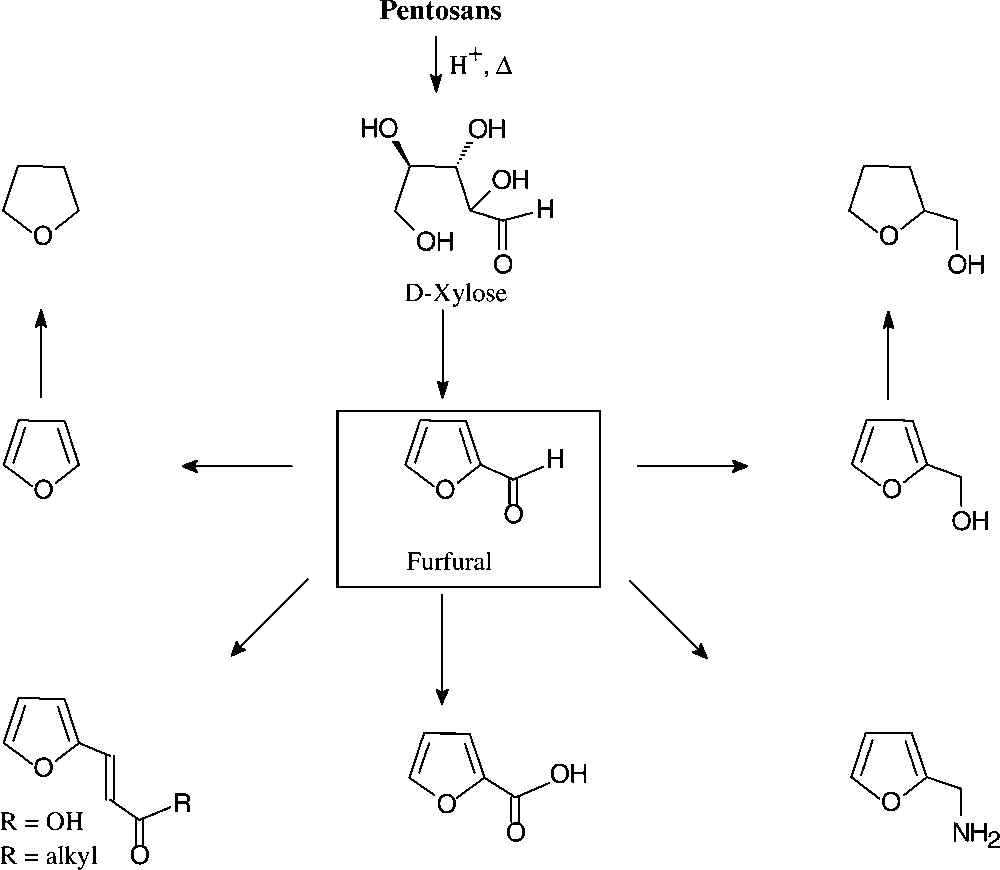
Furanic chemical from pentosans.
The bulk of the furfural produced is used as foundry sand linker, in the refining of lubricating oil, and in condensations with formaldehyde, phenol, acetone, or urea to yield resins of complex, ill-defined structures, yet with excellent thermosetting properties, high corrosion resistance, low fire hazard, and extreme physical strength [135].
2.5 Sugar-derived unsaturated N-heterocycles
Although transformation of sugars into trace amounts of N-heterocycles occurs extensively on exposure of foodstuffs to heat − the nitrogen stemming from amino acids or peptides (Maillard reaction [136]) − and despite the fact that various nitrogen heterocycles have been generated from saccharide derivatives [137], procedures meeting preparative standards are exceedingly scarce. Recent improvements of existing procedures and the development of new methodologies has led to the more ready acquisition of various N-heterocycles from carbohydrates, e.g. imidazoles, pyrroles, pyrazoles, pyridines and quinoxalines, which due to their sugar derivation have hydrophilic side chains.
2.5.1 Pyrroles
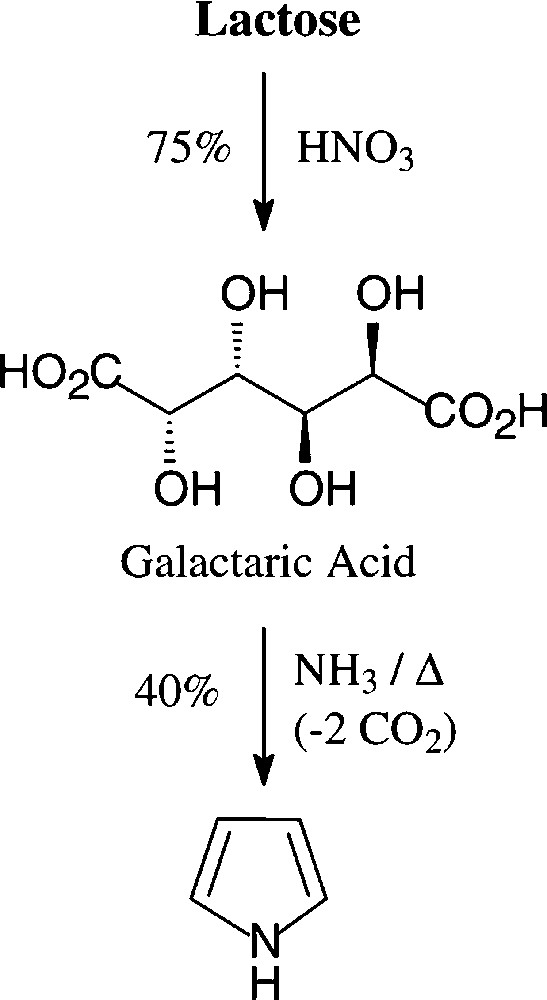
The generation of pyrroles by heating a glycerol solution of lactose-derived [138] ammonium salt of galactaric acid over a free flame [139] appears to be the highest-yielding acquisition (40%) from a carbohydrate source − a process, that in this or modified form does not seem to be utilized industrially (Scheme 8).
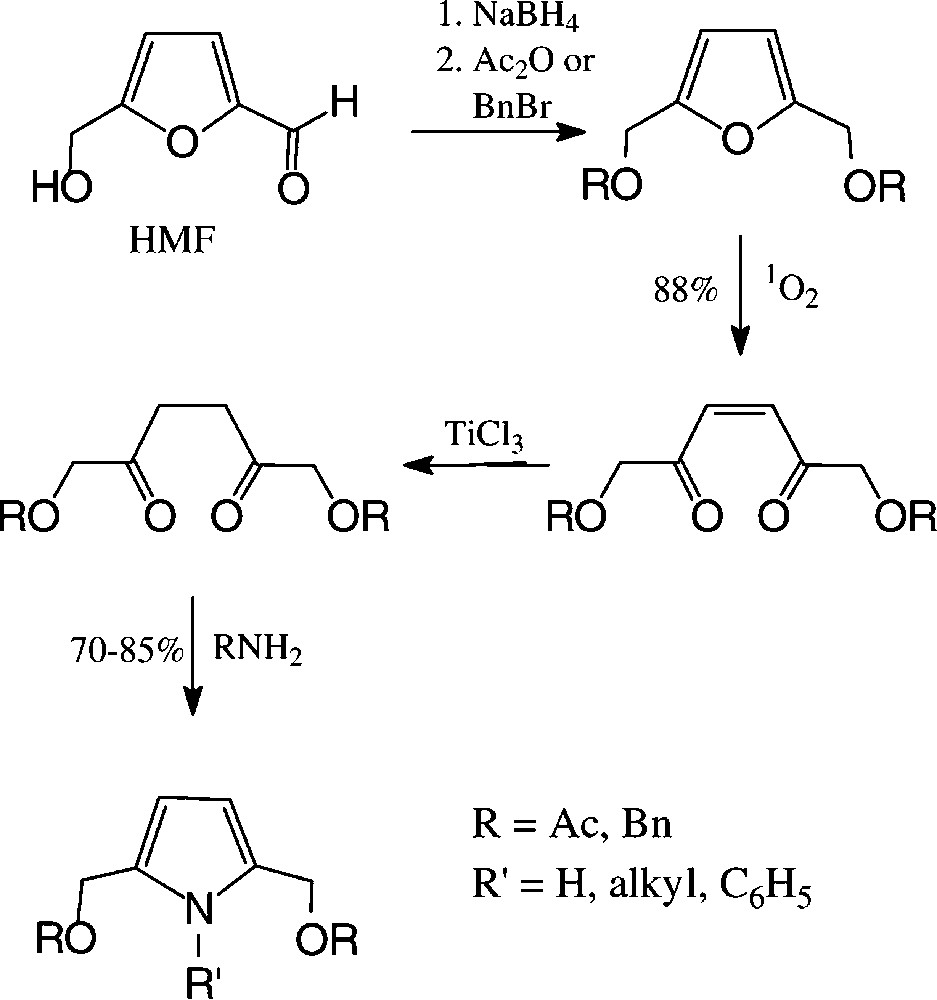
2,5-Disubstituted pyrroles are accessible from carbohydrate sources via HMF in a preparatively straightforward reaction sequence, involving photooxidative furan ring opening and cyclisation of the saturated 2,5-diketones with ammonia or amines [140] (Scheme 9).
These reaction sequences can directly be transferred to GMF, leading to pyrroles carrying an additional glucosyl residue [140]. Pyrroles with hydrophilic tetrahydroxybutyl substituent are available in a one-pot reaction from D-fructose by heating with acetylacetone and ammonium carbonate in DMSO [141] (Scheme 10).
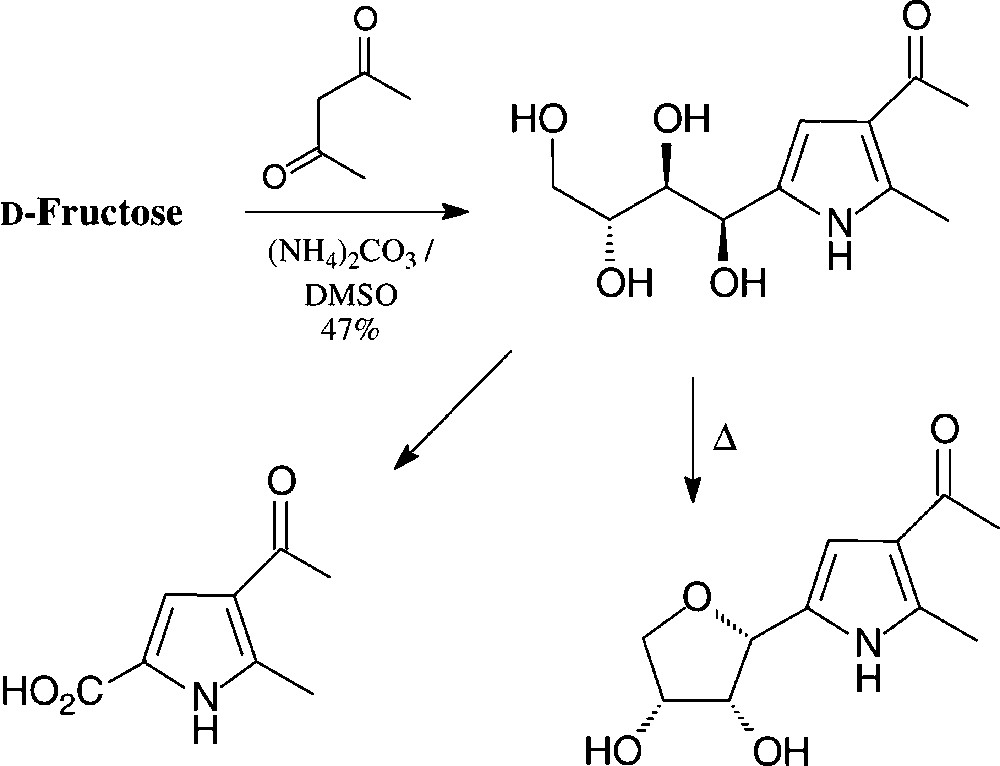
The hydroxylated side chain can, of course, be oxidatively shortened to give a variety of simple pyrrole building blocks, or cyclised to a furanoid ring [142]. These compounds may be considered as C-nucleosides.
2.5.2 Pyrazoles
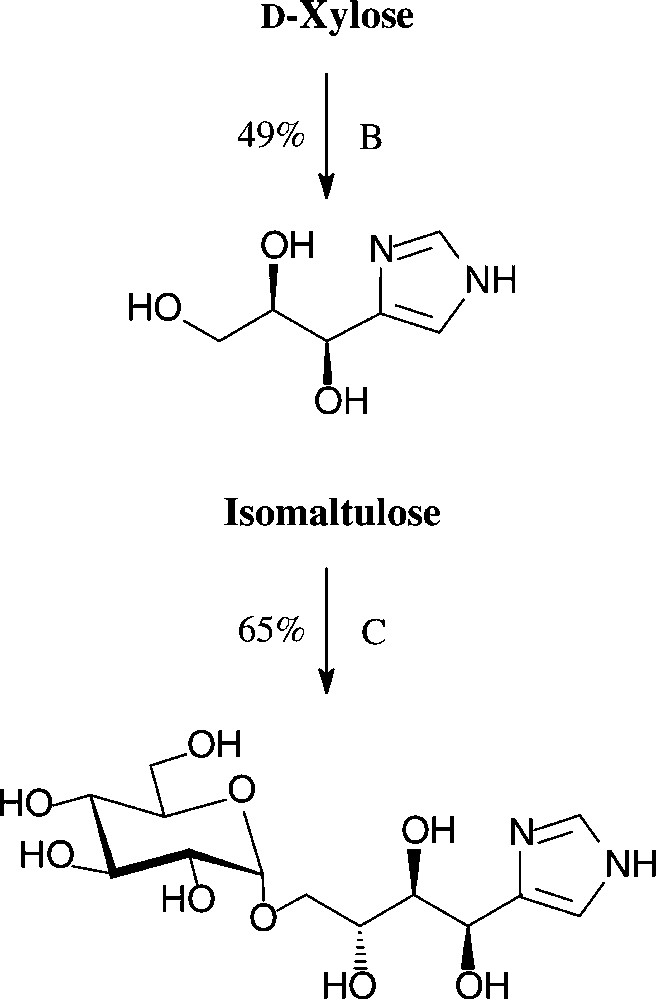
An expeditious four-step approach to 1-phenylpyrazol-3-carboxaldehydes with a 5-hydroxymethyl, 5-dihydroxyethyl, or 5-glucosyloxymethyl substituents has been elaborated starting from D-xylose [143], D-glucose and isomaltulose [144], respectively (Scheme 11).
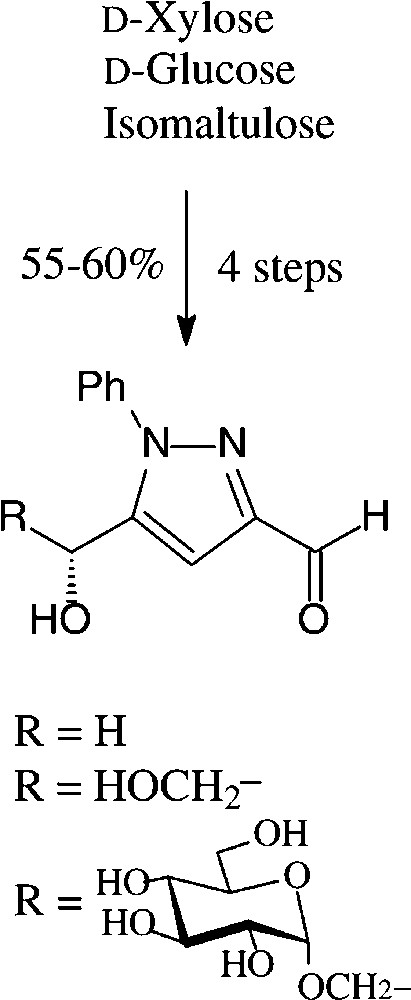
As illustrated for D-xylose, its osazone, nearly quantitatively formed on reaction with phenylhydrazine, straightforwardedly elaborates the pyrazole upon addition to refluxing acetic anhydride. Subsequent removal of the N-acetylphenylhydrazone residue with formaldehyde/acetic acid and de-O-acetylation provides, in 57% yield based on D-xylose [143], a hydroxymethyl-pyrazole-aldehyde (Fig. 17, centre), which constitutes a versatile N-heterocyclic building block for the elaboration of pharmaceuticals or the generation of polyamides and polyesters.
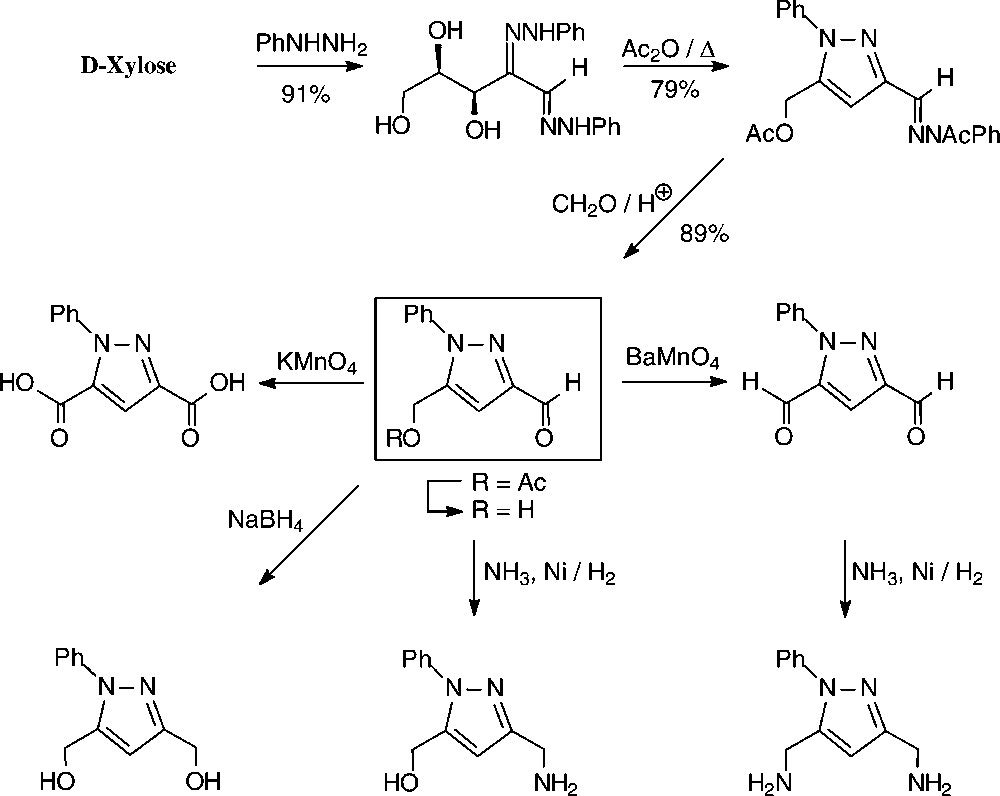
Versatile pyrazoles from D-xylose.
2.5.3 Imidazoles
Various imidazoles carrying hydrophilic substituents in the 4-position are readily accessible from the standard monosaccharides in one-pot procedures. Of those, the formation of 4-hydroxymethylimidazole on Cu(II)-promoted reaction with formaldehyde and concentrated ammonia [145] is rather unique in such as obviously retroaldolization to glyceraldehyde and dihydroxyacetone is involved. The retroaldol fission can be partially suppressed through heating D-fructose with formamidinium acetate in liquid ammonia in a pressure vessel [146] or with formamidinium acetate in the presence of boric acid and hydrazine, obviously proceeding via a boric acid complex of the bishydrazone of D-glucosone [147] (Scheme 12).
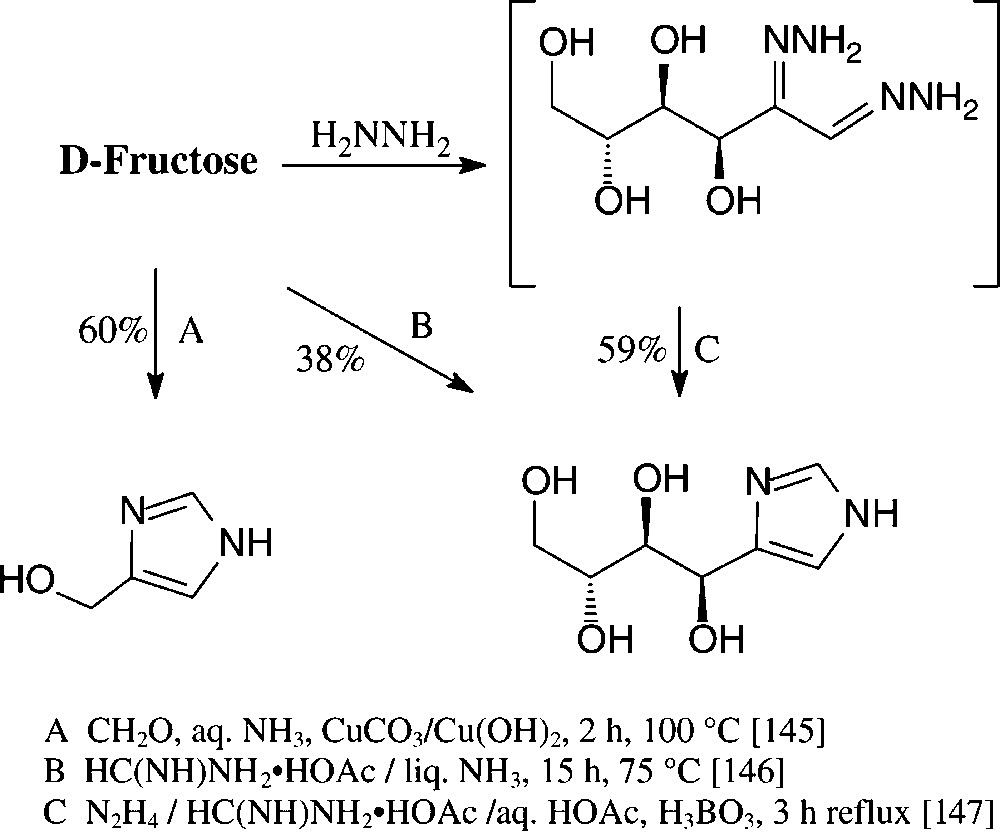
These conditions can be readily applied to pentoses or disaccharides, as exemplified with D-xylose [146] and isomaltulose [147] in for one-pot procedures acceptable yields (Scheme 13).
2.5.4 3-Pyridinols
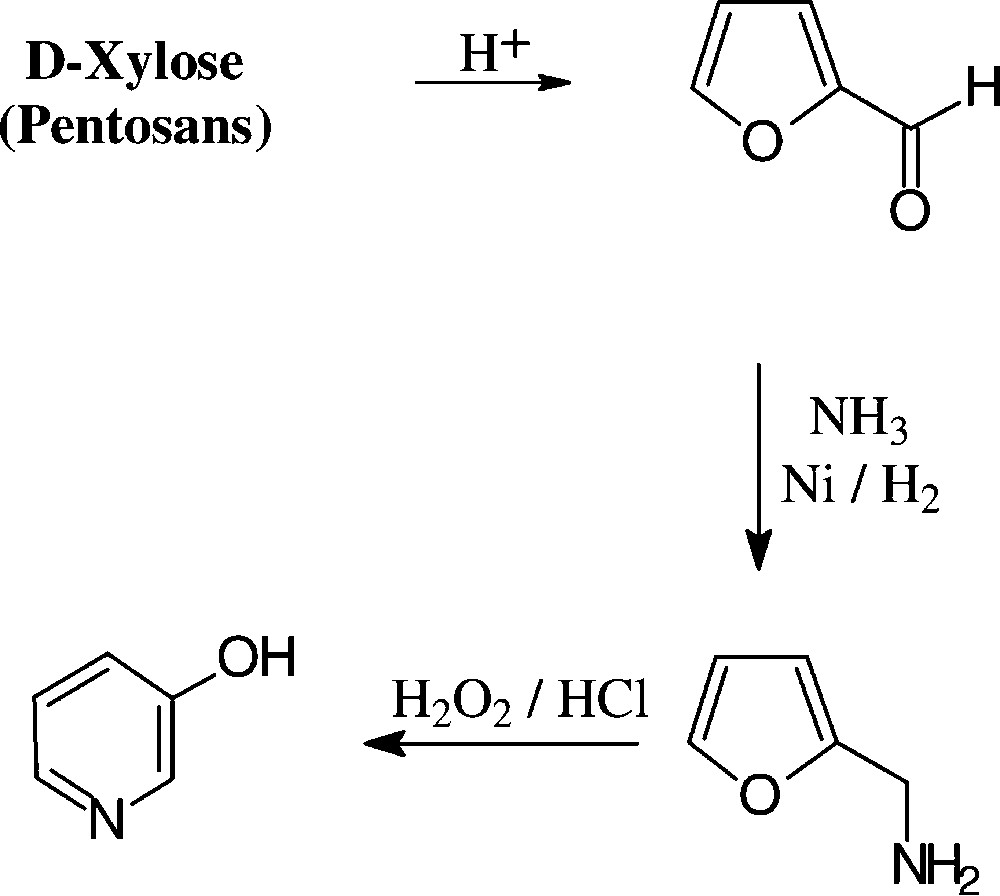
The conversion of pentosans or pentoses into 3-pyridinol can be effected in a practical 3-step sequence, involving acid-induced dehydration to furfural, reductive amination to furfurylamine, and subsequent oxidation wit hydrogen peroxide [148,149], the last step conceivably proceeding through the stage of a 2,5-dihydroxy-2,5-dihydrofurfurylamine, which elaborates the pyridine nucleus via dehydration to a 5-aminopentenal intermediate and intramolecular aldimine formation. The pyridinol figures prominently as an intermediate chemical for the preparation of herbicides and insecticides [150] as well as cholinergic drugs of the pyridostigmine type (Scheme 14).
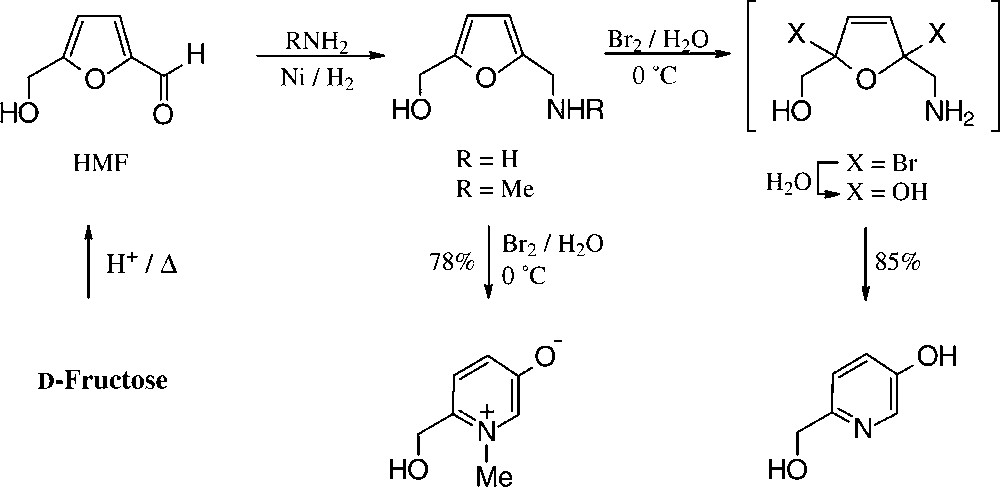
For the conversion of furfurylamines with readily oxidizable hydroxyl groups, e.g. those derived from fructose via HMF/bromine in water/methanol, the entire multi-step process to the hydroxymethyl-pyridinol is effected in a one-pot procedure [151] (Scheme 15).
2.6 Sugar-derived hydrophilic polyamides
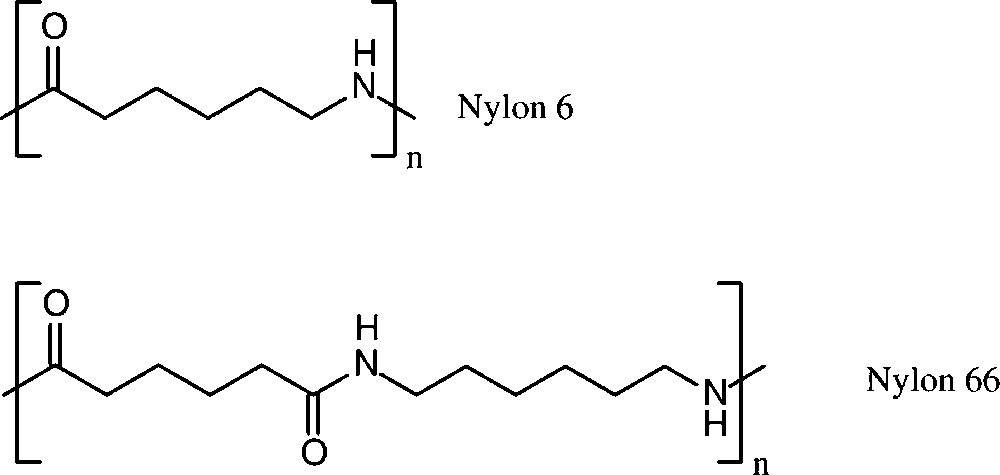
Polyamide production worldwide amounted to about 5.8 × 106 t in 1998 [152]. More than 90% of these polyamides are based on six-carbon monomers, i.e. caprolactam lactam (nylon 6), and adipic acid/hexamethylenediamine (nylon 66) (Scheme 16).
As six-carbon compounds in the form of hexoses are abundantly available in nature, substantial efforts have been made to derive monomers suitable for polyamidation from the bulk scale-accessible hexoses. This approach becomes particularly evident, when considering the large variety of amincarboxylic acids, dicarboxylic acids, and diamines, that are accessible from the common six-carbon sugars [153–155] (Fig. 18).
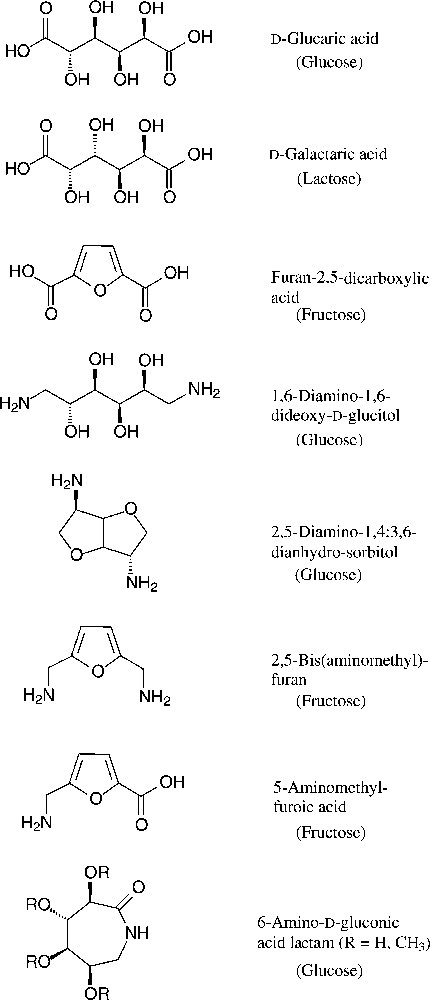
Green six-carbon building blacks for the generation of polyamides (starting materials in parentheses).
Of the myriad of possible combinations of these sugar-derived monomers either with themselves or with the common, petrochemically derived diamines and dicarboxylic acids, an immense number have been realized. Here only a few of these polyamides are exemplarily covered.
Solution or interfacial polycondensation of galactaric acid dichloride in its acetylated form with various aliphatic and aromatic diamines resulted in a series of polyamides [150,152], the one resulting from 1,6-diamino-hexane resembling a nylon-6,6, in which half of the methylene hydrogens of the usual adipic acid are substituted by acetoxy groups (R = Ac). These can be deacylated with aqueous ammonia to give the tetra-hydroxylated nylon-66 (Scheme 17).
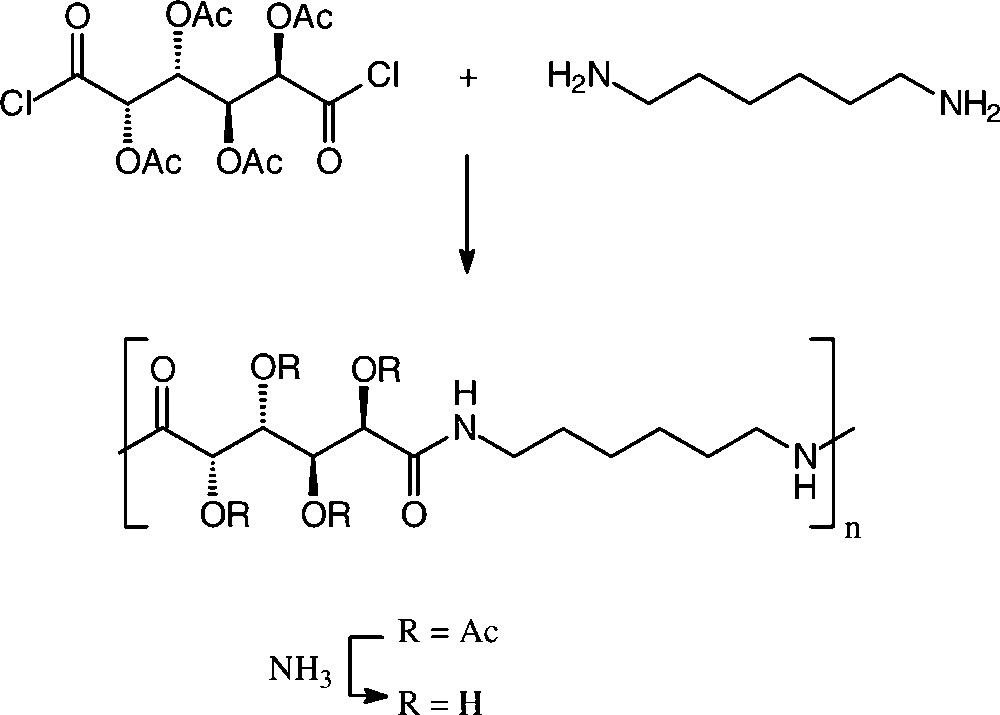
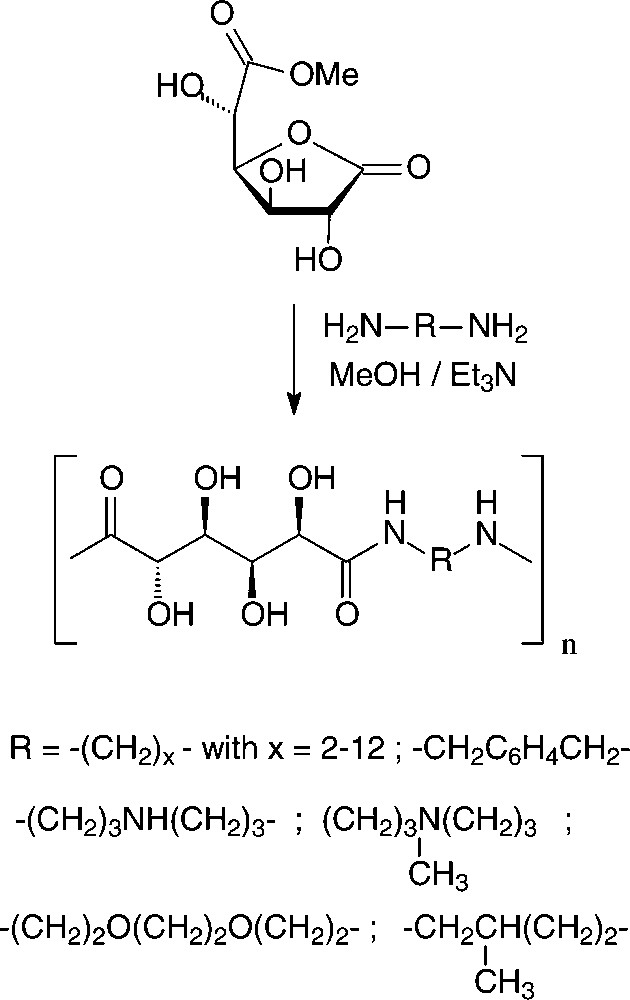
In the case of D-glucaric acid, the use of its 3,6-lactone mono-methyl ester proved advantageous to generate stereoregular polyglucaramides, effected with an impressive array of aliphatic and aromatic diamines [156,157] (Scheme 18).
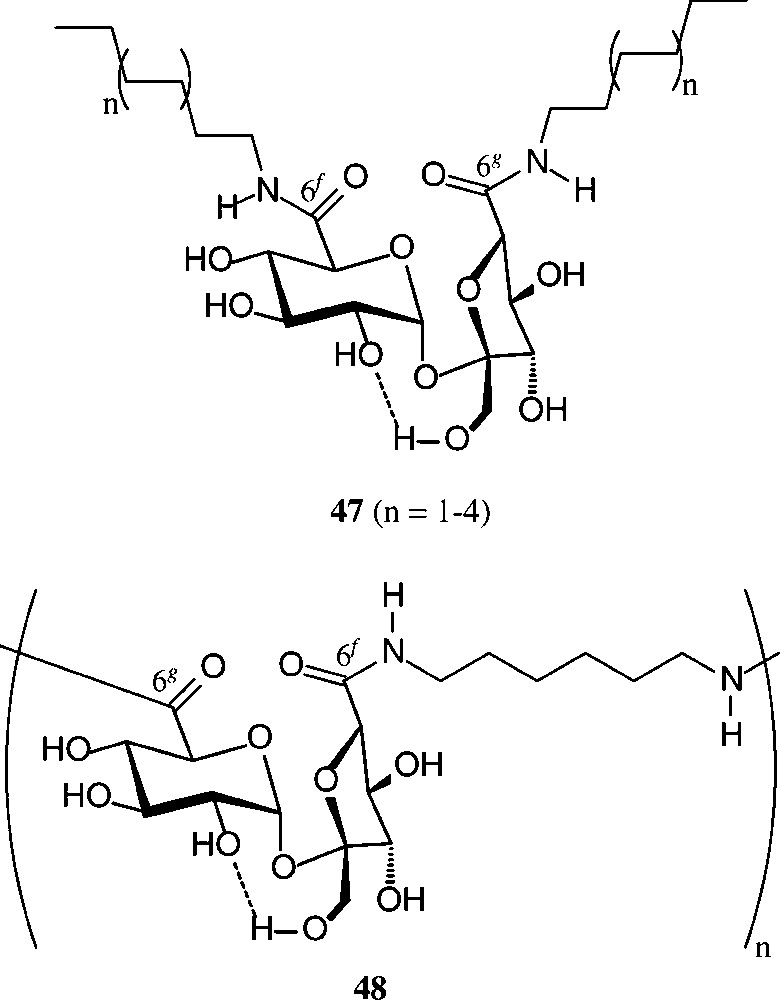
Of similar practical utility is the sucrose-6g,6f-dicarboxylic acid (2), which on amidation of its di-methylester [158] with fat-amines provided surface-active diamines of type 47 with remarkable tensidometric properties, whereas reaction with hexamethylenediamine elaborates a polyamide 48 [159] (Scheme 19).
Sugar-based ‘quasi-aromatic’ monomers for polyamides, i.e. the furan-2,5-dicarboxylic acid, appear particularly relevant as they embody the potential to replace petrochemically derived terephthalic or isophtalic acid in the present industrial products. Similar potential pertains to the furanic 1,6-diamine as a substitute for p-phenylenediamine. Indeed, a series of such furanic polyamides has been prepared [160] using the dicarboxylic acid and aliphatic as well as aromatic diamines. Of these, the polyamide resulting from condensation with p-phenylenediamine, which de facto is an analogue of the commercially introduced Nomex® and Kevlar®, has particularly promising decomposition and glass temperature parameters [161], distinctly better than those found for the all-furanic polyamides (Scheme 20).
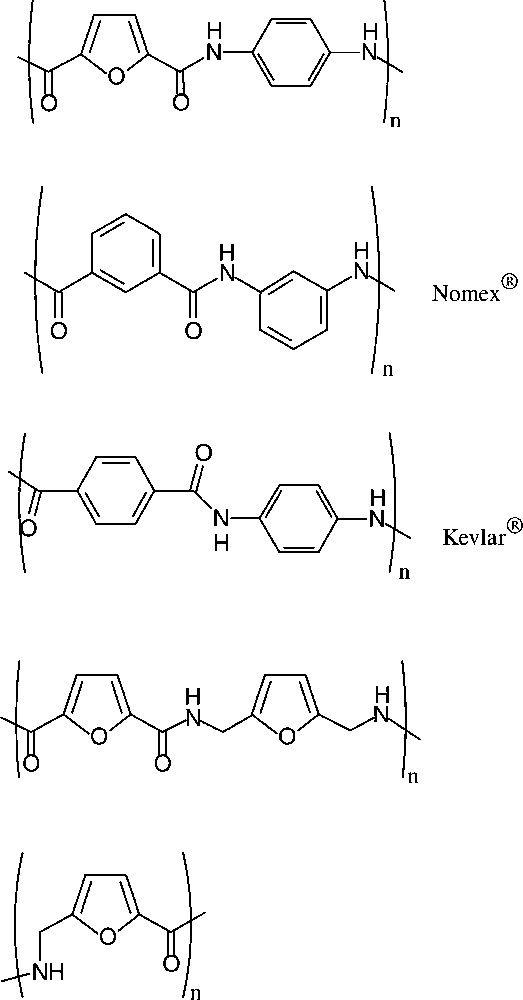
2.7 Sugar-derived polyesters
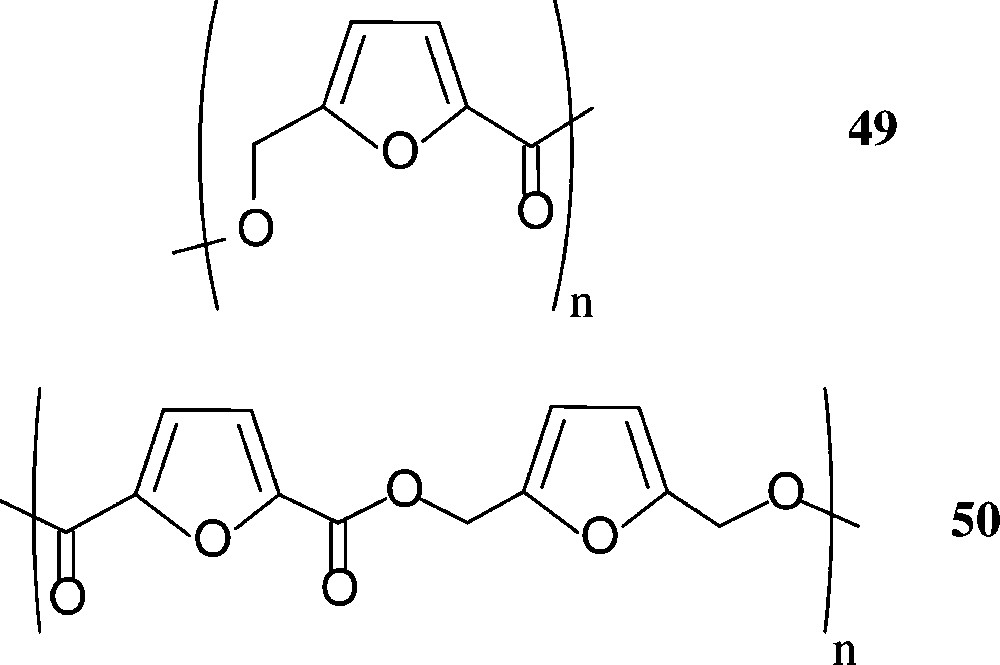
Aside polylactic acid (‘PLA’), that is in use as a benign, biodegradable packaging material (cf. above), other sugar-based polyesters do not seem to have been commercialised – despite of the fact, that HMF-derived furanic polyesters have considerable potential. Thus, the 5-hydroxymethyl-2-furoic acid 42 and the 2,5-dicarboxylic acid 44 have extensively been exploited for the preparation of furanoic polyesters [160], and the corresponding diol 45 has been reacted with various aliphatic and aromatic diacids; the ethyl ester of 42, upon polycondensation, gave a mixture of linear (49) and cyclic products, while the furan-diacid 44 has been polyesterified with a series of aliphatic diols or bisphenols. Even the all-furanic polyester 50 has been successfully prepared from its respective monomeric components [160] (Scheme 21).
In spite of the impressive array of highly useful products polyesters and polyamides − with respect to their application profiles, they compare favourably with the well-known polyamides − none at present, are produced on an industrial scale, the reasons being purely economic as the products derived from fossil raw material sources are still cheaper by a factor of 5 on the average –, a situation that will only change with the progressing depletion of affordable fossil raw materials.
3 Conclusion
At present, the non-food utilization of inexpensive, bulk scale-accessible low-molecular-weight carbohydrates – sucrose, glucose and fructose being the most abundant – is at a rather modest level in terms of large-scale manufactured commodities that are on the market: the APGs (alkyl polyglucosides), PLA (polylactic acid) and furfural, with some of its ensuing products. The unusually diverse stock of readily accessible products described in this account, which cover a wide range of industrial application profiles, lies essentially unexploited − for economic reasons mainly −, as equivalent products based on petrochemical raw materials are simply cheaper. Notwithstanding, a basic change in this scenario is clearly foreseeable. As depletion of our fossil raw materials is progressing, chemical products derived therefrom will inevitably increase in price, such that biobased products will become competitive. Realistic prognoses expect this for 2040 at the latest [7].
In the meantime, it is imperative that carbohydrates are systematically further exploited towards efficient, environmentally benign, and economical process methodologies for their large-scale conversion into industrially viable products, be it bulk or intermediate chemicals, pharmaceuticals, or polymeric organic materials. General conceptual formulations towards this goal are available [2–4,162]; yet, their straightforward implementation, as of now, is exceedingly slow. Thus, it is essential that national and supranational funding institutions – in Europe, the corresponding EU bodies and/or ERRMA (European Renewable Resources & Materials Association [163]) – play a considerably more dynamic role than heretofore. One decisive action, of course, is the generous funding, not only of applied, but of basic research activities in this area, and this over a considerably broader time frame – 5–10 years for promising projects rather than an expectational horizon to deliver marketable products within 3–5 years.