

1 Introduction
Nowadays, there are some major reasons which stimulate research in the field of the sulfoxidation catalytic reactions. Firstly, the oxidation of organic sulfides to sulfoxides or/and sulfones is useful to make intermediates for the synthesis of various chemically and biologically significant molecules [1,2]. Secondly, there is an interest to remove sulfur compounds, from fuels and organosulfur contaminated industrial effluents in order to satisfy the new environmental legislations [3]. For instance, the catalytic oxidative desulfurization (ODS) combined with extraction can be considered as a promising complementary technology to the current hydrodesulfurization process for reducing sulfur in gasoline and diesel fuels [4–7]. In the ODS process, the sulfides and thiophene derivatives are converted into their corresponding sulfoxides and sulfones, which are preferentially extracted due to their increased polarity.
Generally, the sulfoxidation reactions are carried out in the liquid phase, under mild conditions, in the presence of oxidizing agents, such as organic peroxyacids, peroxides, hydroperoxyides, and hydrogen peroxide. Aqueous hydrogen peroxide is an ideal oxidant owing to its high effective-oxygen content, cleanliness (it produces only water as by-product), and enough safety in storage and operation. Consequently, in the last years the importance of hydrogen peroxide as a “green” oxidizing agent has grown considerably [8–10].
Because hydrogen peroxide is a soft oxidizing agent, often it has to be activated by an electrophile catalyst. The catalytic oxidation with hydrogen peroxide can be carried out either in homogeneous [11] or heterogeneous catalysis [12–15]. With respect to the green chemistry principles, the ideal catalytic systems belong to the class of solid materials under heterogeneous catalysis. Consequently, efforts are devoted to the development of processes using highly robust and recyclable catalysts which provide higher atom utilization and minimized pollution levels using greener ingredients. In the last years selective oxidation of sulfur-containing compounds by H2O2 has been carried out using a large number of redox solid catalysts: Ti-containing zeolites (TS-1, TS-2, Ti-beta) [16–19], Ti–mesoporous silica (Ti–HMS, Ti–MCM-41) [20–23], V- or Mo-containing molecular sieves [17,24], V2O5/TiO2 [25] layered double hydroxide (LDH) [26,27].
In our group, significant efforts have been dedicated in the recent years to develop new heterogeneous catalysts for both olefins [28–30] and S-containing compound [26,31–37] oxidation with hydrogen peroxide. Two families of catalytic materials were mainly investigated: (i) LDH-based catalysts and (ii) Ti-containing microporous and mesoporous materials. Recently we reported that layered double hydroxides with Mg2+ and Al3+ cations in the brucite-like layer and WO42−, W7O246−, MoO42−, Mo7O246−, V2O74− or V10O286− anions in the interlayer gallery are active catalysts for the oxidation of sulfides/thiophene derivatives [35,36] and dimethylsulfoxide [37] with H2O2. These results will be briefly reviewed in this article.
We have also studied the oxidation of various sulfides and thiophene derivatives by hydrogen peroxide on TS-1, Ti-beta and Ti–MCM-41, using different solvents. TS-1, the original Ti-containing molecular sieve, was the best catalyst for the selective oxidation of small size molecules, but its activity strongly decreased when bulkier compounds were to be oxidized [31,32]. In contrast, catalysts with large pore size, such as Ti-beta and Ti–MCM-41 were very active for the mild oxidation of large size molecules, such as aryl-sulfides and dibenzothiophene [34]. Among the common organic solvents, the highly polar compounds like lower alcohols and acetonitrile showed the most favourable effect in the oxidation reactions with H2O2.
In this paper we report for the first time about the catalytic potential of the Ti–MCM-48 mesostructured material for the oxidation of various S-containing organic compounds. MCM-48 features the same high surface area as MCM-41 but possesses a three-dimensional array of pores within a 20–30 Å diameter range, which is beneficial with respect to molecular diffusion. Compared to the hexagonal Ti–MCM-41 mesoporous material, only few efforts have been focused on the synthesis and catalytic applications of the cubic Ti–MCM-48 phase [28–48].
2 Experimental
2.1 Catalysts' preparation and characterization
The parent MgAl-LDH (denoted MA-NO3) was prepared by pH controlled co-precipitation of Mg(NO3)2·6H2O and Al(NO3)3·9H2O, according to the method reported previously [35]. Me-containing LDH catalysts (Me = W, V, Mo), denoted as W-LDH, V-LDH and Mo-LDH, were obtained by direct ion exchange of the crystalline MA-NO3 material, using Na2WO4·2H2O, NaVO3 and Na2MoO4·2H2O, respectively [36,37]. Except for the nitrogen adsorption measurements, the materials were used without further thermal treatment.
Ti–MCM-48 was prepared by direct hydrothermal synthesis, using an original method based on fumed silica (Aerosil 200) as silicon source. First, a gel was obtained by adding successively SiO2 and titanium isopropoxide to an aqueous solution containing NaOH and hexadecyl trimethyl ammonium bromide (CTAB). The mixture was stirred for 2 h at 50 °C and then autoclaved without stirring at 150 °C for 24 h. The molar composition of the gel was 1 SiO2:0.38 NaOH:0.175 CTABr:0.0195 TiO2:120 H2O. We note that parallel synthesis performed at longer hydrothermal treatment resulted in lamellar structures. The resulting solid was recovered by filtration, extensively washed with deionised water until neutral pH and dried overnight at 80 °C. The organic template was removed from the as-synthesized material by calcinations under an optimized program. Thus, the solid was heated slowly (0.5 °C min−1) up to 500 °C under nitrogen and maintained at this temperature for 1 h under N2 and 5 h under air.
Suitable physicochemical techniques (chemical analysis, X-ray diffraction (XRD), diffuse reflectance ultraviolet spectroscopy (DRUV), thermogravimetric analysis (TGA) and nitrogen sorption) were used to evaluate the textural and morphological properties, as well as to confirm the presence and the state of the metal species in LDH and MCM-48 frameworks. Powder XRD patterns were collected on a Bruker AXS D8 diffractometer using Cu Kα radiation. The nitrogen adsorption/desorption isotherms (−196 °C) were measured with a Micromeritics ASAP 2010 automatic analyzer. DRUV spectra were recorded on a Varian Cary 05 E UV–vis–NIR spectrometer equipped with an integrating sphere using BaSO4 powders as the reflectance standard. Thermogravimetric analysis was carried out in a Netzsch TG 209C thermobalance. About 15 mg of catalyst was loaded, and the airflow used was 50 cm3 min−1. The heating rate was 5 °C min−1 and the final temperature was 900 °C.
2.2 Typical procedure for oxidation in liquid–solid system
The catalytic experiments were carried out at atmospheric pressure in a three-neck glass batch reactor (50 mL), equipped with magnetic stirrer, thermometer, and condenser, immersed in a thermostated oil bath. In a typical run, the solid catalyst (50 mg) was suspended under stirring (800 rpm) in a mixture containing the organic substrate, hydrogen peroxide (30 wt.% aqueous solution), and 15 mL solvent. Blank experiments were carried out by mixing the reactants in absence of catalyst. Samples of the reaction mixture were periodically withdrawn, filtrated and analyzed on a Varian 3900 chromatograph equipped with a capillary column (DB-1, 60 m, 0.20 mm i.d., 0.25 μm film thickness). The hydrogen peroxide concentration was measured by standard iodometric titration.
3 Results and discussion
3.1 Characterization of catalysts
3.1.1 LDH-based catalyst
The main physicochemical characteristics of solids are summarized in Table 1. They prove that all the oxoanions species were successfully intercalated into the interlayer gallery. Thus, characteristic large bands attributed to different oxospecies [36] were observed in the UV spectra of the W-, V- and Mo-containing LDHs. These spectra were deconvoluted (using Gaussian curves) and two bands were identified for each metal, proving the intercalation of two main oxoanions species (Table 1).
Main characteristics of LDH samples.
| Sample | Mg/Al/Mea (atomic ratios) | UV bands (nm) | Oxo-species | SBET (m2 g−1) | Weight loss 250–500 °Cb (%) | |
| Nature | % | |||||
| MA-NO3 | 2.00/1.00 | 16 | 35.8 | |||
| W-LDH | 1.99/1.00/0.5 | 220 | WO42− | 71 | 25 | 17.6 |
| 260 | W7O246− | 29 | ||||
| V-LDH | 1.43/1.00/0.7 | 310 | V2O74− | 30 | 50 | 11.5 |
| 430 | V10O286− | 70 | ||||
| Mo-LDH | 1.6/1.00/0.6 | 255 | MoO42− | 39 | 45 | 13.8 |
| 325 | Mo7O246− | 61 |
a Me = W, V or Mo.
b By TGA.
The textural properties of parent hydrotalcite were significantly changed upon the exchange process with oxometalates anions. Thus, the BET specific surface areas of W-LDH, V-LDH and Mo-LDH are 25, 50 and 45 m2 g−1, while for the parent MA-NO3 it is only 16 m2 g−1 (Table 1). The N2 adsorption–desorption isotherms indicated that the pore volumes for the W-, V- and Mo-intercalated LDHs were also greater that that of the precursor.
In TGA, the parent and oxometalates-containing LDH samples exhibited two important stages of weight loss, which are the characteristics for the layered double hydroxides. The first stage, between 30 and 250 °C (which is obviously attributed to the removal of interlayer water molecules and adsorbed water molecules) was around 14.0% for all the materials. For the parent layered double hydroxides (MA-NO3 sample), a second stage high weight loss is recorded between 250 and 500 °C, and it corresponds to the dehydroxylation of brucite-like layers and the decomposition of nitrate ions. This weight loss is nearly three times larger than those obtained for the exchanged LDHs (Table 1). In fact, the weight loss observed for the exchanged LDH samples (250–500 °C) corresponds to the dehydroxylation of the brucite-like layers, the removal of residual nitrate ions and especially to the decomposition of metal-containing anions into metal oxides (which remain in the solid phase).
As previously reported [36], the XRD patterns of the exchanged LDHs showed that the basic lamellar structure of the host solid was kept, but the basal distances increased, confirming the replacement of smaller nitrate anions by larger anion species. The basal spacing, calculated from the reflection of [003] plane, was 8.9, 10.5, 10.2 and 11.5 Å, for the MA-NO3, W-LDH, V-LDH and Mo-LDH, respectively.
3.1.2 Ti–MCM-48 catalyst
XRD patterns for the as-synthesized (Ti–MCM-48 nc) and calcined (Ti–MCM-48) materials are reported in Fig. 1. The sets of eight peaks ((211), (220), (321), (400), (420), (322), (422) and (431)) in the low-angle XRD patterns indicate uniform pores arranged in a typical system of a cubic symmetry, which confirms that the calcinations at 500 °C did not change the structure of mesostructured material. Moreover, after calcination, the XRD intensities of the sample are increased, indicating some improvement of the pore ordering, through the surfactant removal. Additionally, the DRX peaks shift to bigger angles after calcination, because of the shrinkage of structure. This modification can be explained by change in the connectivity of the SiO4 network during calcinations.
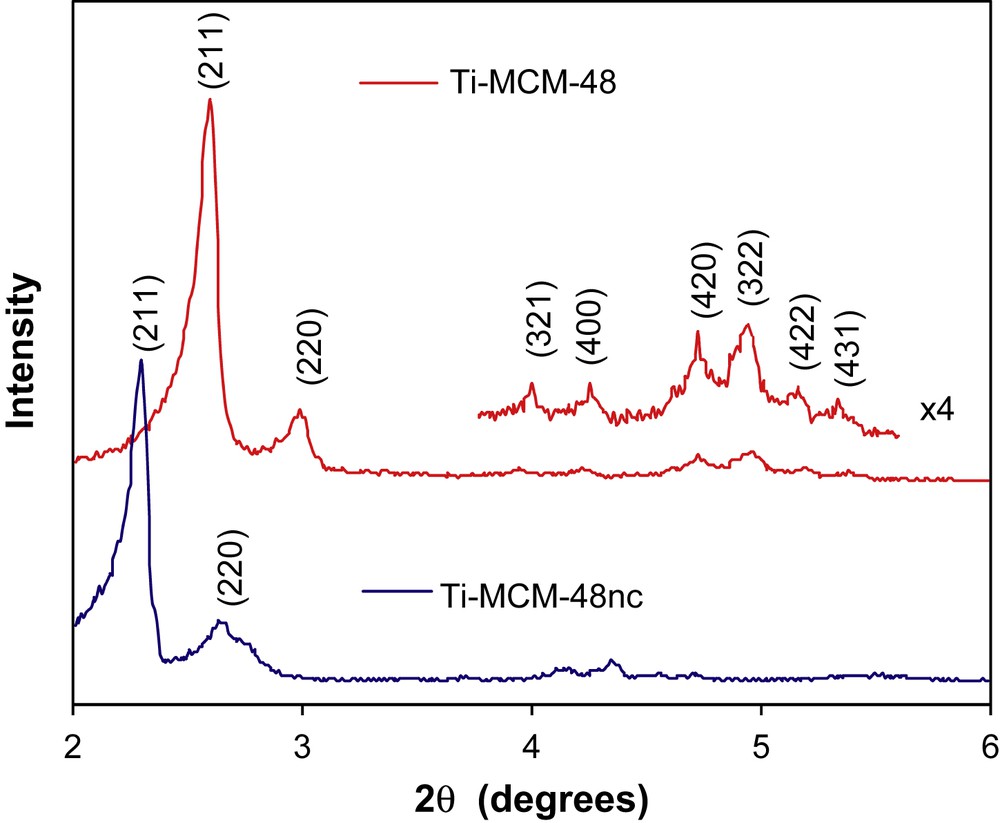
XRD patterns for the non-calcined (Ti–MCM-48 nc) and calcined (Ti–MCM-48) samples.
The N2 adsorption–desorption isotherm and the corresponding BJH pore size distribution based on the adsorption branch for the Ti–MCM-48 sample are shown in Fig. 2. The isotherm is a typically reversible adsorption isotherm of type IV with a sharp step between relative pressure p/p0 = 0.2 and 0.3, indicating that the pores' size is extremely uniform. A very narrow pore size distribution with the average pore diameter of 2.9 nm was obtained using the BJH method (inserted graph in Fig. 2). The surface area and pore volume deduced from the nitrogen sorption measurement were 960 m2 g−1 and 0.69 cm3 g−1, respectively.
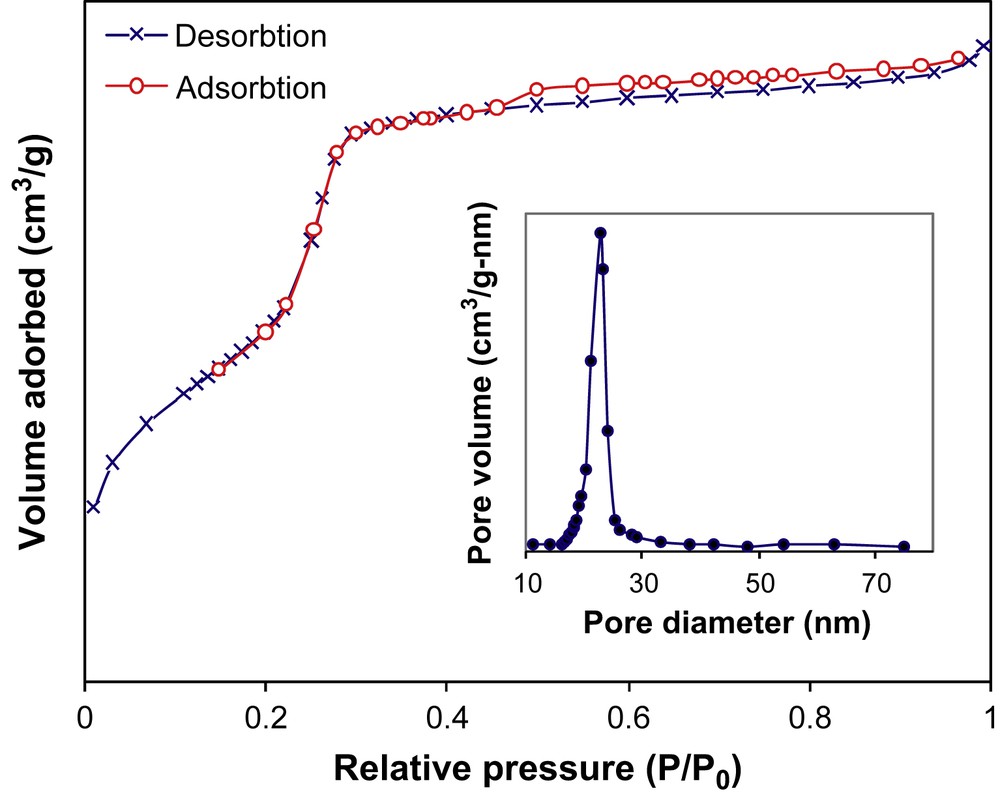
N2 adsorption–adsorption isotherm at −196 °C for Ti–MCM-48.
UV–vis spectroscopy is commonly used for characterizing the coordination state of Ti sites in titanosilicates. The DRUV–vis spectra of the as-synthesized and calcined samples are compared in Fig. 3. Clearly, in the non-calcined sample, which showed a well defined band centred at 210–215 nm, there were essentially Ti atoms in tetrahedral environments. It is well known that this band is attributed to oxygen-to-metal charge transfer at isolated tetrahedral titanium(IV) centers [49,50].
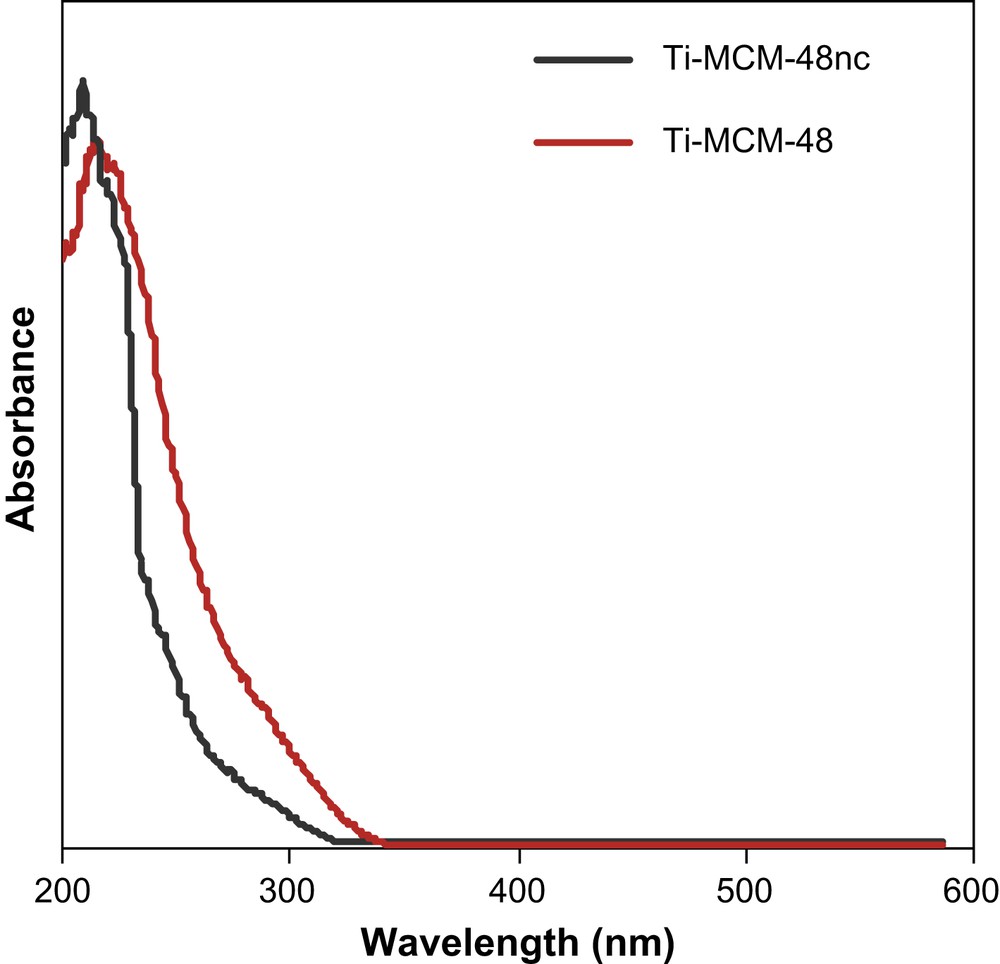
UV–vis spectra of calcined (Ti–MCM-48) and not calcined (Ti–MCM-48 nc) samples.
On the other hand, a slight shift of the band towards higher wavelengths was observed for the calcined sample. The shoulder at 280 nm corresponds to partially polymerized penta- and hexa-coordinated Ti [51–53]. The absence of bands above 300 nm indicates that extra-framework Ti species like TiO2 were negligible in these materials.
3.2 Oxidation of model S-containing compounds
As mentioned in the introduction, the interest for the oxidation reaction of the sulfur-containing organic compounds with H2O2 is multiple: organic synthesis, fuel desulfurization and wastewater decontamination. The catalytic behavior for these applications of the modified LDHs and Ti–MCM-48 was studied by the oxidation of three model molecules (methyl-phenyl-sulfide (MPS), dibenzothiophenes (DBT) and dimethylsulfoxide (DMSO)), as shown in Table 2.
Sulfoxidation reactions of model molecules.
| Reaction | Potential application |
| Sulfoxides and sulfones synthesis | |
| Fuels catalytic oxidative desulfurization | |
| DMSO removal from wastewater |
The oxidation of MPS (a large organic molecule) is a suitable model reaction for the sulfides' conversion into sulfoxides and sulfones. DBT is a major sulfur-containing compound present in liquid hydrocarbon fuels. Oxidative desulfurization is considered as a promising alternative/complementary technology to HDS (current fuels desulfurization technology) for deep fuels desulfurization [4–7,34,35]. DMSO is widely used as solvent in laboratories and industry (e.g., solvent extraction, semiconductors, polymers, and dyes manufacturing processes) and, as a consequence, large amounts of waste containing DMSO resulted. An effective method for removing DMSO from wastewater could be the mild catalytic oxidation of DMSO with H2O2 into dimethylsulfone, a biodegradable compound [37].
The oxidation reaction has been carried out using dilute hydrogen peroxide (aqueous solution, 30%) in the presence of acetonitrile as solvent. The organic solvent forms a single phase with the substrate and hydrogen peroxide solution. The advantage of the oxidation with hydrogen peroxide can be mainly related to the very mild reaction conditions (low temperature and pressure) and to the use of a “clean” oxidation agent.
Under these conditions, the oxidation reaction of phenyl sulfide led to the corresponding sulfoxide and sulfone as major products (Table 2). On the other hand, the kinetic profile of the benzothiophene oxidation indicated only the formation of corresponding sulfone, even for low levels of conversion of the substrate. As expected, DMSO was selectively converted into dimethylsulfone. The direct decomposition of hydrogen peroxide (H2O2 → H2O + 1/2O2) was observed during the oxidation process, but the efficiency of H2O2 (defined as 100 × mol of H2O2 consumed in the formation of sulfones/mol of H2O2 converted) was higher than 95% for all the oxidation tests.
3.2.1 W-LDH, V-LDH and Mo-LDH catalysts
The results obtained in the MPS and DBT oxidation at 40 °C, without catalyst (w.c.) and using fresh W-LDH, V-LDH, Mo-LDH and MA-NO3 samples as catalysts, in the presence of acetonitrile as solvent, are plotted in Fig. 4.
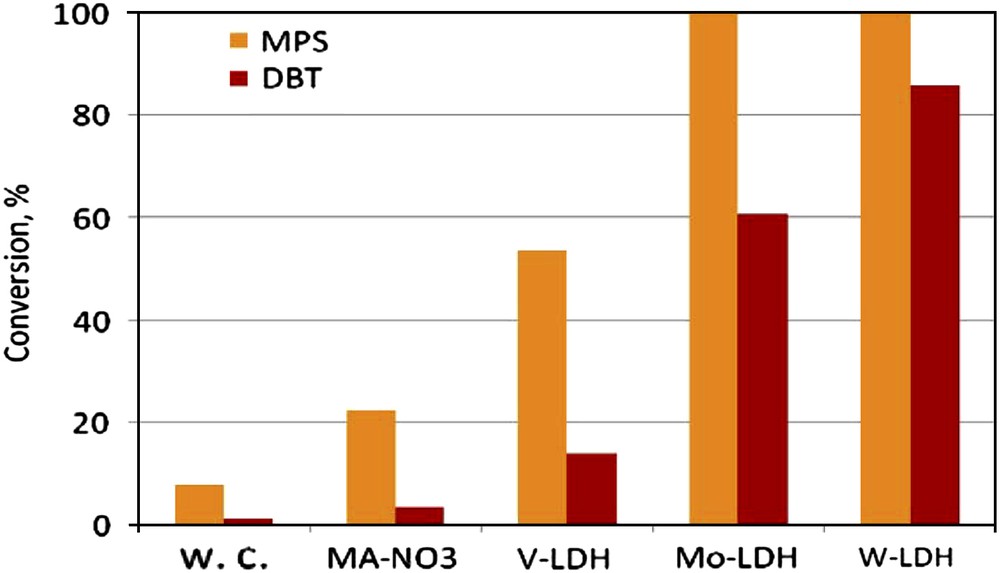
Effect of the catalyst on the organic sulfur compounds conversion; T = 40 °C, H2O2/substrate = 5 (mol/mol); Solvent = MeCN, t = 45 min for MPS, t = 3 h for DBT.
The blank experiments performed in absence of catalyst showed very low conversions for both MPS and DBT. Low conversions (1–11%) were also obtained in the presence of the MA-NO3 sample which does not contain oxometalates species. The conversion of organic sulfur compounds substantially increased when exchanged LDH materials were used as catalysts, showing that the W, Mo and V-oxospecies have an essential contribution to the activation of the oxidizing agent. For example, conversions up to 100% were obtained in the MPS oxidation over Mo-LDH and W-LDH catalysts only after 45 min of reaction. Generally, the conversion strongly depends on the metal nature. The order of the catalytic performance (evidently in the case of the less reactive DBT molecule) was: W-LDH > Mo-LDH > > V-LDH. These results indicate that the intrinsic nature of active sites (oxospecies nature) is a very important parameter for the oxidation activation.
In order to stand out the effect of the reaction temperature on the MPS, DBT and DMSO oxidation with H2O2, it, W-LDH, was used as catalyst. The initial rates and the conversion of the organic substrates are summarized in Table 3. As expected, both the initial rate and the conversion of MPS increased when the reaction temperature increased.
Effect of the reaction temperature on the MPS, DBT and DMSO oxidation over W-LDH catalyst.a
| Temperature, °C | MPS | DBT | DMSO | |||
| Ri | Conversion (%) after 20 min | Ri | Conversion (%) after 180 min | Ri | Conversion (%) after 20 min | |
| 30 | 9 | 92 | 10.6 | 68 | ||
| 40 | 14 | 97 | 0.2 | 46 | 21.9 | 94 |
| 50 | 19 | 100 | 0.4 | 58 | 26.0 | 100 |
a Reaction conditions: H2O2/MPS = 2:1 (mol/mol), H2O2/DBT = 5:1, H2O2/DMSO = 1.7:1; solvent = acetonitrile (for MPS and DBT) and water (for DMSO); Ri = initial rate (mol l−1 h−1 gcat−1).
The results in Fig. 4 and Table 3 show that the MPS and DMSO are oxidized much faster than the DBT, confirming that the reactivity of sulfur compounds is correlated to the nucleophilicity of the sulfur atom. It is known that organic sulfides are oxidized by hydrogen peroxide via a heterolytic process involving the nucleophilic attack of the sulfur atom on oxygen [54,55]. Compared to a sulfur atom in sulphide and sulfoxide, the sulfur atom in thiophene derivatives is less nucleophilic, since its electron pairs participate in the aromatic delocalization and are therefore less available for donation.
A plausible catalytic cycle for the oxidation of sulfides to sulfoxides, which is the first stage of the oxidation process, over W-containing LDH catalyst, is depicted in Scheme 1 [36,56]. As known, the role of the metal atom (W, V or Mo) is to form peroxo-metal species which are able to activate the H2O2 molecule. This mechanism involves in the first step the easy formation of the peroxotungstate species (2) after the interaction of the tungstate anion (1) with the hydrogen peroxide. This species facilitates the transfer of the electrophilic oxygen to sulfide (3), forming the sulfoxide. Consequently, the nucleophilicity of substrate has an important role and thus the order of reactivity mentioned above could be explained.
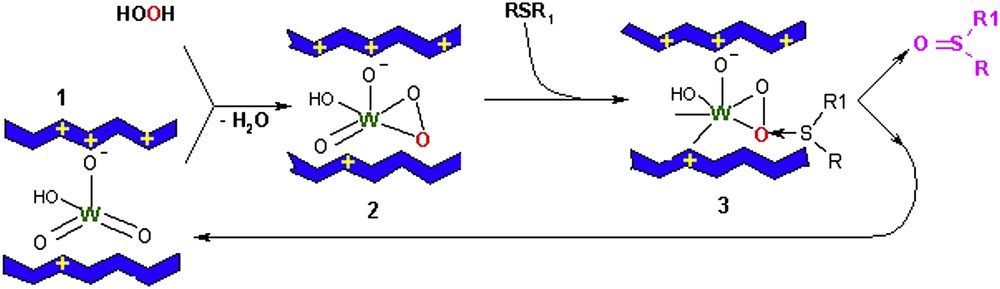
Proposed mechanism for the sulfide oxidation with H2O2 over W-LDH catalyst.
3.2.2 Ti–MCM-48 catalyst
The catalytic potential of Ti–MCM-48 was evaluated in the oxidation reaction of MPS, DMSO and DBT with H2O2 (30 wt.% aqueous solution), at 40 °C in the presence of acetonitrile as solvent. MPS was also oxidized with dry tert-butyl hydroperoxide (5.5 M in decane). Low DBT conversion was achieved at this temperature (only 10% after 3 h), but promising results were obtained in the MPS and DMSO oxidation (Fig. 5). The Ti-containing mesoporous materials (Ti–MCM-41, Ti–MCM-48) are known as hydrophilic and less active in the oxidation with aqueous H2O2 solutions, due to a competitive adsorption of the water molecules and the reactant molecules on the active site [57]. Corma et al. [58] proposed two strategies to improve the activity of Ti–MCM-41: (i) silylation of the surface, which is making the surface more hydrophobic; and (ii) using dry TBHP as oxidant. As expected, the catalytic activity of Ti–MCM-48 in anhydrous medium (MPS/TBHP/acetonitrile) was very good (Fig. 5). Surprisingly, this catalyst proved interesting activity in the oxidation of DMSO and MPS even with H2O2. For all tests, an excellent efficiency (>95%) of the H2O2 towards the catalytic oxidation was observed. However, under the same oxidation conditions, the catalytic potential of the Ti–MCM-48 sample was interior to that of the W-LDH catalyst (Table 3, Fig. 5).
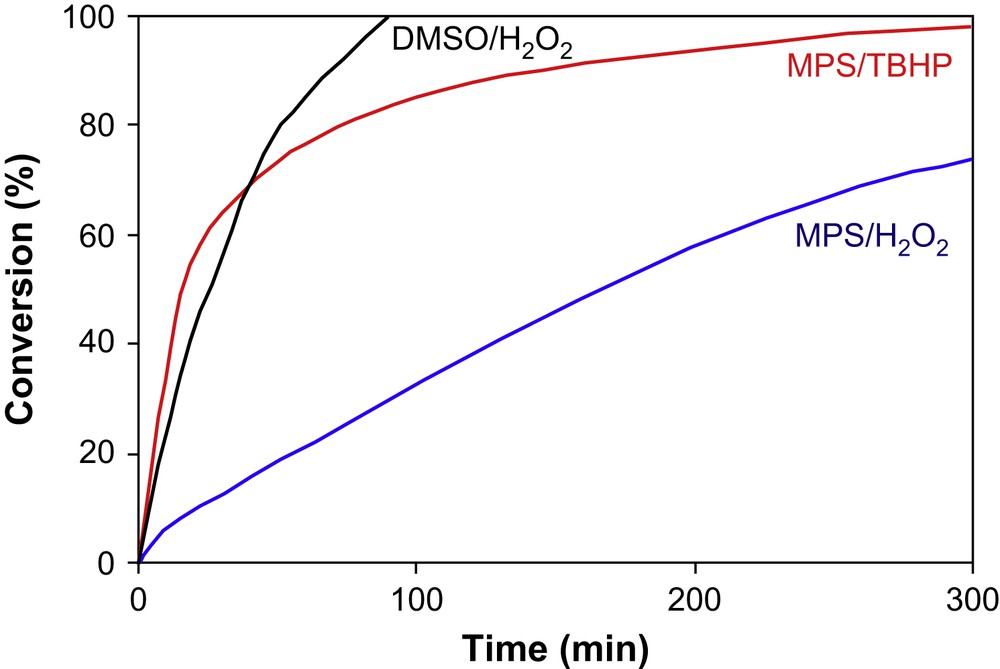
Kinetic profiles for the MPS and DMSO oxidation; Reaction conditions: Oxidant/Substrate = 2:1 (mol/mol), Solvent = acetonitrile, T = 40 °C, catalyst = Ti-MCM-48.
4 Conclusion
W-, Mo- and V-containing LDHs were prepared by direct anionic exchange of MgAl-NO3 LDH. Intercalation of WO42−, W7O246−, MoO42−, Mo7O246−, V2O74− and V10O286− species in the hydrotalcite-like structure has been successfully realized, as confirmed by XRD, N2 adsorption, TGA, and DRUV spectroscopy. All the materials exhibited good catalytic potential for the conversion of sulfides, thiophene derivatives and sulfoxides with dilute H2O2 aqueous solution, at low temperature. In the case of LDH-based materials, the catalyst performance depended on the nature of the anion species in the interlayer gallery, the W-LDH sample being more active than the V-LDH and Mo-LDH samples.
Ti–MCM-48 was successfully prepared by the direct synthesis using fumed silica as silicon source, without forming extra-framework TiO2. Here we investigated for the first time the catalytic potential of this catalyst in the sufoxidation reaction. Ti–MCM-48 proved good behavior as catalyst for the mild DMSO and MPS oxidation with both TBHP and H2O2 oxidants.