

1 Introduction
As early as 1975, de Gennes predicted and discussed qualitatively the extraordinary mechanical and optical properties that should be expected from nematic main-chain liquid crystalline elastomers (MCLCEs), where the mesogenic units are connected within the polymer backbone to build up the polymer network [1].
One of the most outstanding features of nematic elastomers is a reversible change in length at the nematic-to-isotropic phase transformation for monodomain samples that are macroscopically ordered with respect to the director. This change in shape is due to the coupling of the liquid crystalline order with the polymer chain conformation of the network. The coupling has been investigated in detail for LC side-chain elastomers [2,3], where the mesogenic moieties are attached as side groups to the monomer units of the network. However, only few investigations have been performed on main-chain elastomers, as suitable systems are much more difficult to realize chemically. Main-chain LC polymers typically exhibit high clearing temperatures and tend to crystallize [4]. These properties are desired e.g. to obtain highly ordered fibers with exceptional mechanical properties [5,6]. However, for detailed investigations of the mechanics of networks in the nematic state with our experimental setup, clearing temperatures not too far above ambient temperature are required. In order to perform reversible measurements at equilibrium conditions, no decomposition of the organic material should occur.
Several approaches are known from literature to realize crosslinked main-chain polymers. Mainly linear polymers containing a photoactive moiety have been prepared [5–10]. These functional linear polymers were crosslinked after processing. Another method described in the literature is the preparation of linear polymers bearing functional groups that are chemically crosslinked following a conventional polymerization technique [11–14]. Crosslinked materials with a high crosslinking density, the so-called thermosets, have been synthesized in a one-pot reaction. Here the repeating unit of the linear polymer and the crosslinker were reacted together in order to directly form the densely crosslinked sample [15–17]. The one-pot approach has been used as well to obtain weakly crosslinked materials by adding a flexible spacer as co-monomer to the reaction mixture [18–22].
Up to now only few weakly crosslinked nematic networks are known that have been synthesized from main-chain polymers [9,13,14,19,21,23–25]. The first nematic monodomain elastomers with a permanently fixed orientation of the director were presented by Bergmann [13,26]. They enable the coupling behavior between the nematic order and the chain conformation of the network strands to be investigated. For these MCLCEs, a much stronger coupling is expected than for side-chain (SC) elastomers, where the mesogenic groups are decoupled from the network chains by flexible spacers.
Thermoelastic experiments reveal temperature dependent changes in length in the vicinity of the nematic-to-isotropic phase transformation up to 300% [14,23]. Even more pronounced effects are changes in length of up to 500%, as observed in thermoplastic elastomer fibers [27]. The coupling between the LC phase and the network can be quantified by the coupling coefficient U defined in the Landau-de Gennes theory.
If the elastomer synthesis is performed in the isotropic state – e.g. in the isotropic phase at elevated temperatures, or in the presence of an isotropic solvent –, and the sample is subsequently cooled or deswollen avoiding any external field that might cause orientation, a stable polydomain (PD) structure is observed. This nematic polydomain network exhibits macroscopically isotropic properties. It is well known from liquid crystalline SC elastomers that applying external fields on a nematic polydomain network causes a macroscopic orientation of the director that leads to a monodomain (MD) structure [28]. This orientation process results in a plateau in the stress/strain relation that reflects a very low modulus (“soft elasticity”) [29]. As the monodomain is formed, the stress increases again and displays the modulus of the anisotropic network in the direction of the director. The orientation behavior of MCLCEs clearly differs from that of liquid crystalline SC elastomers with respect to the strain required to obtain a monodomain sample. Furthermore, the monodomain structure can already be obtained in the plateau region of the stress/strain curve [13,14]. Additional effects on the mechanical behavior of the elastomers, such as the removal of hairpins, have to be considered as well [30–33].
In the first part of this paper, we summarize some recent synthetic approaches to obtain nematic main-chain elastomers with nematic-to-isotropic phase transformations at temperatures suitable for long-term mechanical measurements. In the second part, the networks are analyzed with respect to deducing the coupling constant U from thermoelastic experiments and measurements of the nematic order parameter as a function of the temperature. Finally, the mechanical orientation behavior of polydomain MCLCEs is investigated.
2 Synthesis of the main-chain liquid crystalline elastomers
For MCLCEs that can be synthesized and examined easily, several aspects have to be considered. Due to the rigid rod-like mesogenic units in the polymer backbone, these systems tend to crystallize and show clearing temperatures where the polymer already starts decomposing. Furthermore, homo-polymers with a regular arrangement of the monomer units along the backbone tend to exhibit smectic or other highly ordered mesophases [4]. Strategies to overcome these problems are the incorporation of kinks into the mesogens, laterally attached side-chains that act as plasticisers, or the use of flexible spacer chains between the rigid mesogenic units. These procedures are described extensively in the literature [34,35].
In 1997, Bergmann [13] applied these concepts to the synthesis of the first nematic MCLCE. He started from a semi-flexible polyether (Scheme 1), the properties of which had been reported in detail by Percec et al. in 1991 [36].
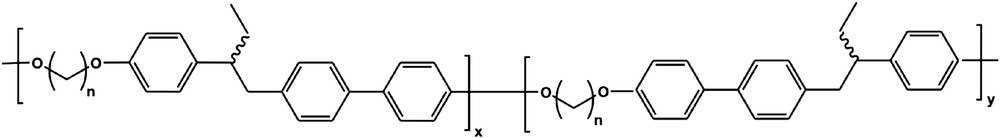
Percec's [36] polyether, incorporating constitutional and conformational isomers of the repeating unit.
The free rotation around the central C–C bonding between the biphenyl unit and the phenyl ring gives rise to two different conformations of the mesogen. This brings about a comparatively low nematic-to-isotropic phase transformation temperature Tn,i of around 120 °C, and suppresses the formation of a smectic phase. The laterally attached ethyl spacer hinders crystallization and decreases both the clearing temperatures and the glass transition. It also creates chirality within the repeating unit and results in four different constitutional possibilities. These factors additionally decrease the crystallization tendency and enhance the solubility in standard organic solvents. With an alkyl spacer length of 10 methylene units, the system shows a broad region of the nematic phase and was chosen for the synthesis of elastomers.
Scheme 2 illustrates the synthetic approach to the elastomers. A functional linear polyether was synthesized, substituted on both ends with a rigid biphenyl moiety containing a vinyl end group. This polyether was then allowed to react with a cyclosiloxane crosslinker via platinum catalysis to yield the main-chain elastomers MD-1 and PD-1. The monodomain elastomers MD-1 were synthesized according to the well-known two-step reaction procedure [37], where a mechanical field is applied to the network just above the gelation point and the reaction is completed in the loaded state. The polydomains PD-1 were synthesized without a mechanical field. It needs to be noted that the exact stoichiometry for the reaction of the polymer with the cyclic crosslinker is hard to determine. Both the measurement of the concentration of functional groups in the pre-polymer and the weighting of the volatile crosslinker bring about considerable errors. Therefore, three different polydomain elastomers PD-1a to PD-1b with slight variations of the crosslinker concentration were synthesized, the objective being to minimize the amount of material not incorporated into the network, the so-called soluble contents.
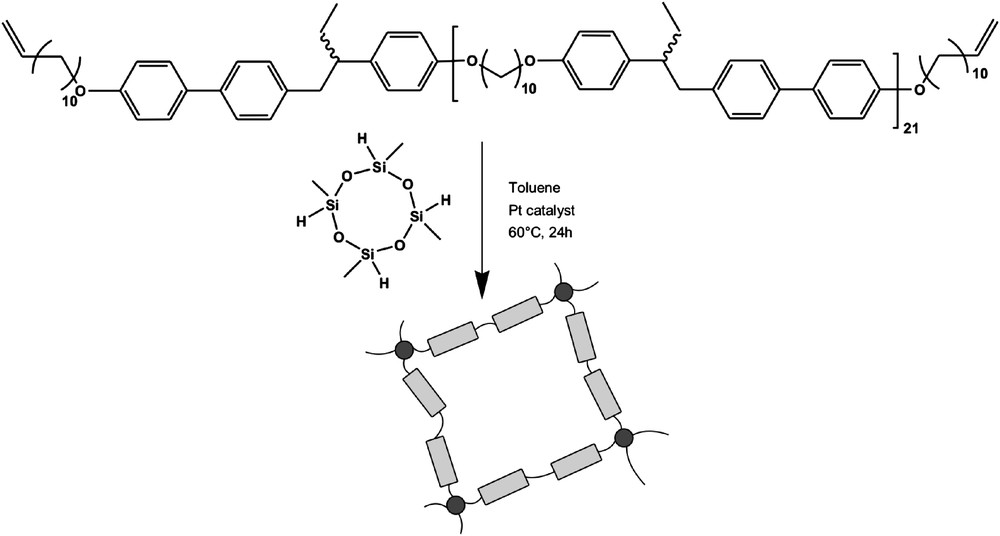
Elastomer synthesis scheme of the systems MD-1 (monodomain) and PD-1 (polydomain).
Table 1 summarizes the thermal properties and swelling behavior of the systems investigated (see chapters 3 and 4). As expected, MD-1 displays a large swelling anisotropy Σ (with Σ = (aperp/apar) − 1, where aperp is the degree of swelling perpendicular to the director and apar is that one parallel to the director) while the polydomain samples swell isotropically. The sample PD-1a has a remarkably high degree of swelling q, indicating a weakly crosslinked structure. This is supported by the large soluble contents of this sample, and by the temperatures of the glass transition Tg and phase transformation Tn,i, which are close to those of the non-crosslinked polymer (where Tg = 28 °C and Tn,i = 99 °C).
Thermal and swelling properties of the MD-1 (monodomain) and PD-1 (polydomain) systems
| Tg | Tn,i | q | Σ | Sol. cont./% | |
| MD-1 | 39 | 122 | 3.2 | 2.8 | 2 |
| PD-1a | 23 | 96 | 8.4 | 0 | 61 |
| PD-1b | 36 | 124 | 1.7 | 0 | 3 |
| PD-1c | 37 | 114 | 3.2 | 0 | 3 |
In order to examine the influence of an LC elastomer's chemical constitution on its physical properties, Wermter [14] developed a co-elastomer system, where he used Bergmann's vinyl-functionalized polymer as a main-chain crosslinker for a side-chain LC polymer. Thus, he was able to control both the crosslinking density and the degree of polymerization of the crosslinker. Also, the side-chain component acts as plasticizer, further decreasing the phase transition temperature. Scheme 3 shows the general synthetic concept of these systems, where MD-2 (monodomains) and PD-2 (polydomains) are synthesized from the side-chain mesogen W1, the main-chain component W2, and the poly(methylhydrogensiloxane) by a platinum catalyzed addition reaction. Table 2 summarizes the composition and thermal properties of the samples, which will be discussed in chapters 3 and 4. With an increasing fraction of main-chain mesogens, both Tg and Tn,i increase. For the monodomain samples, the swelling anisotropy Σ increases as well. The degree of swelling q is almost constant for all samples. It should be noted that the soluble contents of the elastomers were below 2%, which indicates a clean polyaddition reaction.

Synthesis concept of the co-elastomer systems MD-2 (monodomain) and PD-2 (polydomain).
Composition, thermal and swelling properties of the MD-2 (monodomain) and PD-2 (polydomain) systems
| Ratio of W2 in the sample/% | Length of W2/monomer units | Tg | Tn,i | q | Σ | |
| MD-2a | 20 | 13 | 7 | 89 | 6.9 | 1.8 |
| MD-2b | 28 | 13 | 8 | 86 | 6.3 | 2.9 |
| MD-2c | 46 | 20 | 13 | 95 | 6.5 | 4.1 |
| MD-2d | 57 | 26 | 19 | 96 | 6.8 | 4.5 |
| PD-2a | 20 | 13 | 3 | 89 | 6.9 | 0 |
| PD-2c | 46 | 20 | 11 | 96 | 6.5 | 0 |
| PD-2e | 77 | 30 | 24 | 94 | 6.9 | 0 |
A different approach to obtain main-chain LCEs was proposed by Donnio et al. [19]. It is based on the chemistry of linear polymers investigated by Aguilera et al. [38]. Here, dimethyl-siloxane units are introduced to lower the phase transition temperatures. Instead of starting from pre-synthesized main-chain polymers, this synthesis is based upon a simple one-pot polyaddition reaction via hydrosilylation of a divinyl-substituted mesogenic monomer with tetramethyldisiloxane (TMDS) and a cyclic siloxane crosslinker (HD5). This elegant approach facilitates the access to MCLCEs, since it merely requires the preparation of a divinylic mesogenic monomer. However, the first networks prepared by this approach show smectic phases at low temperatures. This is due to the symmetric structure of the mesogenic units and the tendency of the siloxane units to phase-separate.
By varying the structure of the mesogens and the length of the siloxane units, Brandt elaborated Donnio's approach (Scheme 4). Using mesogens with only two aromatic rings shift the phase transition temperature to lower values. In order to avoid the segregation of the siloxane units and the formation of a smectic phase, short vinyl chains and short-chained siloxane co-monomers were used. By co-polymerization of mesogens with different lengths of the vinyl chains, the tendency to arrange in a layered structure was additionally decreased. With these concepts, elastomers with a broad nematic phase, which exhibit practical and experimentally accessible temperatures, were realized.

Synthetic concept of the one-pot approach with two-core mesogens B1–B4.
Table 3 summarizes the composition, thermal and swelling properties of the systems MD-3 (monodomain) and PD-3 (polydomain), which will be described further in chapters 3 and 4. The sample MD-3a only shows a narrow nematic phase above a smectic-C phase and a smectic phase of higher order. MD-3b and PD-3b, which contain mesogens with shorter spacers, exhibit a nematic phase over a broader temperature region, but still have a smectic-C phase at low temperatures. This SC phase is successfully suppressed in MD-3c by co-polymerization of mesogens with a large difference in the vinyl chain length.
Composition, thermal and swelling properties of the MD-3 (monodomain) and PD-3 (polydomain) systems
| Mesogen | Cross-linker/% | Phase behavior | q | Σ | Sol. cont./% | |
| MD-3a | B3 | 8 | g 8 Sx 21 SC 71 n 73 i | 6.2 | 1.7 | 20 |
| MD-3b | B2 | 10 | g 3 SC 31 n 48 i | 9.2 | 1.7 | ∼20 |
| MD-3c | B1:B4 (1:1) | 8 | g −7 n 60 i | 10.9 | 2.2 | 25 |
| PD-3b | B2 | 10 | g −3 SC 28 n 43 i | 6.2 | 0 | 19 |
Although the convenient and simple procedure of this synthesis of MCLCEs has to be emphasized, some problems need to be mentioned. An incomplete conversion of the addition reaction is caused by side reactions of the vinyl groups. These side reactions have been carefully analyzed. Furthermore, traces of mono-functional mesogens may be introduced due to impurities of the vinyl derivative used for their synthesis. The impurity and the side reactions cause a considerable amount of material that can be extracted from the network, the so-called soluble content. To overcome this problem, Krause developed a new mesogenic unit based on Donnio's three-ring central core. The laterally attached ethyl side-chain successfully suppresses a smectic phase. The vinyloxy end groups lead to a clean polyaddition reaction without side-reactions such as double-bond migration. Indeed, this concept results in elastomers with very low soluble contents. Hence, networks with low concentrations of crosslinkers, and therefore with a low elastic modulus, are synthetically accessible.
Scheme 5 shows the general synthetic concept for the MD-4 (monodomain) and PD-4 (polydomain) elastomers. The composition, the thermal and swelling properties, as well as the soluble contents, of the systems investigated here are summarized in Table 4. As expected, both Tg and Tn,i increase with increasing crosslinker concentration for either monodomains or polydomains, whereas the degree of swelling q decreases. The swelling anisotropy Σ is not influenced by the crosslinker content. The soluble content varies between 2% and 14%, and is especially low for those systems with a crosslinker concentration between 6% and 8%.

Synthesis of the elastomers MD-4 (monodomain) and PD-4 (polydomain) with a three-ring mesogen with vinyloxy moieties in the lateral chains.
Composition and properties of the MD-4 (monodomain) and PD-4 (polydomain) systems
| Content of cyclosiloxane crosslinker/% | Tg | Tn,i | q | Σ | Sol. cont./% | |
| MD-4a | 2.5 | −9 | 60 | 12.6 | 1.9 | 14 |
| MD-4b | 4.0 | −8 | 64 | 8.6 | 2.3 | 10 |
| MD-4c | 6.0 | −5 | 72 | 6.3 | 2.3 | 3 |
| MD-4d | 8.0 | −1 | 84 | 5.4 | 2.1 | 2 |
| MD-4e | 12 | 6 | 86 | 4.7 | 2.1 | 9 |
| PD-4a | 2.5 | −7 | 63 | 12.5 | 0 | 10 |
| PD-4b | 4.0 | −5 | 65 | 7.6 | 0 | 13 |
| PD-4c | 6.0 | −4 | 70 | 5.2 | 0 | 5 |
| PD-4d | 8.0 | 0 | 84 | 4.0 | 0 | 2 |
| PD-4e | 12 | 1 | 86 | 3.6 | 0 | 10 |
Over the last decade, the central synthetic goal in the field of MCLCEs has been to obtain systems with a defined and reproducible network structure, i.e. low soluble contents, with nematic phases over the whole mesophase temperature range, and with convenient phase transition temperatures. Bergmann and Wermter used pre-polymerized mesogens as main-chain parts. This approach enables the chain length of the resulting networks to be controlled precisely, but requires a two-step reaction with intermediate work-up. Brandt and Krause made use of a more flexible one-pot reaction, accepting a statistical distribution of the chain lengths and in some cases increased soluble contents.
3 Coupling
Nematic monodomain elastomers exhibit a large change in length in the vicinity of Tn,i, which is proportional to the change of the order parameter. This proportionality was experimentally verified for nematic side-chain elastomers. In order to quantify the coupling between the nematic order and the macroscopic dimensions of a nematic monodomain elastomer, the basic relations can be evaluated in the frame of the Landau-de Gennes [39,40] theory. The free energy F of a nematic network is expanded in a power series of the nematic order parameter S. The order parameter S itself is a product of Snematic, describing the nematic order of the mesogens within the domains, and Sdomains, which describes the order of the domains over the whole sample:
For monodomains, we assume Sdomains = 1 and therefore S = Snematic. According to de Gennes [1,30], the free energy expansion for crosslinked polymers takes the form:
| (1) |
| (2) |
If the external field is non-zero, in addition to the spontaneous deformation another deformation of the network occurs. Minimizing (1) with respect to ɛ under strained conditions for small values of σ yields an equilibrium value of ɛm = (σ + US)/E.
From either equilibrium relation, one can deduce the coupling coefficient U. The plotting of the spontaneous deformation ɛ against the nematic order parameter S should result in a straight line. The coupling coefficient U can then be calculated from the slope of this straight line and the elastic modulus E of the isotropic network at the phase transition. Instead of the deformation ɛ, we use the length of the rubber referred to as the length in the isotropic state, λ = L/Lisotropic, where ɛ = λ − 1.
| (3) |
In the following section we discuss how the coupling coefficient U is influenced by the chemistry of the networks described in the previous chapter. Experimentally the value of S is determined by X-ray scattering using the method described by Lovell, Mitchell and co-workers [42,43]. The elastic modulus is obtained by measuring the elastic moduli in the isotropic phase for several temperatures and extrapolating to Tn,i. Other methods have been used to calculate the coupling coefficient U, for example from the pre-transformation region above Tn,i. The authors find a difference of about one order of magnitude between the values obtained by each method [3]. In order to be able to compare the behavior of different MCLCEs, we will only use the method described above.
The first system is elastomer MD-1 (see Table 1), a nematic main-chain polymer crosslinked via functional end groups. This system shows a spontaneous change in length as a function of the temperature of up to λ = 3.1 (or 210%) under load, as shown in Fig. 1(a). Extrapolating the length of the sample as a function of load, the spontaneous change in length can be evaluated for the unloaded network. The order parameter S follows the spontaneous change in length, as can be seen in Fig. 1(b) and (c). It has to be noted that – in direct analogy to side-chain elastomers – all monodomain networks exhibit a continuous change of the order parameter with temperature, which indicates supercritical behavior [28,44].

(a) Spontaneous change in length λ vs. temperature measured with different loads for MD-1, where λ = L/Lisotropic. (b) Temperature dependence for the nematic order parameter S determined by X-ray for MD-1 without load and (c) nematic order parameter S vs. change in length λ without load.
In order to calculate the coupling coefficient U, the elastic modulus of the network E is required. However, the sample MD-1 has a clearing temperature Tn,i of 122 °C (see Table 1). When measured under equilibrium conditions at this temperature, it decomposes with time and causes non-reproducible stress–strain curves. Hence, it was not possible to measure different moduli in the isotropic state and to extrapolate to the modulus at Tn,i. Extrapolating measurements of the moduli from lower temperatures to Tn,i does not yield the required modulus E, either, because these moduli do not solely represent the rubber elasticity of the network. Rather, the modulus decreases with increasing temperature, reflecting the strong influence of the nematic field and not the temperature dependence of the rubber elasticity. A calculation of E via the ideal network theory is not reasonable due to rather large non-ideal effects of such a system even in the isotropic phase. Therefore, no reasonable quantity of U can be given for MD-1.
These problems clearly indicate the necessity to modify the chemistry of the systems in order to get networks with a temperature regime of the nematic phase that allows obtaining reliable experimental results.
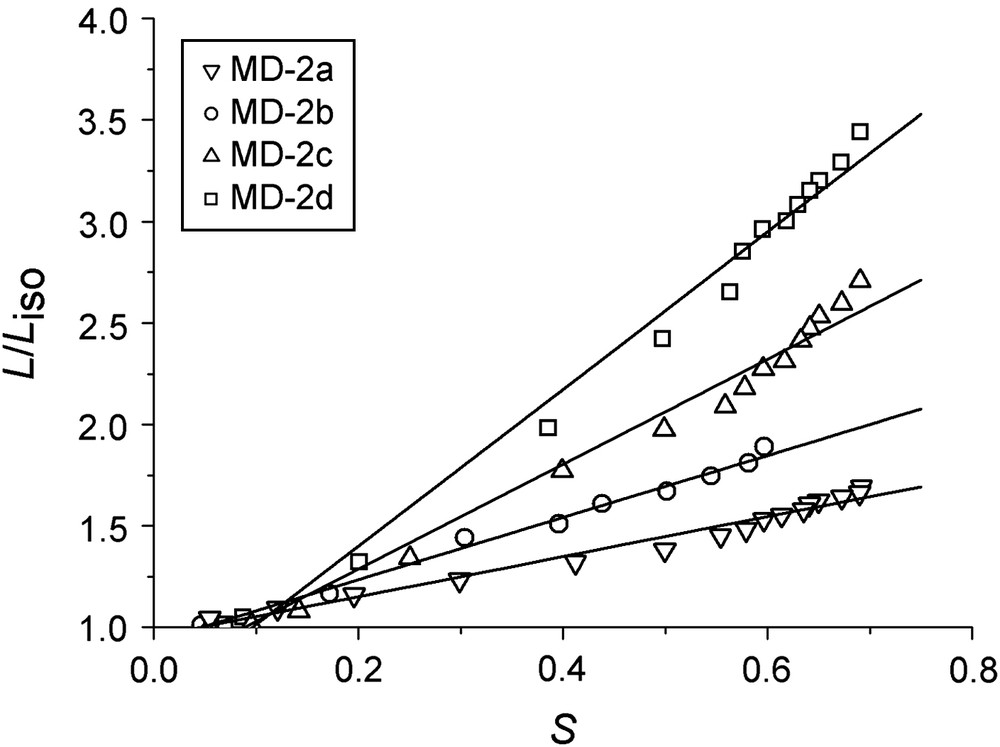
For the elastomers MD-2a–MD-2d (see Table 2), the same main-chain LC polymers are crosslinked by side-chain LC polymers. Here Tn,i can be affected and lowered by the side-chain component. The thermoelastic experiments reveal that both a higher ratio of main-chain polymer and main-chain polymers with a higher degree of polymerization increase the spontaneous change of length to up to λ = 3.8 (or 280%) (see Fig. 2a). The effect of an increasing fraction of the main-chain polymer can be seen when elastomers MD-2a and MD-2b are compared, where the main-chain parts have the same degrees of polymerization. The effect of the chain length becomes evident when the elastomers MD-2c and MD-2d are compared. The amount of main-chain polymer is approximately constant (4 mol% vs. 4.6 mol%), but the degree of polymerization differs by 25%. Also, MD-2c and MD-1 show similar changes of length, and in both cases the main-chain component has the same degree of polymerization of about 20 repeating units.

(a) Spontaneous change of length λ = L/Liso plotted vs. the reduced temperature Tred = T/Tn,i without load for MD-2a, MD-2b, MD-2c and MD-2d; (b) temperature dependence of the nematic order parameter S, as determined by X-ray, for MD-2a, MD-2b, MD-2c and MD-2d without load; (c) nematic order parameter S vs. change of length λ without load for these elastomers.
As can be seen in Fig. 2(b), the nematic order parameter is not affected by the chemical composition of the networks such as the ratio of the main-chain and side-chain parts. Therefore the slope of λ(S) is solely determined by the spontaneous change of length. For all four elastomers MD-2a–MD-2d, a slight deviation from linearity in the plot of λ(S) can be observed, especially at higher order parameters (Fig. 2(c)).
The elastic modulus rises with increasing amount of the main-chain component (Table 5) although the degree of swelling q remains almost constant at a value of q ∼ 6–7 (Table 2). This is remarkable since for an increase of the fraction of the main-chain component, which has a poorer solubility, and for an increase of modulus, a decrease in q should be expected. The topology of the networks, however, is poorly understood and no simple explanation can be provided for their swelling behavior.
Summary of slopes of λ(S), elastic moduli E and coupling coefficients U for all elastomers investigated
| E [105 N/m2] | U [105 J/m3] | ||
| MD-1 | 2.6 | ||
| MD-2a | 1.00 | 0.21 | 0.21 |
| MD-2b | 1.53 | 0.22 | 0.34 |
| MD-2c | 2.63 | 1.00 | 2.63 |
| MD-2e | 3.84 | 1.60 | 6.15 |
| MD-3a | 1.2 ± 0.1 | 3.9 ± 0.2 | 4.6 ± 0.5 |
| MD-3b | 1.5 ± 0.2 | 3.3 ± 0.2 | 4.9 ± 0.7 |
| MD-3c | 1.6 ± 0.2 | 2.1 ± 0.1 | 3.4 ± 0.4 |
| MD-4a | 3.0 ± 0.4 | 1.9 ± 0.2 | 6 ± 1 |
| MD-4b | 2.8 ± 0.4 | 2.4 ± 0.2 | 7 ± 1 |
| MD-4c | 2.3 ± 0.4 | 4.4 ± 0.4 | 10 ± 2 |
| MD-4d | 2.6 ± 0.3 | 5.9 ± 0.5 | 15 ± 2 |
| MD-4e | 2.4 ± 0.2 | 6.9 ± 0.6 | 17 ± 2 |
In the series of elastomers MD-2, both the slope of λ(S) and the elastic modulus E rise upon an increasing ratio and length of the main-chain component, which result in a higher coupling coefficient. This result is reasonable and confirms the assumption that main-chain elastomers exhibit higher coupling coefficients than side-chain elastomers.
The networks MD-3a–MD-3c contain the benzoic acid phenylester, which is a short rigid mesogenic unit with two aromatic units. They are linked together via the tetramethyldisiloxane and alkyl chains of different length. Varying the length of these alkyl chains offers a simple approach to manipulate the phase behavior of the elastomers.
The elastomer MD-3a, which contains a mesogenic group substituted with a hexyl-chain, exhibits a nematic phase only in a narrow temperature region above a smectic-C phase (refer to Table 3). The thermoelastic experiments (Fig. 3a) actually reveal a shoulder in the λ(T) curve and in a narrow temperature regime the network elongates by about 80%. The existence of the narrow nematic phase regime is also shown in detailed X-ray experiments, where the intensity of the small angle reflection as well as the correlation length of the smectic layers as a function of temperature indicate a smectic to nematic phase transformation [45]. If the thermoelastic measurements are correlated with the order parameter (Fig. 5), a linear relationship between S and λ is observed, which even covers the region of the smectic state.
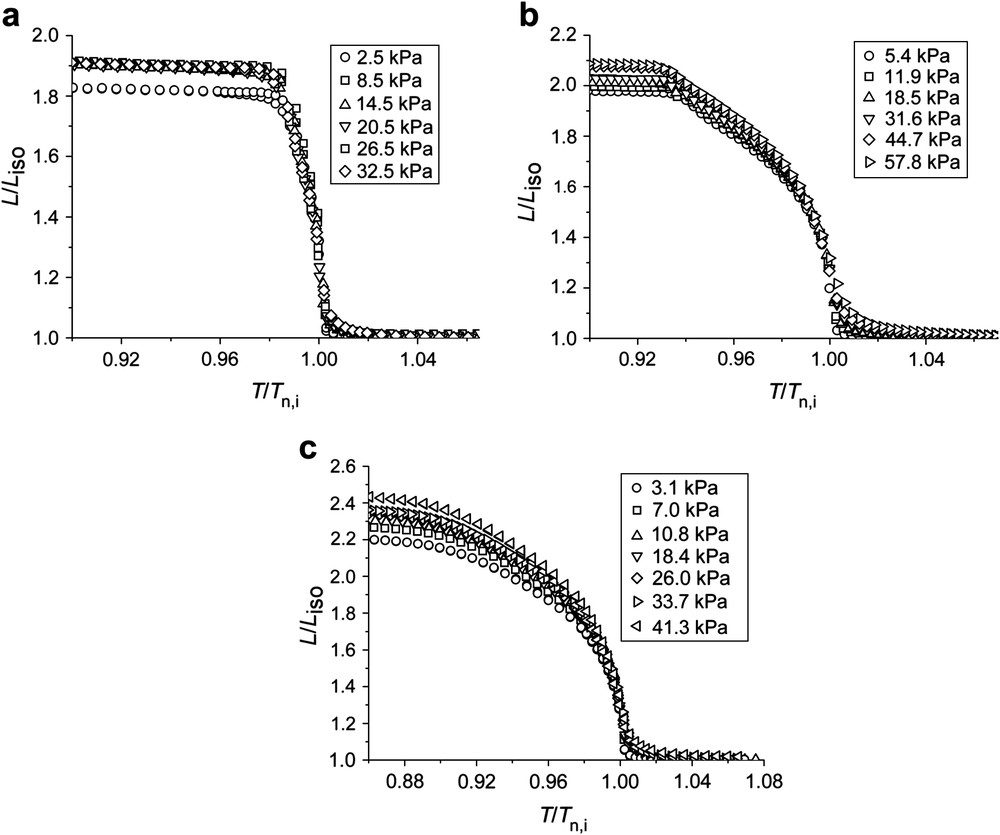
Spontaneous change of length λ = L/Liso vs. the reduced temperature Tred = T/Tn,i measured with different loads for (a) MD-3a, (b) MD-3b and (c) MD-3c. λ is referred to the length in the isotropic phase of each measurement.
Nematic order parameter S vs. change in length λ = L/Liso for the elastomer set MD-3.
The elastomer MD-3b differs from the previous sample by a shorter alkyl chain. Similar to the systematics of low molar mass liquid crystals, the existence of a nematic phase is favored and the temperature regime broadened compared to the previous network. Within the nematic phase the sample changes its length by about 200% (Fig. 3b), which is unaffected by the load. At the nematic to smectic phase transformation, the length of the networks remains almost constant at a decreasing temperature, which is due to the formation of the smectic layers. The change of length with temperature is now mainly determined by the thermal expansion behavior parallel to the smectic layer normal. The order parameter (Fig. 4b) follows the behavior observed for the change of length, and remains nearly unchanged within the smectic phase.
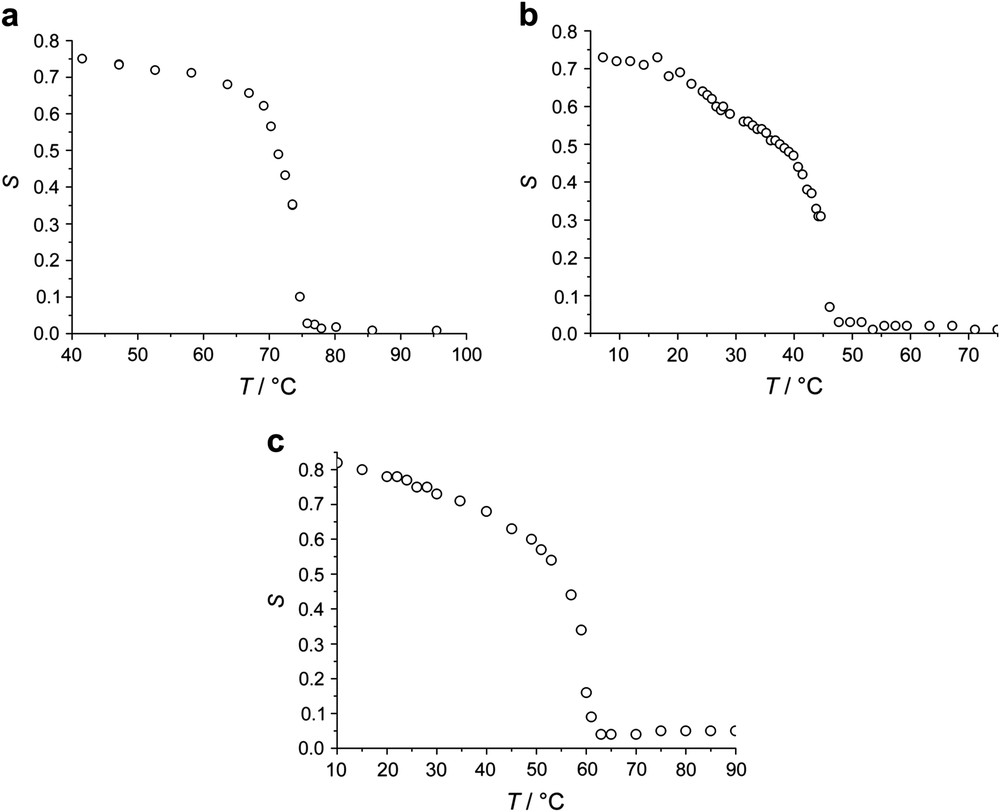
Temperature dependence of the nematic order parameter S determined by X-ray without load for (a) MD-3a (b) MD-3b (c) MD-3c.
Finally, MD-3c only exhibits a nematic phase. The formation of the smectic order is successfully suppressed by the co-polymerization of mesogenic monomer units having different length of the flexible alkyl chains. Starting from the isotropic phase, the sample shows a pronounced change of length at the clearing temperature and continuously elongates with falling temperature (Fig. 3c). The total change of length of λ = 2.2 (or 120%) is not as large as in the case of MD-1 or the MD-2 systems, but clearly exceeds that of side-chain elastomers, where an elongation of λ ≈ 1.6 (or around 60%) was observed. The nematic order parameter, as determined by X-ray, follows the change of length measured by thermoelastic measurements (Fig. 4c). A linear relation between the spontaneous change of length and the order parameter (Fig. 5) therefore enables the calculation of the slope of λ(S) for all elastomers of the MD-3 subset.
In plotting the spontaneous change of length vs. the order parameter, a linear relation is found again. A small deviation from linearity appears in the smectic phase.
For this series of elastomers the slope of λ(S) does not seem to depend on the liquid crystalline phase, and only varies between 1.2 for MD-3a and 1.6 for MD-3c. Considering the elastic moduli (Table 5) at the nematic-to-isotropic phase transformation, the difference in the moduli also corresponds to the difference in the degree of swelling. A larger degree of swelling results in a lower modulus and therefore reflects lower effective crosslinking density. Obviously, for these elastomers the solubility of the networks is not affected by the change of the alkyl chain length, which is in contrast to the results obtained for the elastomers MD-2a–MD-2d. The coupling coefficients, calculated from the slope of λ(S) and the moduli, show very similar values in the range between 3.4 × 105 J/m3 and 4.6 × 105 J/m3. This result is reasonable because the chemistry of the mesogenic units is not modified within this series.
In order to investigate the dependence of the coupling coefficient U on the elastic modulus, five elastomers MD-4a–MD-4e with different crosslinker concentrations were synthesized by the same one-pot reaction used for the subset MD-3, but with mesogenic units that contain three aromatic rings instead of two. This should lead to a stiffer polymer backbone. Additionally, by improving the polyaddition reaction by using vinyloxy-terminated monomers, we are able to vary the crosslinking density and therefore the elastic modulus of the network. This is shown by swelling experiments (Table 4), where the swelling degree increases with lower crosslinker content. The observation from the swelling behavior coincides with the mechanical investigation, where the elastic modulus in the isotropic phase increases with higher crosslinker content (Table 5). It has to be noted that the elastic moduli for the elastomers containing three aromatic unit mesogens, MD-4a–e, range from 1.9 × 105 Pa to 6.9 × 105 Pa, and are on the same order of magnitude as those for the MD-3 series.
Fig. 6(a) shows the spontaneous change in length, measured by a thermoelastic experiment, for the elastomers MD-4a–e with different amounts of crosslinker. As can be seen in the curves, this series of elastomers only exhibits nematic phases. Starting from the isotropic phase, a pre-transformation regime can be seen for all elastomers. The change in length is most pronounced at the phase transformation. On further cooling, a continuous elongation is observed in the nematic phase. Comparing the change in length between the isotropic phase and a reduced temperature T/Tn,i = 0.85 for each sample, one finds a change in length of λ = 2.5 for MD-4a with 2.5% of crosslinker, whereas MD-4e with 12% of crosslinker only shows a change in length of λ = 2.1. Elastomers with longer network strands obviously exhibit larger spontaneous changes of length than highly crosslinked elastomers. The effect of the length of the network strands on the change in length is not as pronounced as in the series of the co-elastomers MD-2a–d.
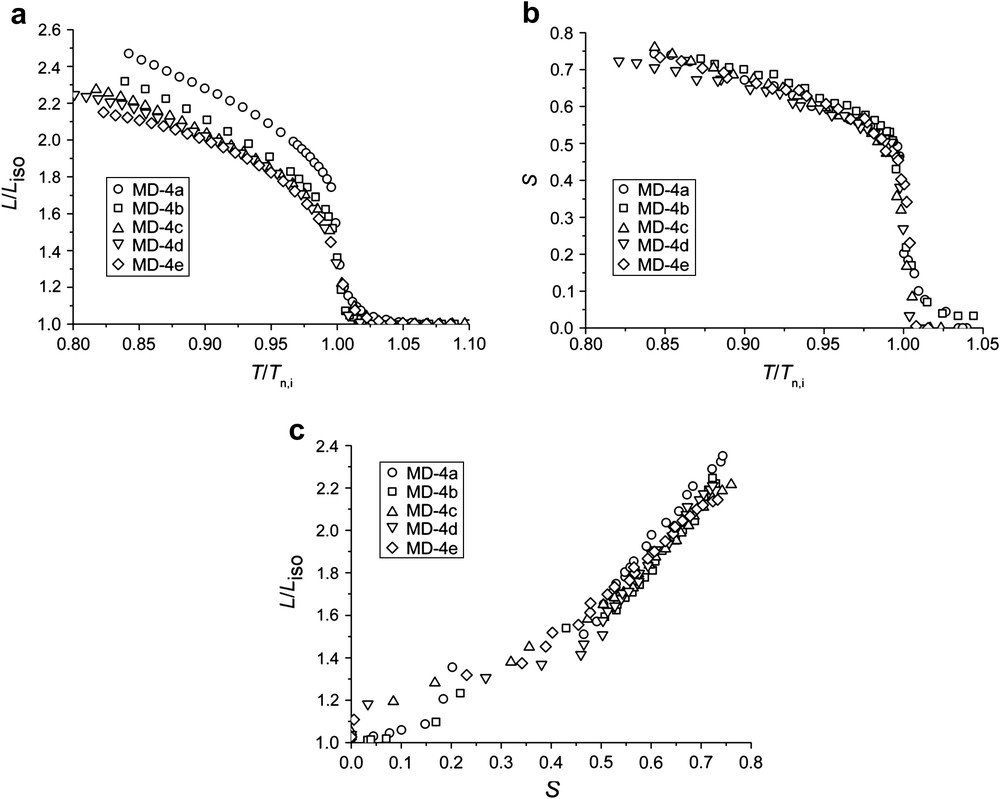
(a) Spontaneous change of length λ = L/Liso vs. reduced temperature Tred = T/Tn,i measured without load for all elastomers MD-4 with different crosslinker content. λ is referred to the length in the isotropic phase; (b) nematic order parameter S determined by X-ray vs. reduced temperature Tred = T/Tn,i for all samples of the subset MD-4; and (c) nematic order parameter S vs. change of length for all samples MD-4.
The temperature dependence of the order parameter S for this series of elastomers is shown in Fig. 6(b). The order parameter increases within a relatively narrow temperature range when cooling from the isotropic phase into the nematic phase. Within the liquid crystalline phase, a further increase of the order parameter is observed. Similar values for the order parameter (S = 0.70–0.76) at T/Tn,i ∼ 0.85 are found. Thus the effect of the amount of crosslinker on the order parameter is rather small. This result is surprising because the cyclic siloxane crosslinker used in these samples was expected to disturb the nematic ordering.
In order to determine the coupling coefficient, the spontaneous deformation λ is plotted against the nematic order parameter, as shown in Fig. 6(c). The plot of λ(S) is not linear over the whole temperature range for the elastomers MD-4a–e. We cannot clearly judge whether there are two linear regions or if λ(S) is a more complicated function. The largest errors must certainly be assumed for small values of S. Additionally, the largest changes in length and in the order parameter are found in the vicinity of Tn,i, and only a small temperature mismatch between the thermoelastic and X-ray experiments will strongly affect the slope of λ(S) in this region. We therefore determine the slope for values of S > 0.3. Comparing the respective values of the slopes of λ(S) for different crosslinking densities, we find the largest value of ∂λ/∂S = 3.0 ± 0.4 for MD-4a with the lowest crosslinking density. For the elastomers MD-4b–e, the slope remains almost constant within the experimental error (see Table 5). The coupling coefficient U is then calculated from the elastic modulus that rises within the series MD-4a–e, and the slope of λ(S), which hardly changes within this series. The coupling coefficients range from U = 6 ± 1 × 105 J/m3 for MD-4a up to U = 17 ± 2 × 105 J/m3 for MD-4e, as summarized in Table 5. The chemical constitution of neither the mesogenic unit nor the spacers is modified for the elastomers MD-4a–e. Only the amount of the crosslinker was varied. Therefore we can conclude that the coupling becomes stronger with increasing elastic modulus, as was predicted by de Gennes [30]. Comparison of the elastomers MD-4a–e with mesogens consisting of three aromatic units with those from MD-3a–c containing shorter mesogens with only two aromatic units show a weaker coupling for the two-ring systems. We speculate that the extended mesogens of MD-4a–e lead to a larger nematic field and a stiffening of the polymer backbone, resulting in a stronger coupling.
Neither here nor in the case of the co-elastomers of the subset MD-2 is the relation of strain λ and order parameter S linear for higher values of S. To investigate this non-linearity in more detail, we measured the birefringence as a function of temperature for the samples with the lowest (MD-4a) and highest crosslinking density (MD-4e), respectively, using the method described by Lim and Ho [46]. A detailed description of this method, which was previously applied for the investigations on SCLC elastomers, can be found in the literature [2]. The stress applied was about 100 kPa for both samples. Fig. 7(a) shows the birefringence (on the left ordinate) and the change in length (on the right ordinate) as a function of the temperature for MD-4a. Starting from the isotropic phase, a distinct pre-transformation behavior is observed for the birefringence, being proportional to the nematic order parameter. Due to the high stress applied to the sample, the phase transformation is very broad. In the nematic phase, the typical continuous increase of the order parameter is observed. The change in length is defined as λ = L/Lσ=0,iso, where Lσ=0,iso is the length of the unloaded sample in the isotropic state, and L is the length of the sample at a given temperature and stress. Comparing the change in length with the temperature dependency of the birefringence as shown in Fig. 7(a), we observe that both coincide well in the pre-transformation region and around the phase transition. However, the curve progression of the birefringence and the change in length clearly differ when going to lower temperatures. This becomes even clearer when we plot the change in length as a function of the birefringence for MD-4a, as shown in Fig. 7(c). At small values of birefringence, one may still observe a linear relation of λ(Δn ∼ S) whereas at higher values of Δn, λ(Δn ∼ S) deviates from linearity.
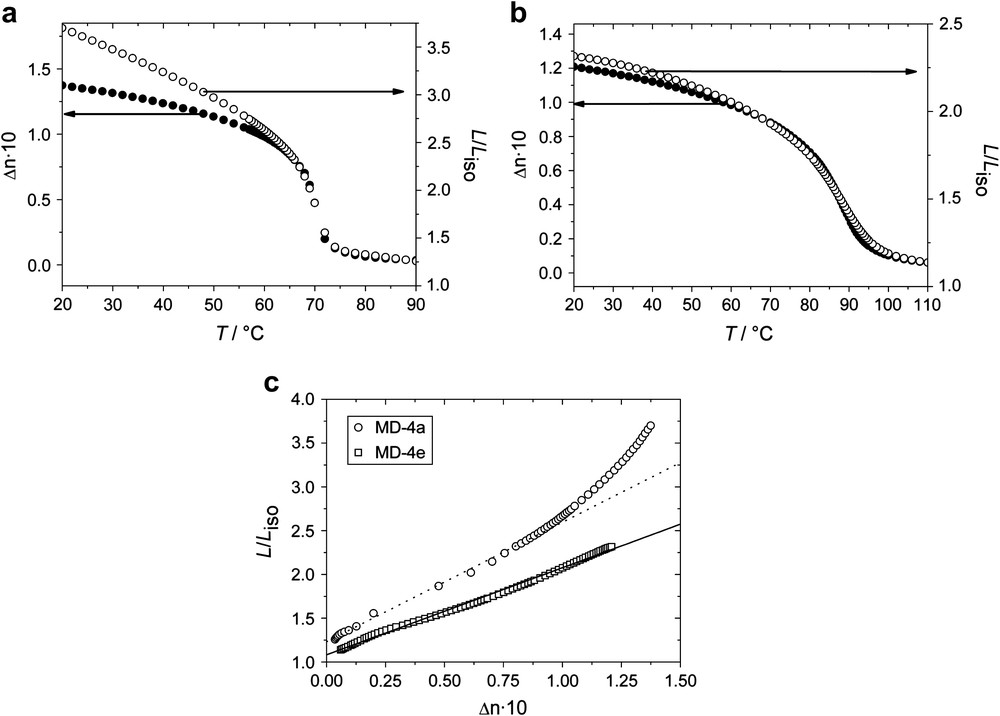
Birefringence Δn (left ordinate) and change in length λ = L/Liso (right ordinate), referred to the length of the unloaded sample in the isotropic phase for (a) MD-4a and (b) MD-4e. The measurements were carried out with an external stress of ∼100 kPa. (c) Spontaneous change in length λ = L/Liso from thermoelastic experiments vs. the birefringence Δn from temperature dependent measurements for MD-4a with the lowest and for MD-4e with the highest crosslinker content.
MD-4e differs from MD-4a in the crosslinking density. Fig. 7(b) again compares the temperature dependency of the birefringence (left ordinate) with the change in length (right ordinate) for this sample. For both measurements, a very pronounced pre-transformation region passes into the broadened phase transformation. These measurements indicate that we are here far above the critical stress. Nevertheless, both curves nicely coincide. The proportionality of the change in length and the birefringence for MD-4e is shown by plotting λ as a function of Δn in Fig. 7(c). The result is similar to the observations made in SCLCEs. Obviously it depends on the crosslinking density or on the length of the network strands in LCMCEs whether the coupling is linear or not. We assume that in elastomers with long network strands, and especially at higher temperatures, the network will tend to recover some entropy by forming hairpin defects. This backfolding of the polymer backbone was first predicted by de Gennes [30], and theoretically described by Warner et al. [31,32,47,48]. The first experimental evidence of this particular chain conformation in linear polymers was obtained by neutron scattering experiments [33]. By cooling an elastomer with low crosslinking density below the phase transformation temperature, some chains may form hairpins. By further cooling, these hairpins are removed due to the external field leading to an additional extension of the sample. This could explain the non-linearity in coupling behavior observed for MD-4a, while in the sample MD-4e with a high crosslinker content the network strands are too short to form hairpins, and therefore a linear coupling is observed.
3.1 Discussion of the coupling behavior
The elastomer prepared from an end-functionalized linear polymer, MD-1, shows a remarkable change in length of λ = 3.1 (210%). A similar change in length is observed for the combined MC/SC elastomer MD-2c prepared from MC polymers with a similar degree of polymerization. Within the series MD-2, one can clearly see that with increasing either the molecular weight or the concentration of the main-chain component, larger changes in length are achievable (up to λ = 3.8, or 280%). This effect of longer network strands on the change in length is also observed in the elastomers MD-4a–e, prepared via a one-pot reaction, but here it is less pronounced. An explanation may be the polydispersity of the network strands in the latter system. If most of the strain is taken up by deforming short chains, the deformation of longer chains becomes less important. Another aspect is the role of the crosslinker in the mixed system MD-2a–d. The large deformations observed in these systems could be explained by considering the side-chain component to be a highly anisotropic, multifunctional crosslinker that contributes to the anisotropy of the network chains of the whole sample. Such a contribution is not possible for the isotropic crosslinker used for MD-4a–e. For the elastomers MD-3a–c, elastomers with different liquid crystalline phases were investigated. The changes in length of these elastomers are almost independent of the phase behavior.
For all elastomers, similarly high values of the order parameter S between S = 0.7 and S = 0.8 were observed at lower temperatures. In the combined MC/SC elastomers, the order parameter is almost independent of the amount of main-chain component. Also the concentration of the cyclic crosslinker does not affect the state of order, as seen in the series MD-4a–e. This is surprising because an isotropic impurity should be expected to lower the nematic ordering. In addition, the liquid crystalline phase that the elastomer exhibits – whether smectic or nematic – has little impact on the order parameter, as shown for the elastomers MD-3a–c.
Considering the independence of the order parameter of these factors, the slope ∂λ/∂S is mostly determined by the elastomer's change in length. We observe the same slope ∂λ/∂S when we compare the end-linked elastomer MD-1 and the combined elastomer MD-2c, but this might be coincidental. The more main-chain mesogens the elastomers of the series MD-2a–d contain, the larger the slope ∂λ/∂S is. But already in this series a deviation from linearity of λ(S) is observed. A more detailed investigation by means of birefringence for two elastomers with different crosslinker content, MD-4a and MD-4e, was therefore performed. It shows that λ(S) clearly deviates from linearity for an MC elastomer with long network strands, whereas a linear relation is observed for the elastomer with short network strands. We assume that the deformation of hairpins has to be taken into account here. For a main-chain system that additionally shows smectic phases, MD-3a–c, the linear relation of λ(S) holds even in the smectic phase.
Comparing the coupling coefficients for the combined elastomers MD-2a–d, we find a stronger coupling with increasing contents of main-chain mesogens. The coupling coefficients range from 2.1 × 104 J/m3, a value typical for side-chain elastomers, to 6.15 × 105 J/m3. The largest coupling coefficients, however, are observed in the elastomers synthesized via a one-pot reaction, where values as high as U = 1.7 × 106 J/m3 were found. We therefore conclude that main-chain elastomers generally show a stronger coupling than side-chain elastomers. The coupling is not affected by possible smectic phases at lower temperatures (series MD-3a–c).
If we compare elastomers synthesized via a one-pot reaction, the chemical structure of the mesogens plays an important role. Elastomers with mesogens that consist of three aromatic units (series MD-4a–e) show a stronger coupling than elastomers with mesogens of two aromatic units (series MD-3a–c), independent of the crosslinker content. We explain this observation with a larger nematic field for more extended mesogens that leads to a stiffer polymer backbone and a stronger coupling. By varying the crosslinker content, and thereby the elastic modulus in series MD-4a–e, we find that the coupling is mostly determined by the elastic modulus of the network as predicted by de Gennes.
4 Orientation behavior
In contrast to the transparent monodomain samples, polydomains are optically opaque, indicating a microscopic structure on a length scale of the wavelength of visible light. The term polydomain itself may be misleading because typically, no sharp domain boundaries are observed, but rather a continuous change of the orientation of the local director. From side-chain LCEs it is well known that by applying a mechanical field, a macroscopic orientation of the local directors occurs. In mechanical experiments on these systems, this orientation process causes a soft plateau with a very low modulus up to an elongation λ2, where the modulus rises again. Measurements of the order parameter for polydomain SCLCEs show that λ2 is equal to the elongation λM where the sample achieves the structure of a monodomain [28]. Also, in the case of SCLCEs, the width of the plateau decreases with increasing temperature [28,49]. In this chapter, we explore the orientation behavior of MCLCEs.
In Fig. 8(a) the stress is plotted against the imposed strain for the polydomain elastomer PD-1a, prepared from end-functionalized main-chain polymers by Bergmann et al. For deformations up to λ2 ∼ 5 (or 400%), a soft response with a low modulus is observed. This exceeds by far the width of the soft plateau observed in side-chain elastomers, which show values of about λ2 ∼ 1.6 [50]. In Fig. 8(b), the plot of the order parameter S, as determined by X-ray measurements, vs. the imposed strain λ shows a saturation of S, i.e. a monodomain structure, already at a deformation of λM ∼ 2.5. Thus, in this main-chain system, λ2 considerably deviates from λM, quite in contrast to the observations made in side-chain elastomers. An explanation for this remarkable behavior may be that the additional deformation with a low modulus originates in the presence of hairpin defects. Hairpins can be straightened into fully stretched chains by an external strain with a low modulus and without significant increase of the order parameter.
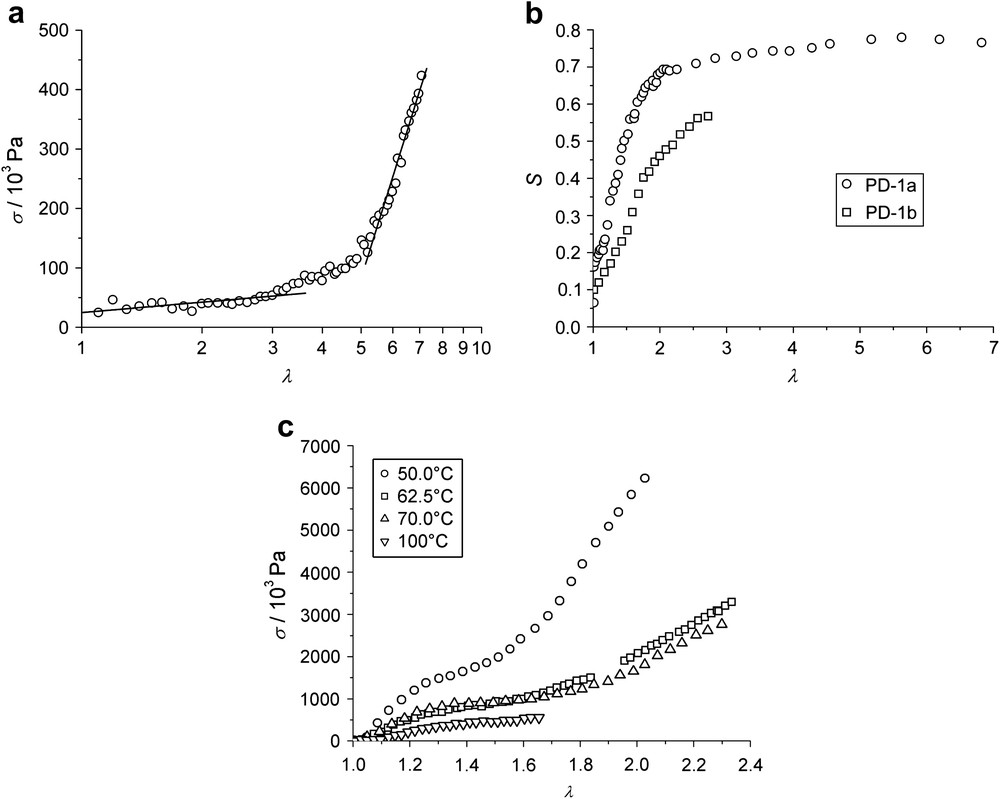
(a) Nominal stress σ vs. imposed strain λ = L/L0 for PD-1a at 70 °C in the nematic phase; (b) nematic order parameter S determined by wide angle X-ray scattering vs. imposed strain λ for PD-1a and PD-1b; (c) nominal stress σ vs. imposed strain λ for PD-1c at 50 °C, 62.5 °C, 75 °C and 100 °C.
The temperature dependence of the stress–strain measurements differs from that observed in side-chain elastomers as well. Fig. 8(c) shows for the case of PD-1c that with increasing temperature, the plateau of soft deformation broadens. As the anisotropy of the network coils is a function of temperature via the order parameter, one should expect a decrease of the plateau width at rising temperatures. If hairpins are present, however, they may partially retain the entropy of the polymer backbone, and a larger number of hairpins and a longer plateau should be expected at higher temperatures. Considering the temperature dependence of the plateau width, one may therefore conclude that for the elastomer PD-1c, the effect of the hairpins dominates the effect of the order parameter.
The co-elastomers PD-2 synthesized by Wermter et al. (see Scheme 3 and Table 2) also exhibit a broad soft plateau up to a deformation of about 300%. Fig. 9(a) shows the plots of the nominal stress against the imposed strain for three polydomain samples. In these elastomers, two effects affect the width of the plateau: the content of main-chain polymer and its molecular weight. The increase of either of these leads to broader plateaus. We find values of λ2 = 2.3 for the sample PD-2a, λ2 = 2.7 for PD-2c, and λ2 = 4.0 for PD-2e. In Fig. 9(b), the order parameter S under deformation, as determined by X-ray investigations, is shown. The elongations necessary for a monodomain, λM, are 1.7, 1.8, and 1.9 for the samples PD-2a, PD-2c, and PD-2e, respectively. They are determined as the intersection points of the fits to the two linear regions of the curves. Obviously, λM is almost independent of the composition of the system. Just like in the system PD-1a discussed above, there is large discrepancy between λ2 and λM for these co-elastomer systems.
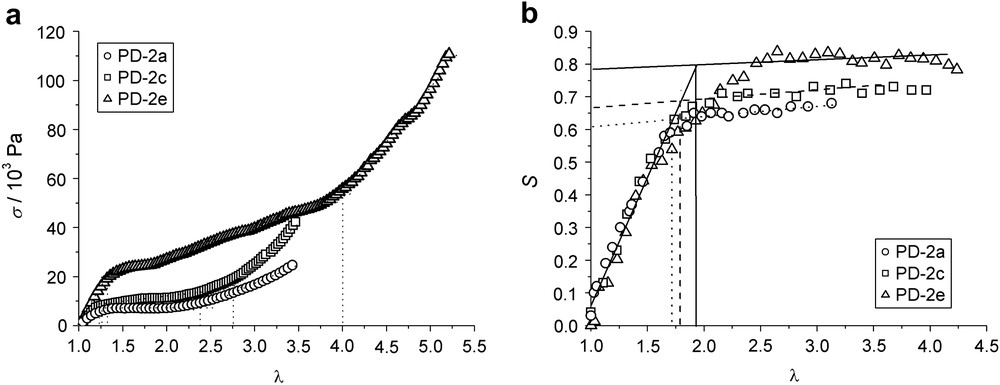
(a) Nominal stress σn vs. imposed strain λ = L/L0 dependent on the quantity of main-chain component in co-elastomers PD-2a, PD-2c and PD-2e at Tred = T/Tn,i = 0.950. (b) Nematic order parameter S determined by wide angle X-ray scattering vs. imposed strain λ for PD-2a, PD-2c and PD-2e.
As for PD-1c, the temperature dependence of the plateau width was investigated for the sample PD-2a by X-ray and stress–strain measurements. Fig. 10(a) shows the stress–strain plots for different reduced temperatures Tred = T/Tn,i between 0.892 and 1.00 (the phase transformation temperature). At a low temperature of Tred = 0.892 (which is still 40 °C above the glass transition), no plateau was found in the mechanical investigation. Probably the mechanical relaxation of the sample becomes so slow that the measurements were not carried out under complete equilibrium conditions. Upon increasing the temperature to Tred = 0.92, a slight non-linearity in the stress–strain curves indicates the occurrence of a plateau. Measurements at higher temperatures then clearly show a mechanical plateau that broadens with increasing temperature from λ2 = 2.0 at Tred = 0.947 up to λ2 = 2.4 at Tred = 0.975. The progression of the order parameter under deformation is shown in Fig. 10(b). Again, λM is determined as the intersection points of the fits to the two linear regions of the curves. S is independent of temperature and a saturation is always observed at λM = 1.7. One may speculate that the increase of the values of λ2 with increasing temperature is again associated with hairpin defects in the main-chain component of the network, which are more likely to occur at higher temperatures.
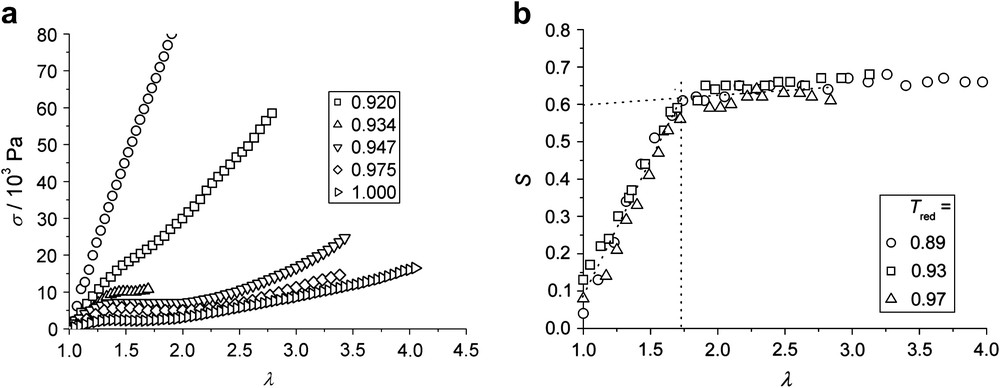
(a) Nominal stress σ vs. imposed strain λ = L/L0 at different temperatures of PD-2a. (b) Nematic order parameter S determined by wide angle X-ray scattering vs. imposed strain λ for PD-2a at different temperatures.
Fig. 11(a) shows the results of stress–strain measurements at different temperatures of the elastomer PD-3b synthesized in a one-step reaction by Brandt et al. The order parameter S as a function of imposed strain is shown in Fig. 11(b), with λM again determined as the intersection of fitting lines to the linear regions of the curves. For this sample, the plateau width is much smaller than for the elastomers prepared from the end-functionalized polymers by Wermter (PD-2) or Bergmann (PD-1), but λ2 is still larger than λM. At 35 °C, S saturates at λM = 1.4 and the modulus starts rising again at λ2 = 1.7, and at 40 °C we observe λM = 1.3 and λ2 = 1.5. The deformation λM here decreases with higher temperature. Remarkably, the end of the plateau λ2 decreases with rising temperature as well, in contrast to the observations from the PD-1 and PD-2 systems. As the plateau width is determined, in part, by the anisotropy of the network strands, which is in turn associated with the order parameter, the effect of the network anisotropy on the mechanical properties seems to be larger for this elastomer than the effect of hairpins. In fact, the probability of hairpin backfolding depends strongly on the length of the network strands. As PD-3b has a higher elastic modulus and was synthesized with a large amount of crosslinker, short strands and few hairpins should occur, and a behavior like the one observed is indeed expected.

(a) Nominal stress σn vs. imposed strain λ = L/L0 of PD-3b at different temperatures indicated in the figure; (b) nematic order parameter S determined by wide angle X-ray scattering vs. imposed strain λ for PD-3b at 35 °C and 40 °C.
The elastomers of the series PD-4 were prepared via the same one-pot synthesis as those of the PD-3 subgroup. The main difference is a significantly lower amount of crosslinker for the case of PD-4a than for all samples of PD-3. Fig. 12(a) shows the stress–strain behavior for PD-4a as a function of temperature. The plateau length decreases with higher temperatures (from λ2 = 2.05 at 30 °C to λ2 = 1.6 at 60 °C), following the tendency observed for PD-3b. Assuming long network strands due to a low crosslinker concentration, and therefore the presence of hairpins, we would expect the opposite temperature dependence. Obviously, the question how the network is formed – either by using polymers that are subsequently crosslinked, or by reacting all components in one step – is very important for the system's orientation behavior.
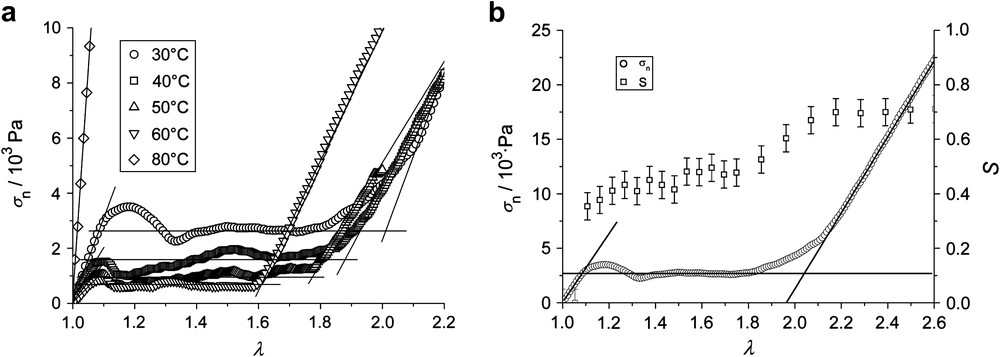
(a) Nominal stress σn vs. imposed strain λ = L/L0 of PD-4a at different temperatures indicated in figure. (b) Nominal stress σn (ordinate to the left) and nematic order parameter S determined by wide angle X-ray scattering (ordinate to the right) vs. imposed strain λ for PD-4a at 30 °C.
In Fig. 12(b), the mechanical stress and the progression of the order parameter S are plotted against the imposed strain λ for a temperature of 30 °C. Already at a deformation of only about 10%, the order parameter reaches a value of about 0.3. In the plateau region of the stress–strain curve, a slight increase of the order is found. The full orientation, where the order parameter remains constant within the experimental error, corresponds to the end of the plateau. In contrast to all types of main-chain elastomers discussed above, we here find λ2 = λM. If hairpins are at all present in these elastomers, the two processes of director orientation and hairpin removal cannot be separated, but rather happen simultaneously.
The influence of the crosslinker content on the orientation behavior is shown in Fig. 13. The width of the plateau decreases with increasing crosslinker content from λ2 = 2.04 for PD-4a with the lowest crosslinking density to λ2 = 1.79 for PD-4e with the highest amount of crosslinker. Thus the amount of crosslinker only seems to have a small impact on the width of the plateau, and therefore on the orientation behavior of the systems.
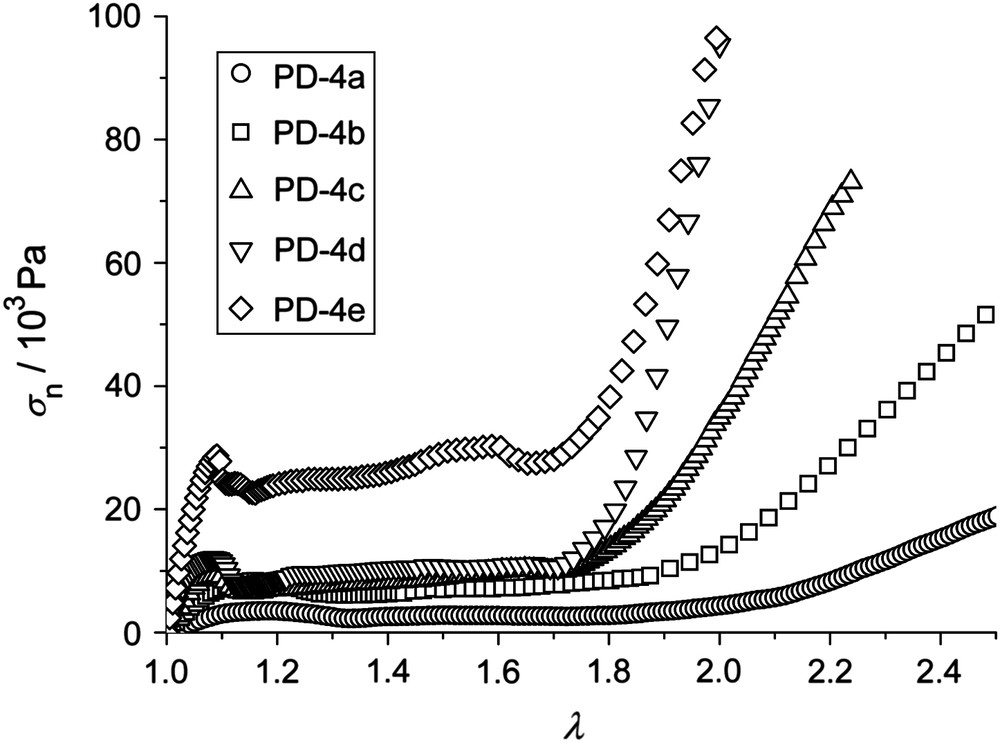
Nominal stress σn vs. imposed strain λ = L/L0 of the series with different crosslinker content PD-4a, PD-4b, PD-4c, PD-4d, and PD-4e at the reduced temperature T/Tn,i = 0.91.
4.1 Discussion of the orientation behavior
For all investigated MCLCEs, we found much higher values for the end of the soft plateau, λ2, than for side-chain elastomers. The networks synthesized from linear polymers, crosslinked either by a multifunctional crosslinker or by side-chain polymers, show similar orientation behaviors: the orientation of the local directors to a monodomain at the elongation λM is already achieved clearly before the stress rises again at λ2. This soft plateau is narrower for elastomers synthesized via a one-pot reaction. Furthermore, the two one-pot systems we investigated differ. A difference between λM and λ2 was seen for the PD-3b elastomer, but for the elastomers with a low crosslinker content from the PD-4 subset, the full orientation and the end of the soft plateau coincide. If the finding that λM is unequal to λ2 is indeed related to hairpins, two different processes might occur. In the first one, the orientation of the domains happens first, and subsequently the hairpins are removed without further increase of the order parameter. In the second one, both mechanisms take place simultaneously. Thus, the orientation behavior cannot merely be related to the network topology, but other aspects such as the crosslinking process or the curing temperature have to be considered as well.
The temperature dependence of λ2 obtained from stress–strain experiments shows a more general trend. Elastomers synthesized via the one-pot strategy exhibit shorter plateaus than elastomers synthesized via linear polymers that are subsequently crosslinked. In the first case, where we assume a broad distribution of the length of the network strands, the anisotropy of the network governs the length of the plateau. For the latter case, likely with a more homogeneous distribution of the network strands, hairpins seem to play a more important role.
5 Conclusion
We reported on the synthesis, coupling and orientation behavior of four series of MCLCEs developed in our group. Crosslinking end-functionalized main-chain polymers yielded the first fully nematic main-chain elastomers with moderate clearing temperatures. Co-elastomer systems where the main-chain part acts as a crosslinker of variable length and concentration showed extraordinarily high changes in length at phase transformation of up to 280%. A one-pot hydrosilylation reaction was developed as a more elegant approach to MCLCEs. Very thorough reactions and large changes in length could be achieved via this route when vinyl ethers were used as polymerizable moieties. By this means, it became possible to vary the crosslinking density in a very defined manner.
The coupling of the nematic order and the rubber network was quantified by the coupling constant U. The order parameter S as a function of temperature showed almost the same behavior for all systems investigated, so the slope of λ(S) was mainly determined by the change in length λ of the samples. We showed that non-linearity in the progression of λ(S) is observed for systems with long network strands and may be related to hairpin defects. For the co-elastomer systems, the slope of λ(S) becomes steeper with increasing content of main-chain mesogens, as does the elastic modulus E. For the one-pot systems, an increasing value of E coincides with a decrease in the slope of λ(S). In this case, however, E compensates for λ(S), and thus the coupling constant U shows the same progression as E for all systems investigated. We were also able to show that λ(S) is independent of the particular liquid crystalline phases the elastomer exhibits, and remains linear within a smectic phase. We observed higher coupling constants in one-pot systems with three aromatic rings in the mesogens than in those with only two, reflecting the higher stiffness of these mesogens and the larger the nematic field they cause.
In orientation experiments on polydomain samples, we found much broader plateaus in the stress–strain curves than for side-chain systems. For the subsets synthesized from functionalized polymers, we observe a large difference between the point where the modulus rises again, and the point where a monodomain is achieved. We ascribe this behavior again to the presence of hairpin defects. This difference is considerably smaller for the one-pot system with two aromatic cores, and completely vanishes for the vinyloxy system, probably because the orientation and removal of hairpins happen at the same time and not successively.