

1 Introduction
Acetylene-containing natural products are common components of terrestrial plants and have been known since the dawn of organic chemistry [1–3]. More recently, biologically active acetylenic natural products have also been isolated from marine organisms [4–8]. Naturally occurring polyyne compounds are an intriguing subclass of acetylene-containing natural products. We have used the term polyacetylene in the past in this context [9], which can be confused with polymerized acetylene. Following the lead of Tykwinski [3], the term polyyne is used here to indicate compounds with two or more conjugated acetylene units. The characteristics of the polyyne compounds include their unusual structural features and their wide spectra of biological activities. Enantioselective syntheses of polyyne natural products were relatively rare due to the high instability of the polyynes [2]. Attracted by the diverse biological activities [10–15] and the challenges of introducing stereocenters into reactive compounds, we commenced our first synthesis of a naturally occurring acetylenic alcohol, (S,S)- and (R,R)-adociacetylene B (1), in 2001 [16]. After the inaugural synthesis of adociacetylene B (1), graduate student Hamilton Dickson took on the tetrayne target, minquartynoic acid [17], and Ryan Fox, an undergraduate at the time, took on the polyyne glycosides, bidensyneosides [18,19]. This account describes the syntheses of these two groups of polyyne natural products first followed by the synthesis of C2 symmetric acetylenic compounds.
2 Minquartynoic acid and its derivatives
Minquartynoic acid (2) was first isolated in 1989 by Farnsworth and coworkers from the stem bark of Minquartia guianensis [20], which was one of the most potent traditional anthelmintics used by the Quijos Quichua people of Ecuador's Amazonian lowlands. In 2001, two additional polyyne natural products (3, 4, Fig. 1), were isolated along with minquartynoic acid from a chloroform extract of the twigs of Ochanostachys amentacea from southeast Asia [21]. In vitro tests showed that minquartynoic acid (1) displayed broad cytotoxicity against 10 different tumor cell lines [21].
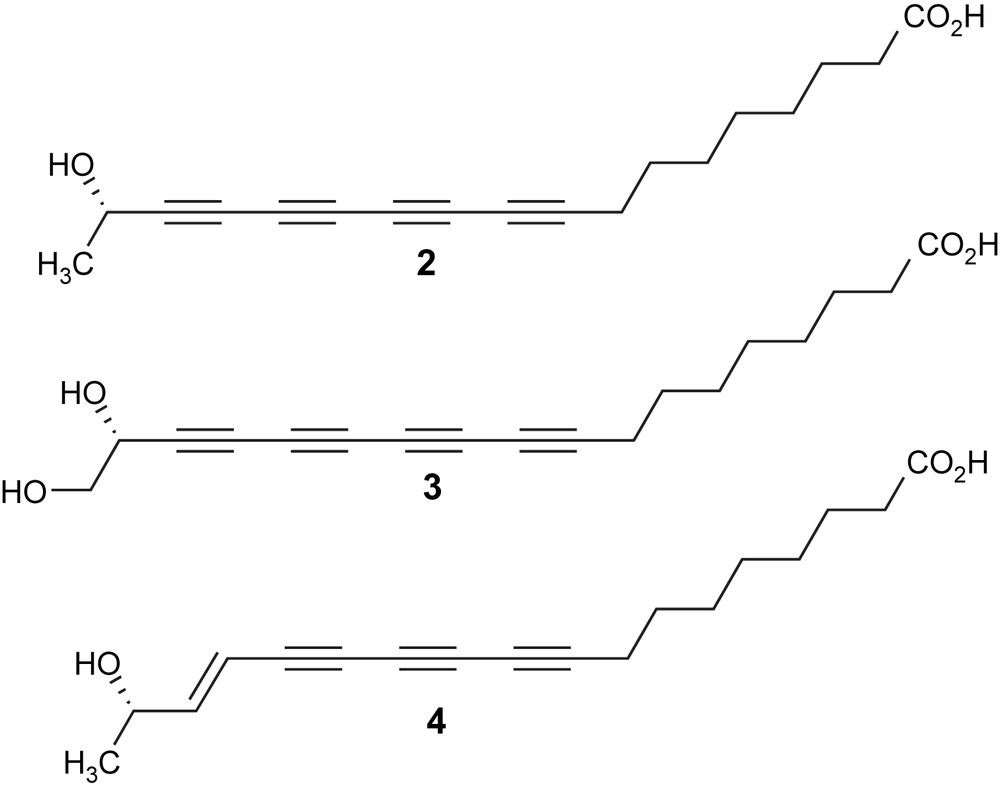
Cytotoxic polyyne natural products from O. amentacea. (S)-Minquartynoic acid 2, 18-hydroxyminquartynoic acid 3, and E-15,16-dihydrominquartynoic acid 4.
Independent isolation and anti-HIV activity of 2 were also reported by two other groups [22,23]. A strong cytotoxicity of 2 against the P-388 (leukemia) cell line with an ED50 of 0.18 μg/mL was reported [20]. Compound 4 exhibited the most potent activity among the three polyynes against the KB, LNCaP (prostate cancer) and SW626 (ovarian cancer) cell lines [21].
The well-known instability of the terminal polyyne intermediates presents a considerable challenge in the syntheses of polyyne natural products [10,24–26]. Our strategy for the synthesis of S-minquartynoic acid is depicted in Scheme 1 [17]. The tetrayne unit was envisaged to be constructed by the powerful Cadiot–Chodkiewicz cross-coupling reaction through a combination of bromoalkynes 5 and 7 and butadiyne 6 [2,27]. The plan was to avoid the isolation of any reactive terminal diyne and triyne intermediates and to obtain the relatively stable internal tetrayne in a one-pot reaction. The bromoalkyne 5 was prepared from (S)-methyl lactate 8. Butadiyne 6 is known and can be prepared in one step from commercially available 1,4-dichlorobutyne [27]. Bromoalkyne 7 was obtained from commercially available azelaic acid monomethyl ester 9.

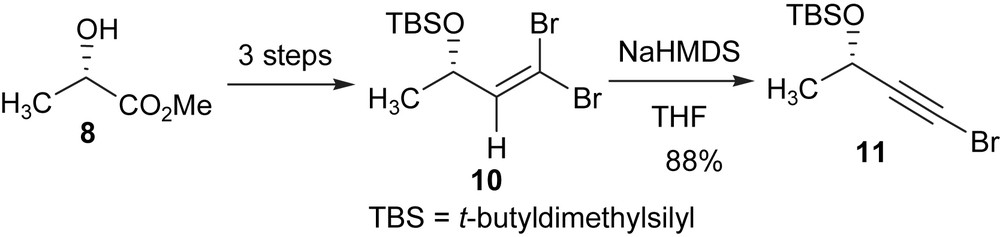
The preparation of the left hand fragment is shown in Scheme 2. Dibromoolefin 10 was prepared from lactate 8 following a modified literature procedure [28] with (1) protection of the –OH group, (2) reduction of the ester function to an aldehyde group, and (3) formation of the dibromoolefin [29]. To avoid loss of the t-butyldimethylsilyl (TBS) protecting group (Scheme 2) during the Corey–Fuchs procedure more hexanes was used to partition the product in the work-up process. Elimination of one molar HBr from 10 was achieved with NaHMDS to give bromoalkyne 11 in high yield [30].
The preparation of the right hand fragment had to use a different protocol (Scheme 3). The aldehyde 12 was obtained uneventfully by the following sequence of reactions: (1) selective reduction of the carboxylic acid function in 9 using 1 equiv of BH3·THF complex in anhydrous THF at 0 °C and (2) oxidation of the resulting primary alcohol with PCC. Aldehyde 12 was treated with the Ohira–Bestmann reagent to yield alkyne 13 in excellent yield [31,32]. The conversion of alkyne 13 to the bromoalkyne 7 was also achieved in excellent yield using (1) AgNO3 and NBS in acetone [33] and (2) hydrolysis of the ester to its corresponding carboxylic acid using LiOH in a mixture of THF and H2O. The Cadiot–Chodkiewicz reaction is known to proceed best with polar substrates [27].

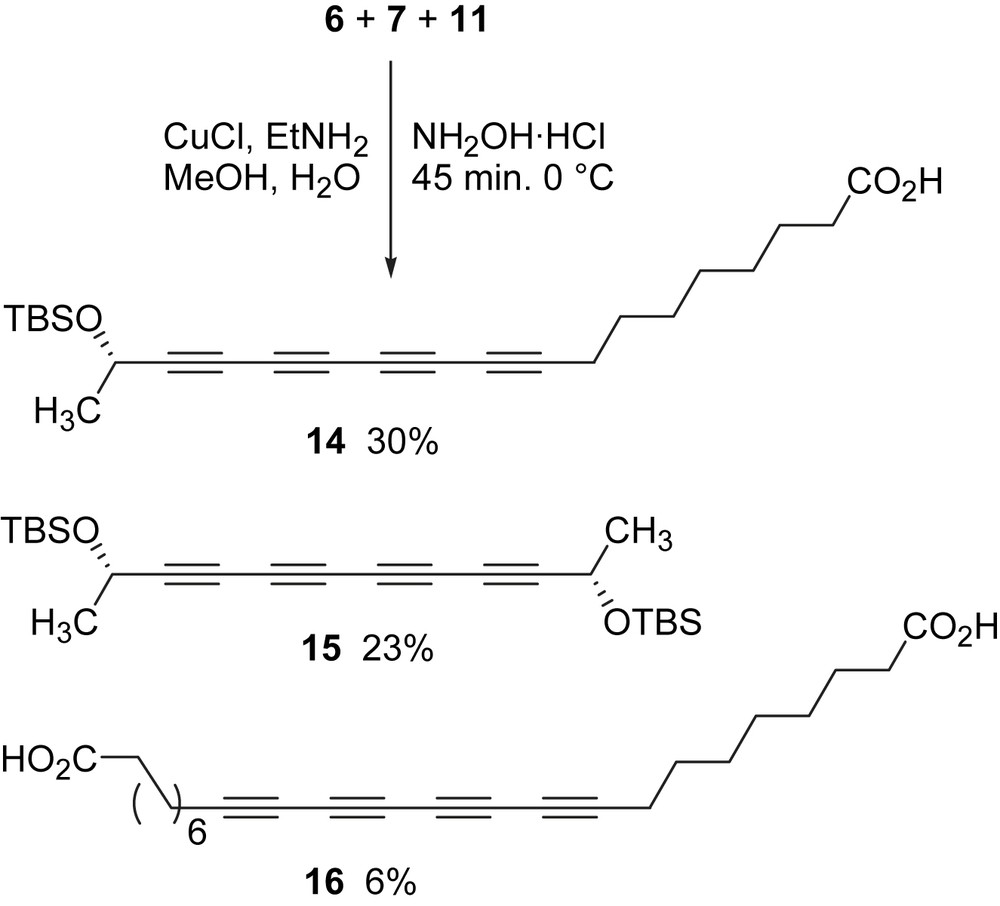
With bromoalkynes 7 and 11 in hand, the stage was now set for coupling via the Cadiot–Chodkiewicz reaction. A one-pot three-component coupling strategy was employed, in which 1 equiv of each of the reactants (6, 7, and 11) was loaded into the flask along with other reagents. A typical literature procedure for the coupling reaction is usually conducted at room temperature [17]. However, CuCl caused butadiyne 5 to immediately decompose at room temperature. The general procedure was modified by first charging the flask with all reactants and then adding CuCl to the mixture at 0 °C (Scheme 4). Three tetrayne products (14–16) were isolated in a combined yield of 59%, 30% of which was the desired cross-coupling product 14. The stability of these internal tetraynes is greater than the terminal triynes isolated in the initial two-component couplings. All three tetraynes (14–16) were purified by column chromatography and were stable during 1H and 13C NMR measurements. The TBS protecting group of 14 was removed using HF·Pyr complex in 72% yield to give a product identical to the reported S-minquartynoic acid (2) in all spectroscopic determinations.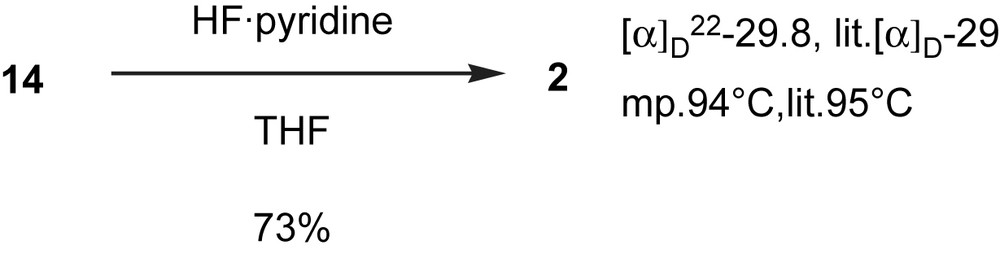
After our success in the synthesis of (−)-minquartynoic acid (2), it was hoped that (−)-18-hydroxyminquartynoic acid (3) would be available by the same strategy from a three-component cross-coupling of butadiyne 6 and bromoalkynes 7 and 19. The bromoalkyne 19 was prepared from d-glyceraldehyde 17 via the dibromoolefin intermediate 18 (Scheme 5) [34].
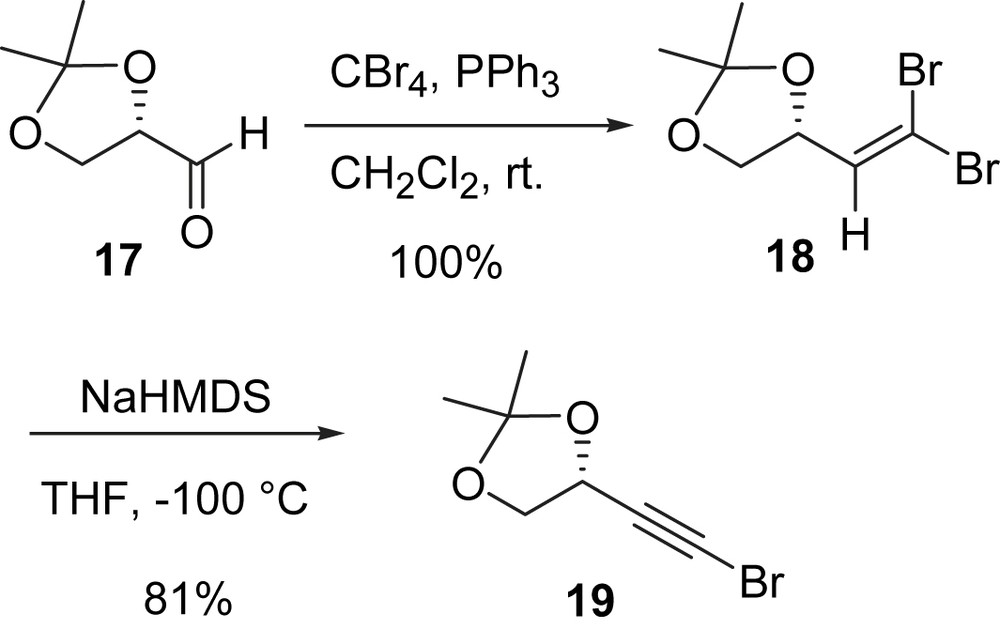
Bromoalkyne 19, butadiyne 6, and bromoalkyne 7 were subjected to a Cadiot–Chodkiewicz coupling [35]. Disappointing results were initially obtained using the same conditions for the synthesis of (−)-minquartynoic acid (1 mmol of each of the three reactants were dissolved in a 2 mL 1:1 mixture of methanol and 70% ethylamine aqueous solution with 5 mol% of NH2OH·HCl and 5 mol% of CuCl and the mixture was stirred at 0–25 °C for 3 h). Bromoalkyne 19 undergoes rapid cross-coupling with butadiyne followed by the addition of 1 equiv of ethylamine. The enamine product 20 was consistently obtained in good yield (Scheme 6). There is apparently a large difference in the reactivity between bromoalkyne 19 and bromoalkyne 7.
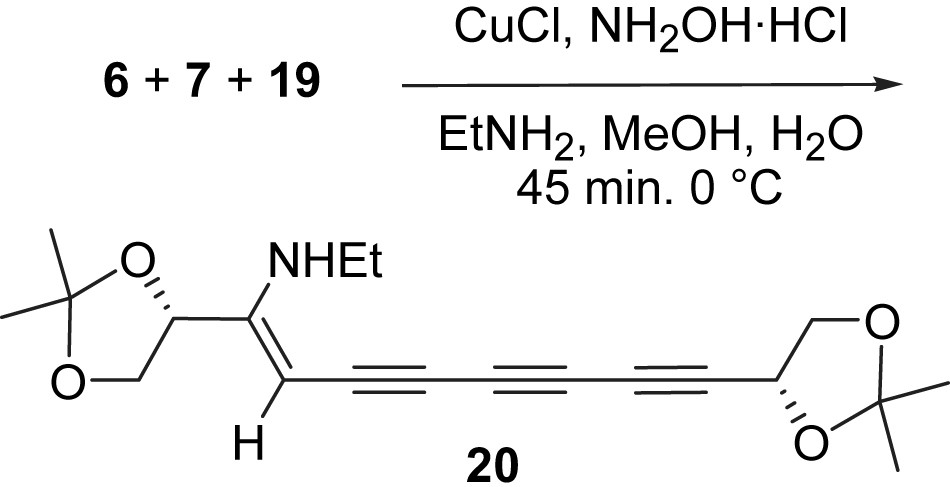
The conditions for the three-component couplings were therefore modified based on this analysis [35]. To avoid the addition of the EtNH2 to the tetrayne intermediate, ethylamine was reduced from 1.0 mL to 0.3 mL for one mmol of each component. It is known that polar terminal alkynes usually afford higher yields in the Cu(I) catalyzed cross-coupling reactions [2,36], it was surmised that polar 1-bromoalkynes might also react better in polar solvents. The acetal group was removed from bromoalkyne 19 to give diol 21 with the treatment of a catalytic amount of p-TsOH in MeOH.
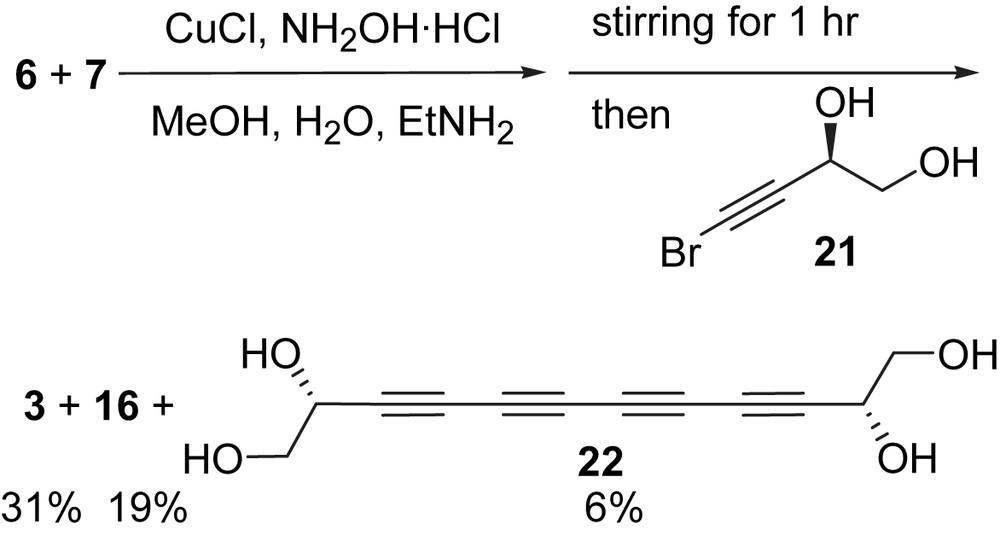
Furthermore, the less reactive bromoalkyne 7 was allowed to react with butadiyne 6 for 1 h before bromoalkyne 21 was added to the reaction mixture (Scheme 7). A solid (mp. 135 °C, 3) was obtained in 31% yield after column chromatography, which gave identical spectroscopic characteristics with that reported for the natural (S)-18-hydroxyminquartynoic acid. Even under these conditions, the symmetric coupling product tetraol 22 was isolated in a substantial 19% and the known diacid 16 in just 6% yield [35].
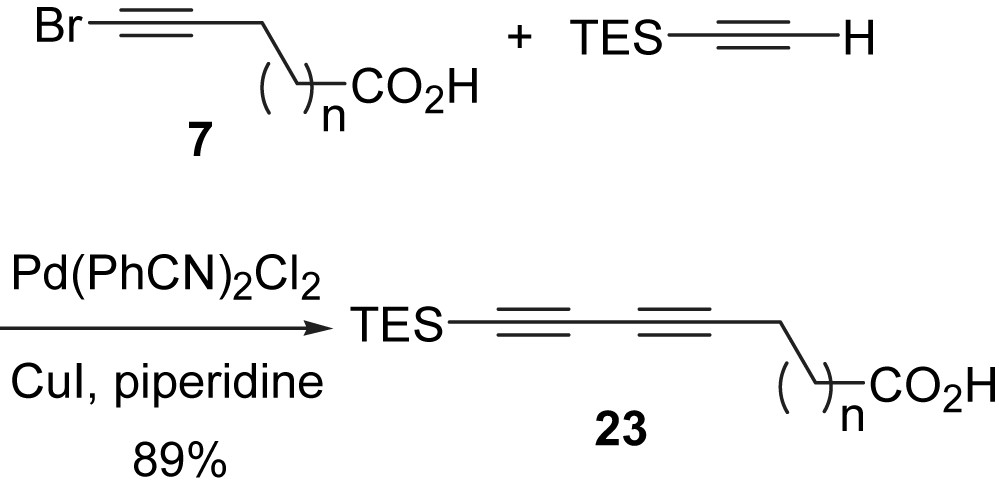
The remaining member yet to be synthesized at the time was E-15,16-dihydrominquartynoic acid (4). While all three compounds showed anti-cancer activities, E-15,16-dihydrominquartynoic acid (4) showed the most potent activity against the human hormone-dependent prostate and ovarian cancer cell lines [21]. E-15,16-dihydrominquartynoic acid (4) is similar to (−)-minquartynoic acid (2) [20,22] and 18-hydroxyminquartynoic acid (3), in that it contains a chiral secondary alcohol and a carboxylic acid functionality, Fig. 1, but differs in that it contains a conjugated enetriyne function, rather than a tetrayne function. Our three-component one-pot Cadiot–Chodkiewicz cross-coupling approach succeeded in giving reasonable yields for otherwise statistical coupling reactions. Although both natural products were successfully synthesized, the statistical nature of the final coupling step was unsatisfactory. For the total synthesis of the third member in this group of natural products, (E)-15,16-dihydrominquartynoic acid (4), a convergent and non-statistical approach was carried out [37]. The required reactive diyne intermediate involved in the total synthesis was obtained in a triethylsilyl protected form (23, Scheme 8).
The other half in the convergent approach towards (E)-15,16-dihydrominquartynoic acid (4) was the enyne bromide 27. The preparation of enyne bromide 27 is depicted in Scheme 9. The known aldehyde 24 was prepared from (S)-methyl lactate in two steps [28], which was then treated with a suspension of CrCl2 in CHCl3 according to the procedure of Takai to give a high yield and high diastereoselectivity (E/Z ratio = 95:5) of vinyl chloride 25 [38]. The cross-coupling of 25 with trimethylsilylacetylene proceeded smoothly under the modified Sonogashira conditions (Pd(PhCN)2Cl2, CuI, piperidine) reported by Alami and Linstrumelle [39,40]. In situ desilylation/bromination of enyne 26 to bromoalkyne 27 was accomplished under mild conditions [41].
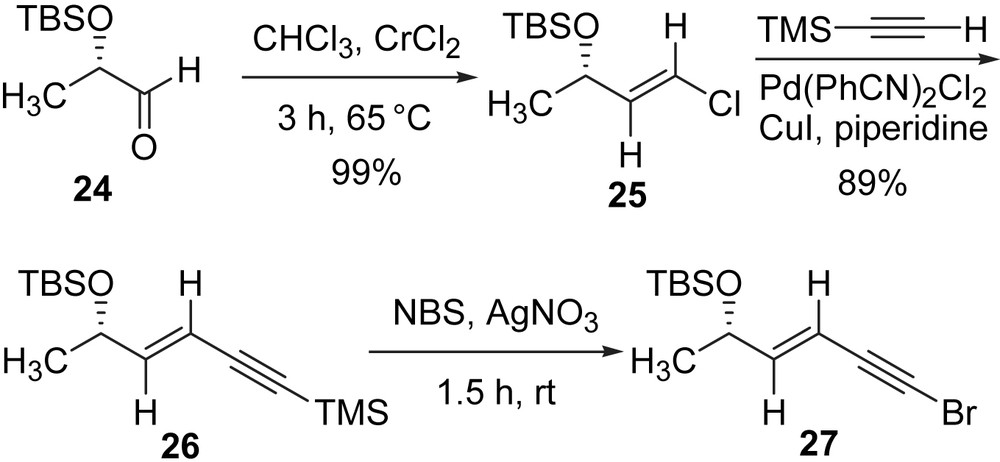
To avoid the isolation of reactive intermediate, a one-pot desilylation/oxidative coupling of trialkylsilyl-protected terminal alkynes was attempted to prepare the final product (Scheme 10). After a few trials with different proportions of reagents and different solvents, the desired cross-coupling product 28 was obtained in 60% yield by treatment of 23 with 1 equiv of n-BuN4F in THF for 1 h followed by the addition of 27, CuCl, HONH2·HCl, EtNH2, MeOH and stirring at 0 °C for 3 h. The final step is the removal of the t-butyldimethylsilyl ether protecting group using HF–pyridine complex to yield (E)-15,16-dihydrominquartynoic acid (4) in 77% yield [37].
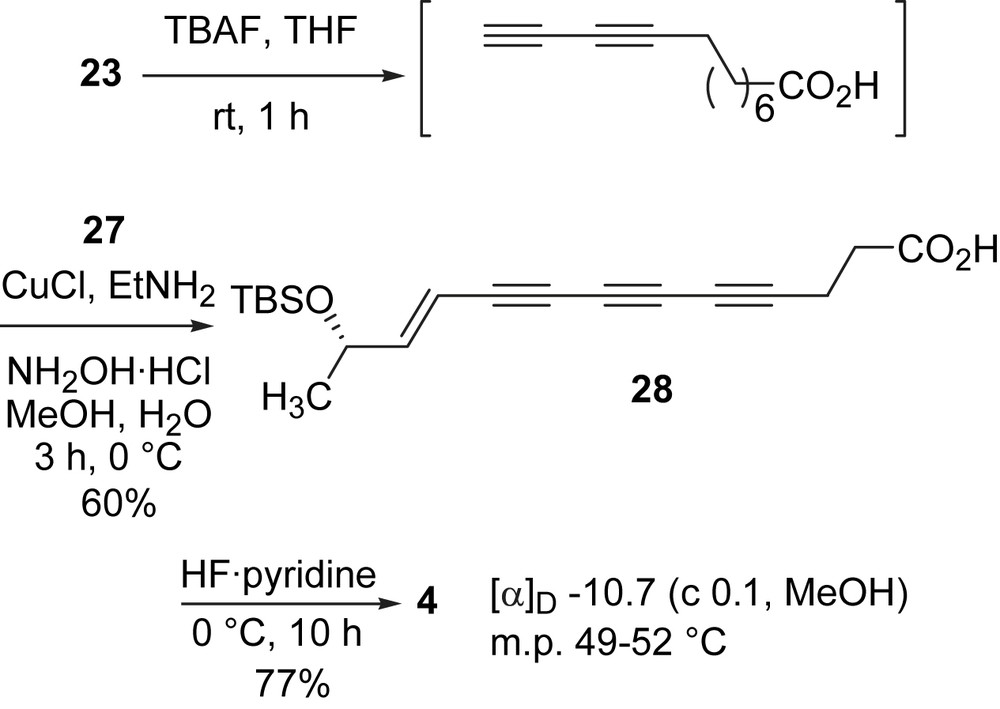
Soon after the first synthesis of (E)-15,16-dihydrominquartynoic acid (4) was reported [37], Kim and coworkers reported the synthesis of this natural product (Scheme 11) with an innovative iterative approach [42]. The iterative strategy entailed a two step acetylene homologation sequence: (1) in situ desilylative bromination of alkynylsilanes and (2) cross-coupling with a trialkylsilyl acetylene. It was found that a combination of the reagents and solvent (AgF/NBS/CH3CN) system was effective in the one-pot conversion of a TIPS-protected terminal acetylene to a bromoalkyne. When using this combination of reagents, the desired bromoalkyne was obtained at room temperature. Conjugated polyynes can be constructed with ease when this one-pot desilylation/bromination procedure is coupled to a Pd-catalyzed coupling of the resulting bromoalkyne with a TIPS-protected acetylene. Thus from bromoalkyne 29, a coupling reaction to TIPS-acetylene followed by a one-pot desilylation/bromination yielded bromodiyne 30. An iteration of the same sequence of reactions produced triyne bromide 31 in 47% for two cycles. A Stille coupling of the triyne bromide 31 and the vinylstannane followed by hydrolysis of the methyl ester gave (E)-15,16-dihydrominquartynoic acid (4) in 63% yield for two steps [42].
A report in 2006 described an approach to all three minquartynoic acid derivatives using the Cadiot–Chodkiewicz coupling reaction as the key step [43]. It is somewhat surprising that this report contains uncapped terminal diyne intermediates throughout both oxidation and reduction sequences, in contrast to previous reports on the instability of these intermediates [10,17,24–26]. No discussion was presented regarding the behavior and handling of these reactive intermediates either [43].
3 Bidensyneosides
Naturally occurring polyyne glycosides have been shown to possess a host of different biological activities although the synthesis of these glycoconjugates has not been widely investigated [44]. Bidens parviflora is a plant genre that has been used in traditional Chinese medicine as an antipyretic, anti-inflammatory, and antirheumatic remedy [45]. B. parviflora contains a wide variety of bioactive compounds including sterols, monoterpenes, flavonones, flavonoids, polyacetylene glucosides, chalcones, aurones, and flavonol glycosides [45]. Over the years, several species of the Bidens family have been investigated by chemical methods [46–50]. Polyacetylenic glucosides isolated from B. parviflora, the bidensyneosides (33–37, Fig. 2) have been shown to inhibit histamine release in rat mast cells and the production of nitric oxide. Their interesting biological activities and chemical structures have started to attract attentions in their synthetic studies [18,19,51].
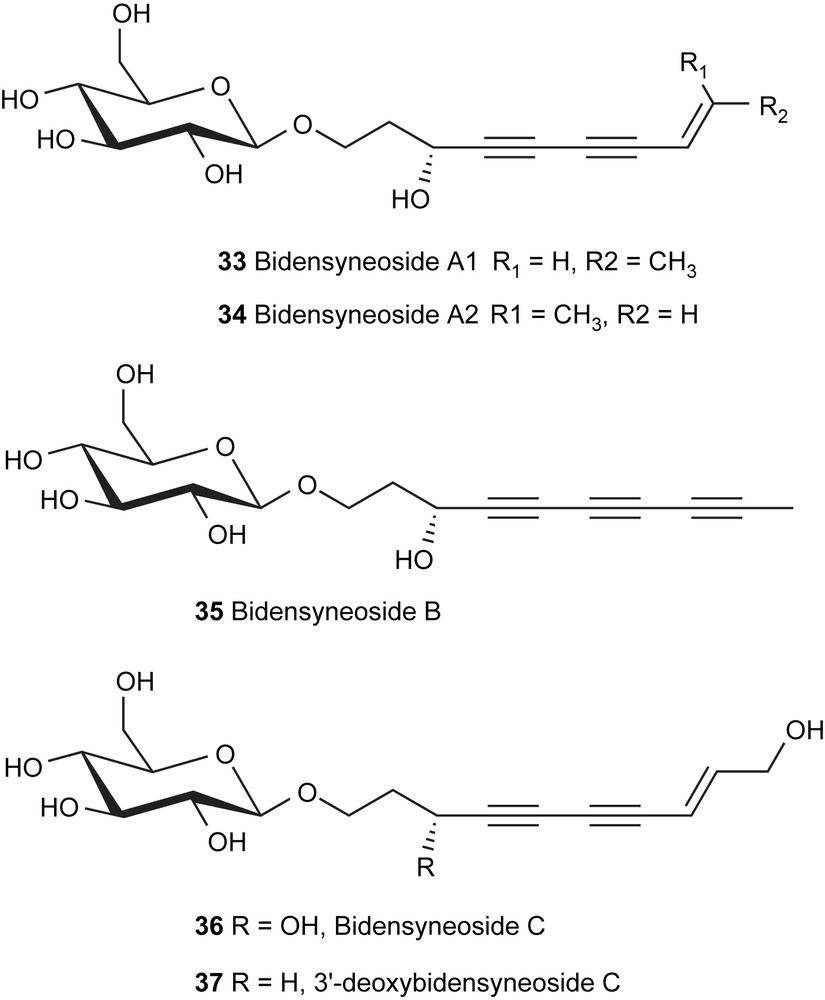
Structures of bidensyneosides from Bidens parviflora WILLD.
Since the bidensyneosides differ among themselves mainly in the aglycon side chain, the development of a strategy allowing for the convergent assembly of different side chain analogs was of importance in our synthetic planning. To study the glycosylation of glucose with an acetylene-containing alcohol, 3′-deoxybidensyneoside C (37) was chosen to start our synthetic study in this series of natural products.
Our initial study was the glycosylation reaction between glucose pentacetate (38) and 4-pentyn-1-ol in the presence of BF3·OEt2 [52]. Both starting materials are available commercially. The desired product 39 was obtained in 30% yield (Scheme 12). The low yield of the desired product 39 was probably due to an achimerrically-assisted deglycosylation, as reported for 4-pentenyl glucosides [53]. Bromo enyne 40 was prepared from commercially available (E)-2-penten-4-yn-1-ol via (1) hydroxyl protection as the t-butyldimethylsilyl ether, and (2) bromination with NBS in the presence of AgNO3 [54]. The coupling of bromoalkyne 40 and glucoside 39 was carried out under Cadiot–Chodkiewicz conditions which, proceeded with concurrent removal of acetate groups by EtNH2, leading to the polar glucoside 41 in 31% yield. Attempted coupling with the unprotected bromo enyne resulted in loss of product during work-up due to difficulties in separating the extremely polar 37 from the solvent. The problem was alleviated by using a TBS ether protecting group. Glucoside 41 could be extracted from the aqueous solution and purified on a silica gel column. Removal of the TBS protecting group from 41 was achieved in THF with the HF·pyridine complex. To avoid the loss of the product, the work-up consisted of adding solid NaHCO3 and evaporating the solvent to slurry, which was transferred to a silica gel column and eluted with a mixed solvent system (MeOH/CHCl3, 10:90) to give 51% of 3-deoxybidensyneoside C (37) as a white solid.
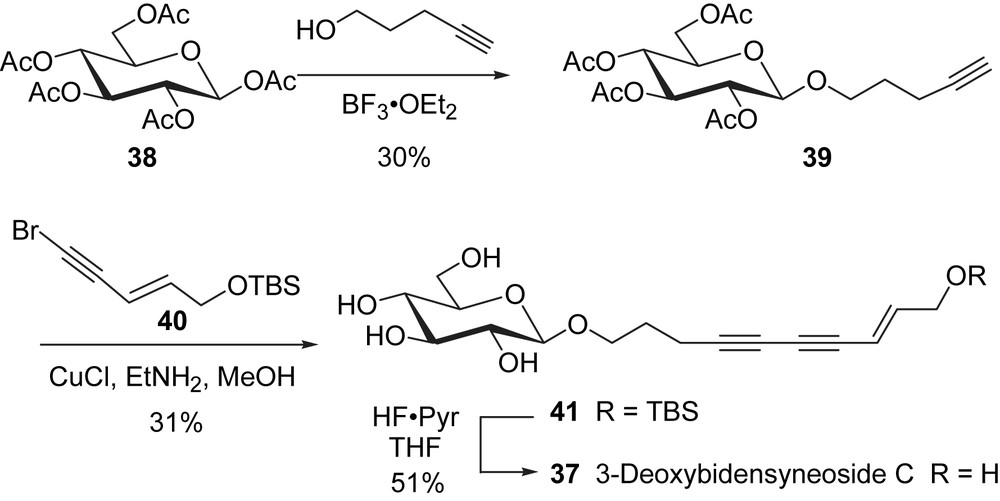
Having prepared a member of the bidensyneoside family of natural products, we turned our attention to the remaining glucosides (33–36, Fig. 2). One structure feature in common for these polyyne glucosides is the stereocenter on the side chain. This chiral center on the side chain was introduced by using a convenient enzymatic resolution approach (Scheme 13). The THP and TBS protected aldehyde 43 were prepared from 1,3-propanediol (42). Oxidation of 43 under the conditions of Mancuso and Swern [55] and in situ addition of ethynylmagnesium bromide to the resulting aldehyde afforded the racemic propargyl alcohol 45 in 61% yield [56]. The hydroxyl protecting group at C1 of rac-45 has a profound influence on the lipase-mediated kinetic resolution. The TBS ether group, unlike the THP, provides the steric bulk required for an efficient resolution and material of greater than 95% ee was obtained when this ether was treated with Amano lipase AK in the presence of vinyl acetate in hexanes [57]. After separation of the alcohol R-45 from the acetate 46 by column chromatography, both enantiomers were obtained in high optical purity as confirmed by the 1H NMR spectra of their O-methyl mandelic esters [58]. The final two steps in the preparation of the side chain involved the acylation of the secondary OH group in R-45 and the removal of the TBS ether with HF·pyridine complex to afford the optically pure alcohol (R)-47.
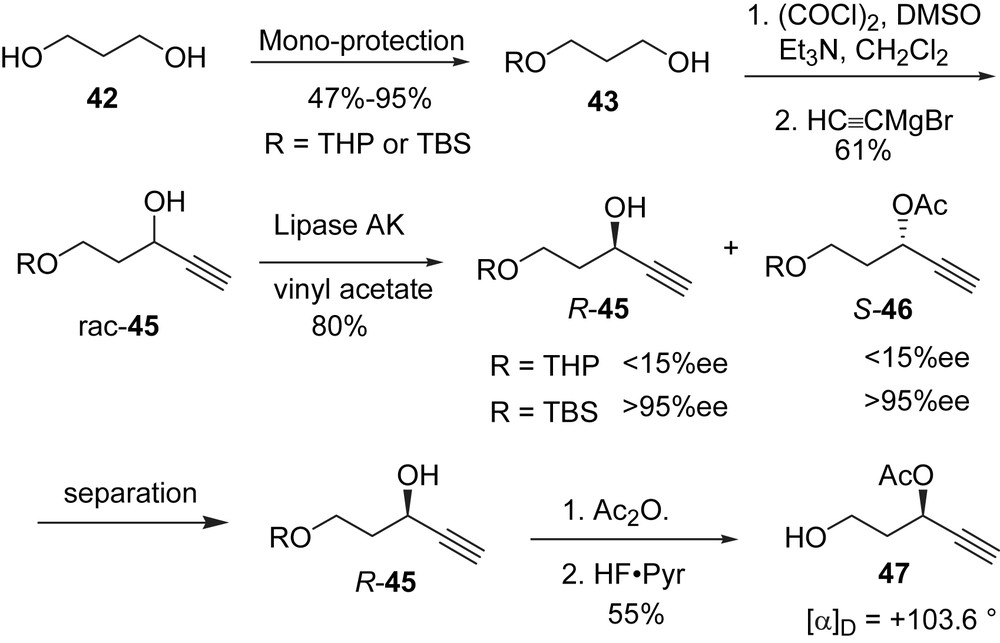
The reason for the low yield of glycosylation product during the synthesis of 3′-deoxybidensyneoside C (37) was attributed to achimerrically-assisted deglycosylation for 4-pentynyl glucosides [53]. Since it took five steps to prepare the optically pure side chain fragment 47, a low yield in the glycosylation step would reduce the efficiency of the synthesis. A more reactive glycosyl donor than glucose pentaacetate (38) was needed to allow mild conditions for the union of the glucose and the side chain. We chose thioglucosides as the glycosylation donors because it is known that these donors are stable, and they couple with a variety of acceptors under mild conditions [59].
The thioglucoside 48 with four acetate groups (Scheme 14) was not reactive enough as a glycosyl donor in the presence of the promoter dimethyl(methylthio) sulfonium triflate (DMTST) [60]. Some of the acetate protecting groups was replaced with the more electron-donating TBS ethers to increase the reactivity of the glycosyl donor. Thus, thioglucoside 48 was deacetylated with K2CO3 in MeOH (Scheme 14) and the crude tetraol 49 was treated with either one or 2 equiv of TBSCl and imidazole in DMF. This led to the selective formation of either the 6-TBS ether 52 or the 3,6-di TBS ether 53, respectively. Bidensyneosides have the β-configuration at the anomeric carbon. A C2-acetate group is necessary to provide neighboring group assistance for control of stereochemistry in the glycosylation step [61]. According to a study by Wong, replacing a C6 acetate group with TBS increases the glycosyl donor reactivity by a factor of 3 to 5× [62].
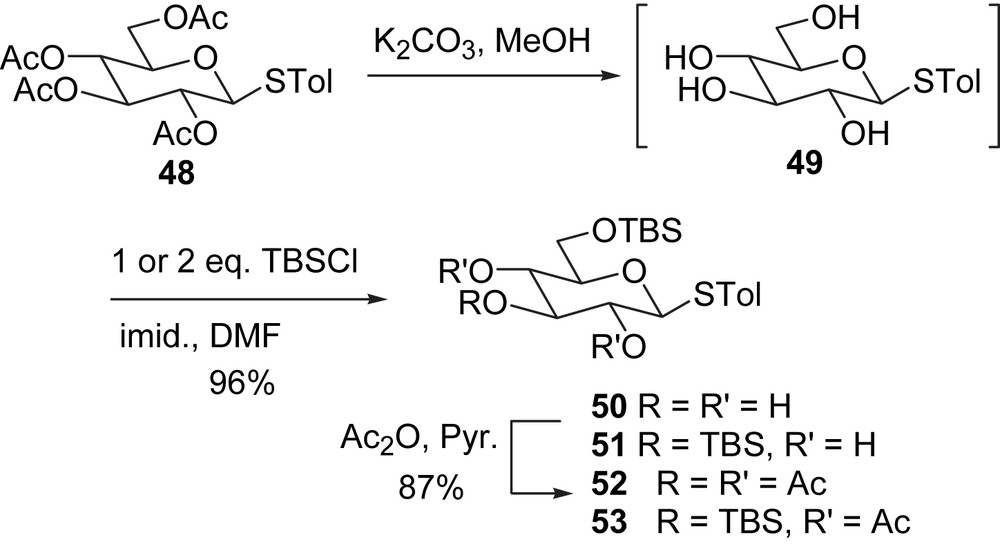
We first examined the behavior of the glycosyl donor 52 (with one TBS ether group) (Scheme 15). When thioglucoside 52 and the side chain alcohol (R)-47 were allowed to react in the presence of DMTST for 1 h at room temperature, surprisingly the orthoester 54 was isolated in 74% yield, rather than the normal anomeric coupling product (Scheme 15). This product suggested an internal nucleophilic attack of the anomeric carbocation by the neighboring acetate carbonyl oxygen, which is followed by the attack of the alcohol 47 on the acetate carbonyl carbon to give orthoester 54.
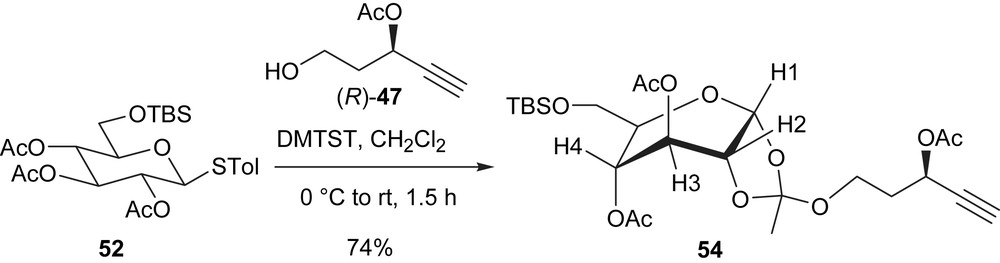
When the more reactive glycosyl donor 53 (with two TBS ether groups) was allowed to react with 47, the reaction was instantaneous in the presence of DMTST (Scheme 16). Within 5 min at 0 °C, TLC analysis indicated no starting material remaining and the glycosylation product 55 was isolated in 68% yield with no orthoester formation. The dramatic difference between the two donors 52 and 53 originates from a single protecting group at C3. An explanation is that both reactions proceed through the initial formation of the orthoester, which rearranges more rapidly in the case of 53 to the normal glycoside [63].
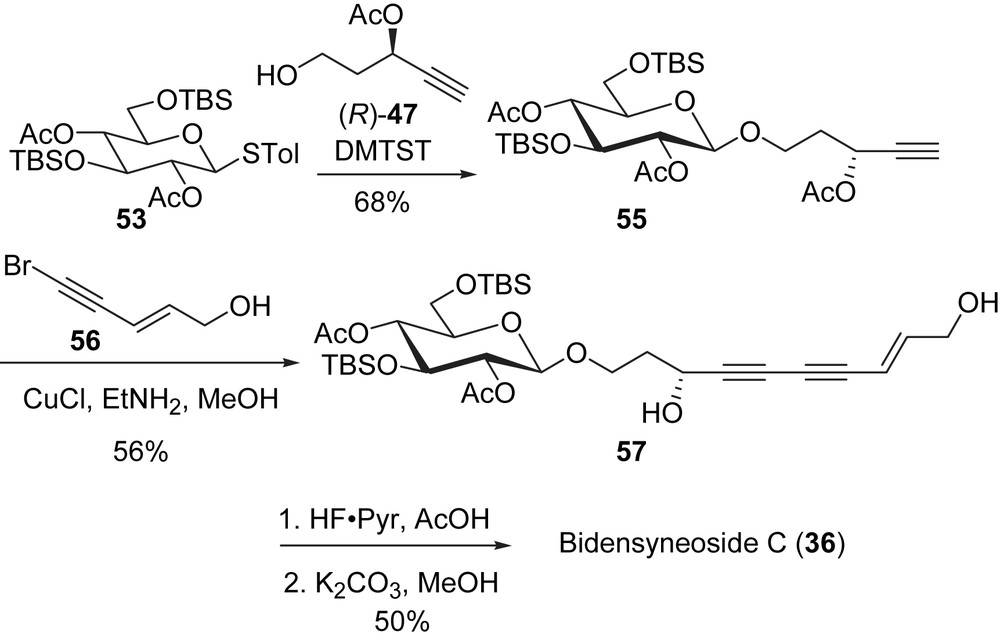
The copper promoted coupling reaction between 55 and the bromoalkyne 56 was carried out under the Cadiot–Chodkiewicz conditions, which furnished bidensyneoside C in its protected form. Interestingly, only the side chain C3 acetate group was removed during the reaction; the two acetate groups on the glucose ring remained intact. The steric bulk of the TBS ethers in 57 may have prevented EtNH2 from attacking these esters. Removal of the protecting groups was undertaken in the sequence shown in Scheme 16 to give the natural product bidensyneoside C (36).
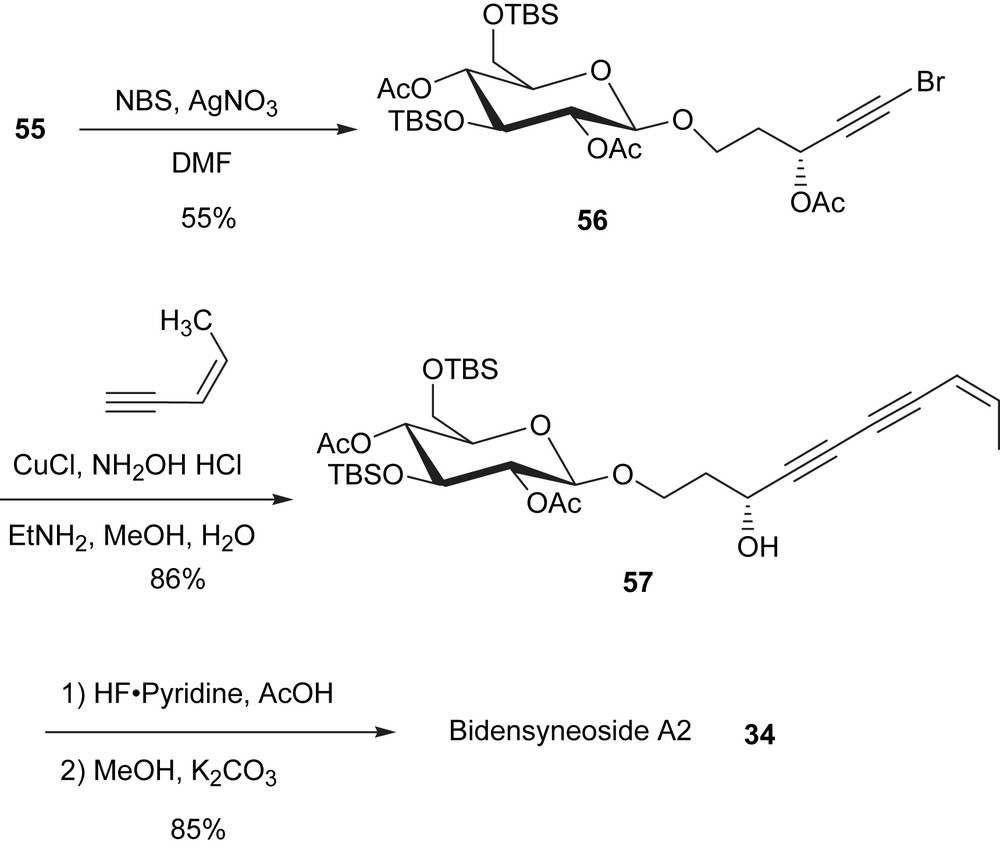
Once the appropriate glycosyl donor was found, the synthesis of the remaining members of the bidensyneoside family was considered a matter of preparing the side chains and couple to the glycosyl donor 53. However, it was operationally easier to use compound 55 as the left hand piece because the volatility of the low molecular weight fragments such as (Z)-3-penten-1-yne. Thus, compound 55 was treated with NBS and AgNO3 to convert the terminal alkyne to a bromoalkyne (Scheme 17). In the bromination reaction of the terminal alkyne 55, the preferred solvent was DMF, instead of acetone, to minimize the breakage of the glycosydic bond. The copper-catalyzed coupling of bromoalkyne 56 with (Z)-3-penten-1-yne proceeded smoothly to produce the protected bidensyneoside A2 in 86% yield. This is consistent with the observation that a propargylic oxygen substitution in the bromoalkyne enhances the rate of the copper-catalyzed cross-coupling reaction to produce conjugated diynes [35]. Without a propargylic oxygen substitution, homocoupling of the bromoalkyne sometimes dominates. The removal of the TBS ether and the acetate protecting group was performed in that order affording compound 34 in 85% yield for two steps. The synthetic sample was identified by spectroscopic methods and the results are consistent with the reported natural product, bidensyneoside A2.
The corresponding enyne fragments for bidensyneoside A1 (33) and B (35) are (E)-3-penten-1-yne and 1,3-pentadiyne, which are not available commercially. Bidensyneoside A1 (33) differs from its closest relative bidensyneoside A2 (34) only by the configuration of the double bond in the aglycone side chain. Since the left hand main fragment 56 was already prepared [18], to prepare (−) bidensyneoside A1 (1) and C (3), the task at hand was to prepare (E)-3-penten-1-yne and 1,3-pentadiyne, respectively. There are several reported procedures for 1,3-pentadiyne. However, the preparation of the (E)-isomer of 3-penten-1-yne turned out to be more difficult than expected.
The preparation of (E)-3-penten-1-yne started from the readily available 1,1-dibromo-1,3-pentadiene (5) [64], which was prepared from (E)-crotonaldehyde using Corey–Fuchs protocol (Scheme 18) [29]. A solution of 5 in hexane was treated with 2.2 equiv of n-BuLi at −78 °C and quenched with saturated NH4Cl solution after 1 h (Scheme 18). The volatility of the desired product made its isolation difficult for small scale operation, which was overcome by bubbling nitrogen through the solution of the product and the desired product was collected in MeOH solution. In the experiment, a stream of N2 was allowed to pass through the hexanes' layer and travel through two traps containing MeOH at −78 °C. The concentration of the (E)-3-penten-1-yne in MeOH solution was determined by integration of the relative peak heights in 1H NMR spectrum. The MeOH solution was used directly in the cross-coupling reaction.
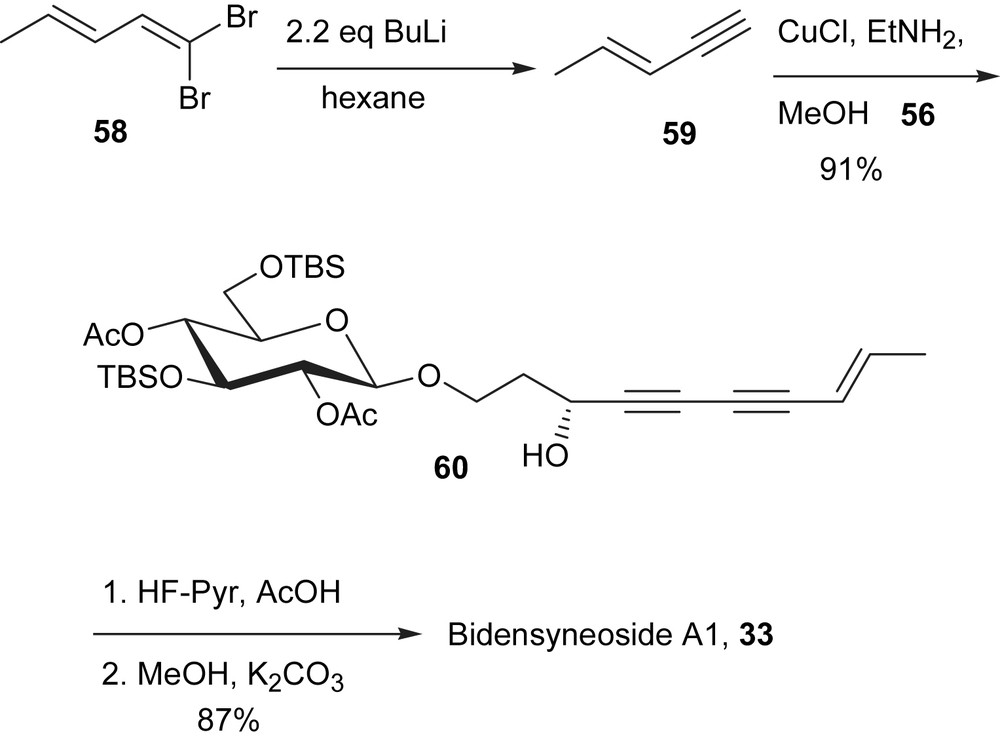
Bromoalkyne 56, which was available as shown in Scheme 17 [18], along with (E)-3-penten-1-yne (59) were then subjected to the copper-catalyzed cross-coupling reaction to give the desired compound 60, the precursor to bidensyneoside A1 (33), in a remarkable 91% yield. The removal of the protecting groups of 60 was carried out by first removing the TBS groups with HF·pyridine complex in acetic acid, followed by removal of the acetate groups using a catalytic amount of K2CO3 in methanol. These two steps led to the isolation of (−) bidensyneoside A1 (33) in 87% yield (Scheme 18). All spectroscopic data of the synthetic sample are consistent with that reported for the natural product [45].
Bidensyneoside B containing a conjugated triyne was the most intriguing target of this series of compounds due to the highest degree of unsaturation on its polyacetylenic side chain. The method for preparing 1,3-pentadiyne reported by Brandsma was chosen, which involved a double dehydrohalogenation of 1,4-dichlorobut-2-yne with sodamide in liquid ammonia followed by in situ methylation with methyliodide [65]. With 1,3-pentadiyne and the advanced intermediate 56 in hand, a cross-coupling reaction under Cadiot–Chodkiewicz conditions (Scheme 19) led to the desired triyne 61 [27].
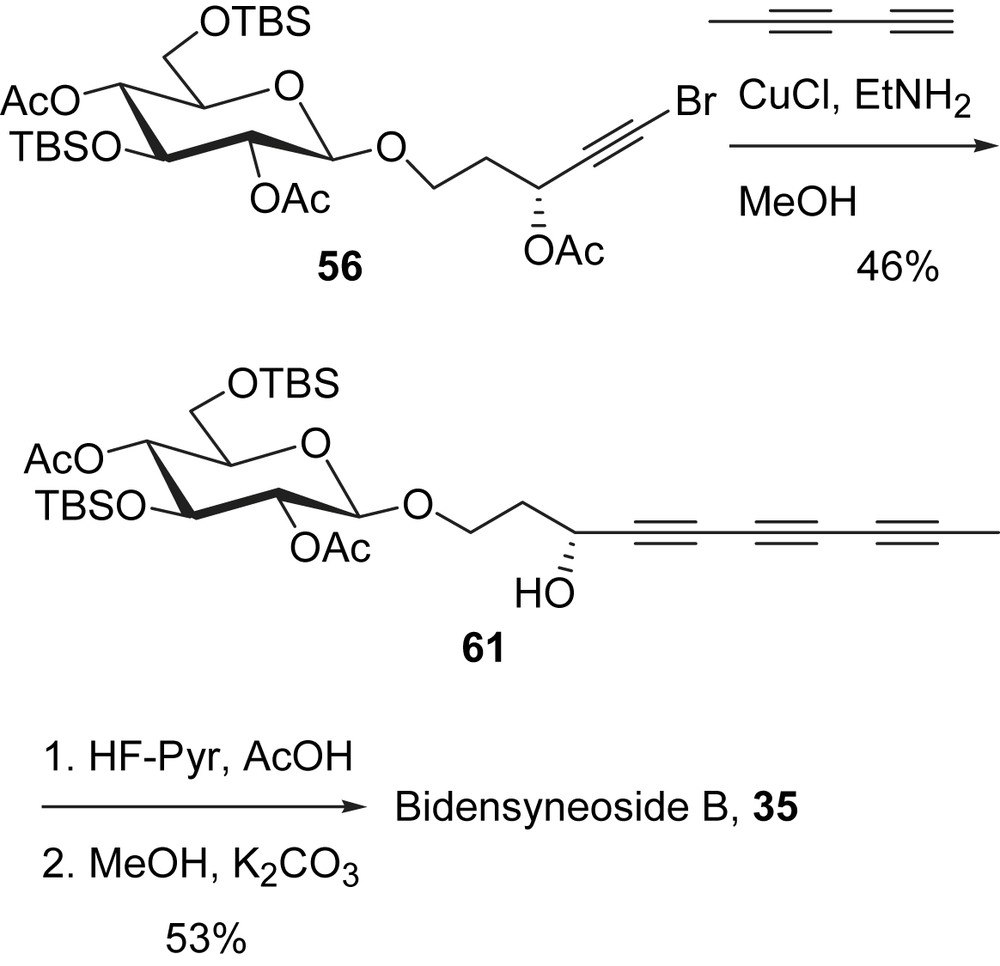
The triyne moiety in 61 appears to suffer from decomposition because the yield of the coupling reaction was not as high as in the previous cases. The removal of the protecting groups on 61 was carried out analogously by first removing the TBS groups and then the acetate groups, which led to bidensyneoside B (35) in 53% yield. All spectroscopic data of the synthetic sample are consistent with that reported for the natural product [45]. During the purification of the final product, a surprisingly polar property was observed for bidensyneoside B (35). The structures of bidensyneoside B (35) and bidensyneoside C (36) differ in their side chains only. Endiyne (36) has a primary OH group while triyne (35) has a methyl group. However, 35 is considerably more polar than 36. In fact, 35 is so polar that the mobile phase for TLC and column chromatograph must contain 5% H2O in a mixed solvent system (H2O:MeOH:CHCl3 = 5:35:60) in order to elute 35 successfully. This unusual polarity cannot be attributed to the usual polar functional groups, such as OH, because the structures of 35 and 36 are identical except for the polyacetylenic side chains. One possible explanation invokes the CH–O hydrogen bonds. The three conjugated triple bonds are so electronegative that the terminal methyl group becomes acidic and forms CH–O hydrogen bonds with silica gel. Even though a CH–O bond is much weaker than an OH–O bond, the three CH–O bonds in the side chain of 35 might have a greater affinity for silica gel than a single OH–O interaction.
4 C2 Symmetric acetylenic natural products
Acetylenic natural products possessing a C2 symmetry are a fascinating class of compounds that have inspired a theoretical study regarding their greater chance of appearance in nature [66]. It has been shown that natural products with a C2 symmetry are present at a higher proportion in nature and are likely to arise by dimerization of a simpler compound. Density functional theory was employed to demonstrate that more energy per connecting bond is evolved to form dimers than to form the corresponding trimers or tetramers. This was proposed to be a guiding parameter that may adjust molecule growth [66]. Organic chemists have always been intrigued by nature's symmetry and strategies have been developed to synthesize symmetrical molecules with greater efficiency [67]. Barrett and coworkers reviewed the syntheses of C2 symmetric natural products and have classified the syntheses into two broad categories: ‘core expansion’ and ‘dimerization’ [68]. It will be seen that our syntheses of C2 symmetric acetylenic natural products (Fig. 3) involved both ‘core expansion’ and ‘dimerization’ [16,69].

C2 symmetric acetylenic natural products: adociacetylene 1, duryne 62, and dideoxypetrosynol A 63.
4.1 Adociacetylene B (1)
Adociacetylene B 1 was a cytotoxic constituent of an Okinawan Adocia sp. that exhibited activity in an in vitro endothelial cell-neutrophil leukocyte adhesion assay [70]. This natural acetylenic alcohol became our inaugural synthesis in 2001 [16].
At the time when our inaugural synthesis of adociacetylene B (1) began, the majority of approaches to the synthesis of chiral acetylenic alcohols involved numerous functional group transformations. In contrast, our approach avoided using protecting groups and only limited number of steps was employed in our synthesis. A two-directional strategy was employed to take advantage of the C2 symmetry [71,72]. This is also known as the ‘core expansion’ according to Barrett [68]. The starting materials used in our synthesis were 2,5-dibromofuran 64 and 8-nonyn-1-ol 65, Scheme 20.
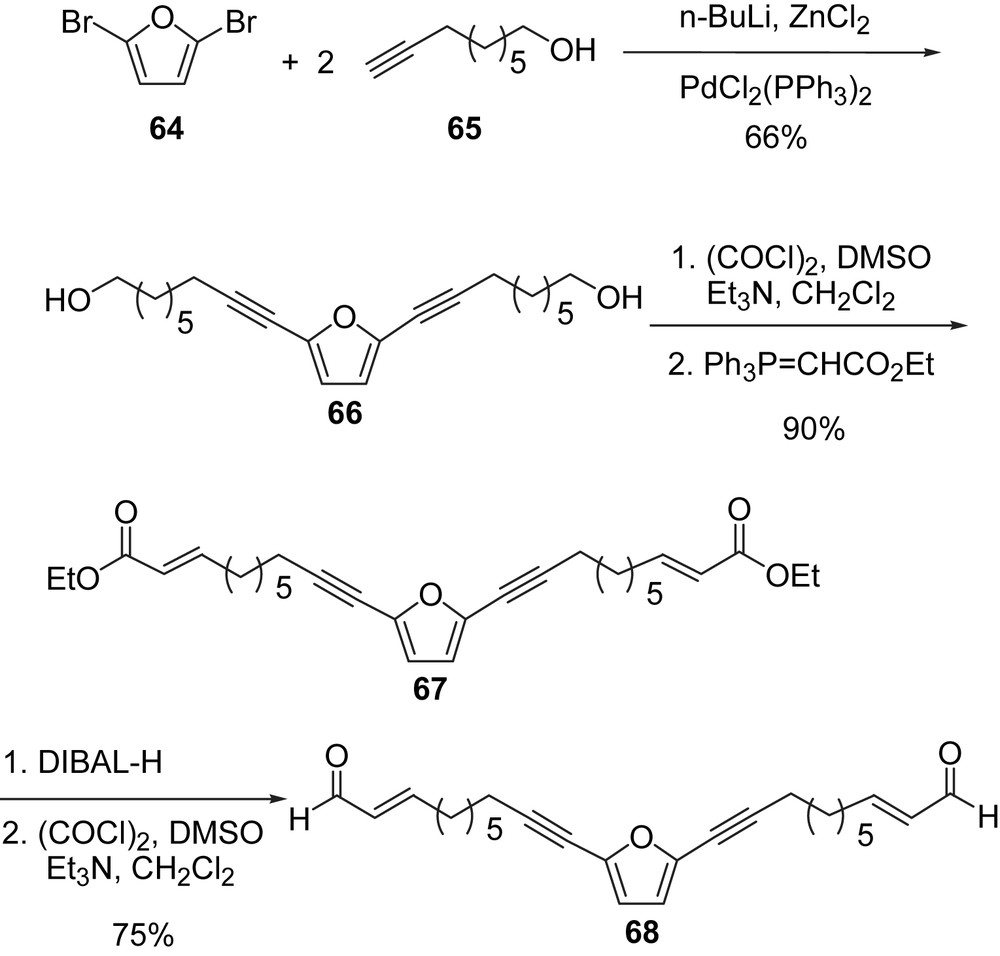
The coupling of the terminal acetylene 65 with 2,5-dibromofuran 64 was successfully carried out using conditions reported by Negishi [73], which involve metalation of the terminal acetylene with n-BuLi/ZnCl2 followed by palladium-catalyzed coupling. Swern oxidation of the primary alcohol 66 followed by in situ Wittig reaction using an ester-stabilized ylide afforded the (E)-isomer of the conjugated ester 67 as the major component of a 9:1 E/Z mixture. The minor (Z)-isomer was not separated, but appeared to have isomerized to the (E)-isomer after the Swern oxidation. The α,β-unsaturated aldehyde 68 was obtained through a diisobutylaluminum hydride (DIBAL-H) reduction of 67 followed by a Swern oxidation of the resulting alcohol.
The next step should have been an enantioselective addition of a terminal alkyne to aldehydes. If a method of direct asymmetric addition of acetylene to aldehyde 68 was successfully employed, the stereocenters and the terminal acetylene groups would be introduced in a single step. Unfortunately, our attempts of using Carreira's method [74] with Et3SiCCH were unsuccessful despite numerous trials with up to three-fold of reagents. Et3SiCCH and α,β-unsaturated aldehydes are apparently not good substrates for this method.
Next we turned our attention to enzymatic resolution. The racemic adociacetylene B (1) was obtained along with the meso isomer by the addition of acetylenic magnesium bromide to aldehyde 68 (Scheme 21). In a study of enzymatic resolution of secondary alcohols by the lipases from Pseudomonas sp. Burgess proposed a simple active site model for predicting enantioselectivity [75]. This model predicts that alcohols resolved most efficiently have one small and one relatively large group attached to the hydroxylmethine functionality. Adociacetylenes turned out to be excellent substrates for these lipases.
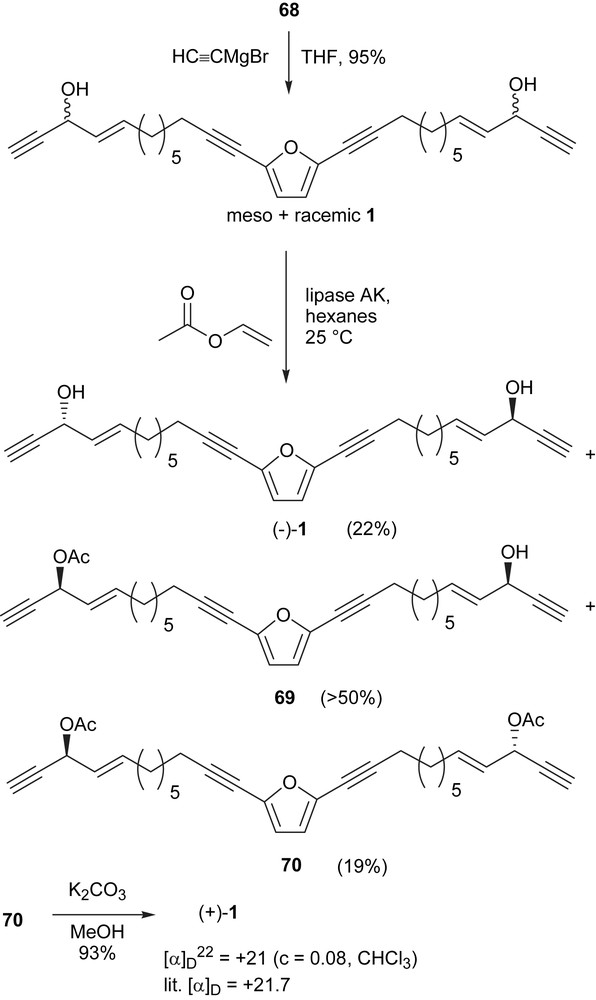
For most secondary alcohols, the rate of acylation is faster for the (R)-configuration than for the (S)-configuration. However, for the acetylenic alcohol 1 the opposite is true because the small acetylenic group has a higher priority in the nomenclature system.
Kinetic resolution of racemic and meso 1 mediated by lipase AK from Amano was performed by stirring the propargyl alcohols in hexane with 2:1 enzyme to substrate mass ratio and 4 equiv of vinyl acetate. The reaction was monitored quantitatively by 1H NMR, and qualitatively by TLC. The hydroxymethine proton is characteristic of the change between the starting material 1 and the mono-acylated product 69 and the diacetate 70. Molecular sieves were added to remove traces of water in the enzyme mediated reaction. Work-up of the reaction consisted of simple filtration, rotary evaporation, and flash column separation of the diacetate 70 from the monoacetate 69 and the diol (−)-1. Adociacetylene B (1) diacetate 70 was treated with K2CO3 in methanol to provide the diol, whose optical rotation was found to be [α]D22 = +21. The reported optical rotation for natural adociacetylene B (1) was [α]D = +21.7 [70].
When our synthesis of adociacetylene B (1) was in press, we became aware that this natural product had been previously isolated from Petrocia sp. [76] and was named petrofuran. An enantioselective synthesis of petrofuran (so was it named) was accomplished by Garcia and coworkers in 1999 [77]. A similar strategy was used to prepare the carbon frame. The stereocenters was set by an enantioselective reduction using Corey's oxazaborolidines [78] (Scheme 22).
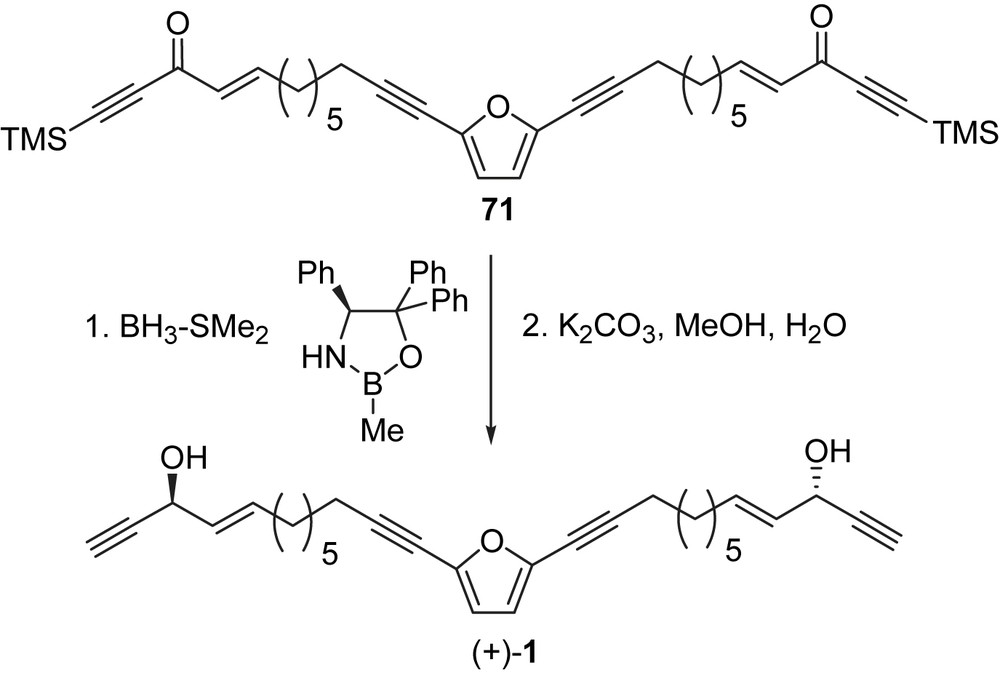
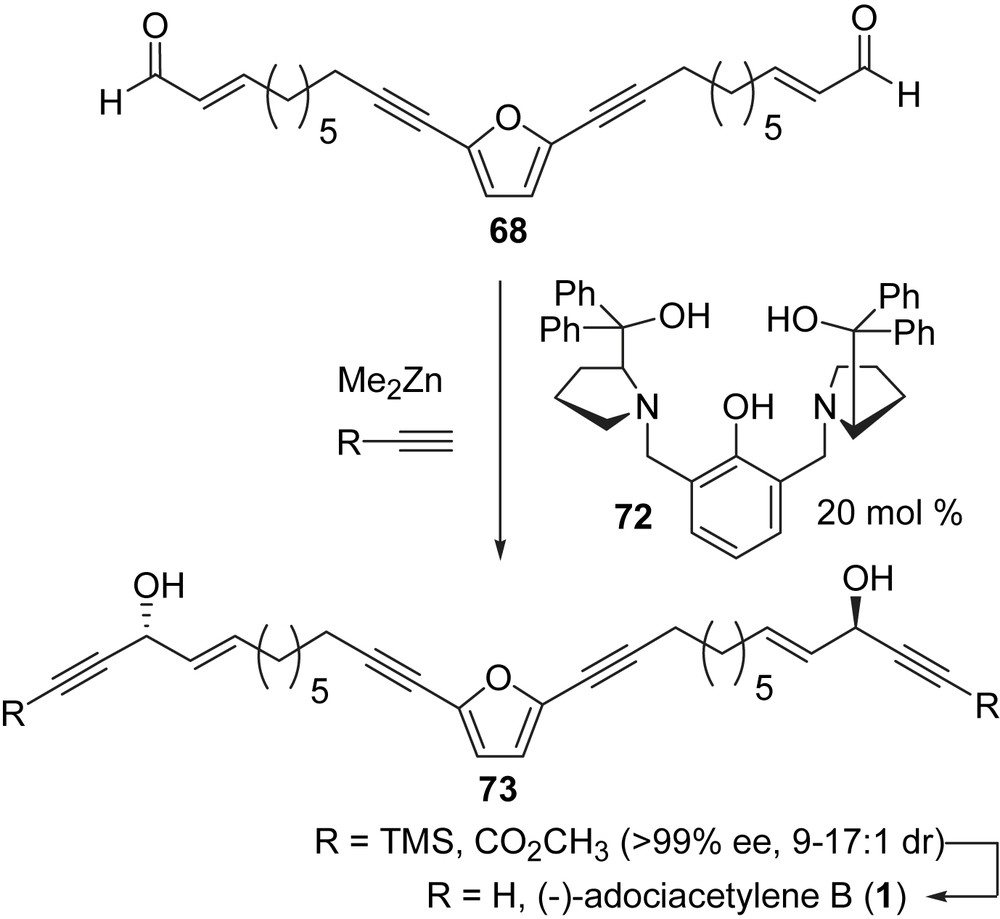
In 2006, Trost and Weiss reported a catalytic enantioselective total synthesis of adociacetylene B (1) in five steps [79]. The efficiency of this synthesis was enabled by an asymmetric zinc alkynylation catalyzed by the proline-derived ligand (72, Scheme 23). The chiral ligand, 72, was prepared in four steps from p-cresol via 2,6-bis(bromomethyl)-p-cresol (obtained by reaction of p-cresol with formaldehyde followed by HBr) [80]. 2,6-Bis(bromomethyl)-p-cresol was then treated with the hydrochloride of methyl prolinate in the presence of triethylamine in methylene chloride at room temperature, which was followed by the addition of phenylmagnesium chloride in THF at room temperature to yield 72. Chiral ligand 72 has found applications in many fields [79–81] and if it becomes commercially available it will probably find many more applications in acetylenic natural product synthesis.
4.2 Duryne
Duryne (62) is a C30 polyacetylenic alcohol with C2 symmetry. Despite its potent cytotoxicity [82], its central double bond geometry and the absolute configuration of the chiral centers were not determined till our recent total synthesis [69]. Two previous synthetic studies have been published. None was able to correlate the stereochemistry of the synthetic sample with the natural duryne. The first study by Deshpande produced racemic mixtures of duryne (62) [83]. The second study by Sharma and Chattopadhyay utilized the mono-protected diacetylene alcohol to prepare (15E,R,R)-isomer of duryne (62) [84]. The C2 symmetry of the molecule renders the two protons on the central double bond equivalent; hence no indicative coupling constant is available. All other spectroscopic data seem to provide no definite conclusions.
Our synthetic effort for duryne (62) started with the known (Z)-alkene diol (74) [85,86], which was prepared in three steps from the commercially available ω-bromoundecanoic acid (Scheme 24). The ω-bromoacid was converted to the corresponding methyl ester, which was treated with Ph3P in acetonitrile to yield the phosphonium salt using a reported procedure [87]. The (Z)-geometry of the central double bond was established by the autooxidation of the Wittig reagent generated in situ as reported by Poulain and coworkers [85]. Oxygen was bubbled through the mixture during the reaction as reported by Capon [88]. The diol 74 was converted to the dialdehyde 75 through a two step sequence: (1) PCC oxidation to the corresponding dialdehyde and (2) Wittig reaction of the dialdehyde with Ph3PCHCHO to yield dial 75. The addition of acetylenic magnesium bromide reagent to dial 75 produced the meso and racemic 62 for enzymatic resolution.
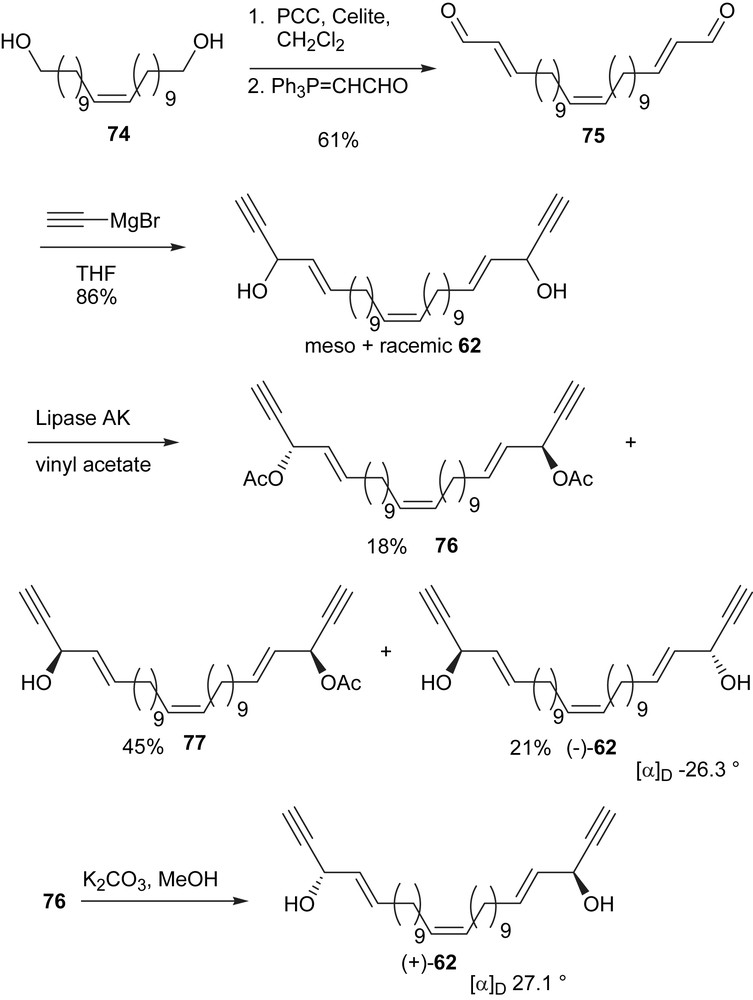
The general procedures previously described for the resolution of adociacetylene B (1) were followed. The separation of the diacetate 76, monoacetate 77, and the diol (−)-62 was done by column chromatography. Diol (−)-62 has a melting point of 41–42 °C and an optical rotation of [α]D22 −26.3. The removal of the acetate group from 76 yields diol (+)-62, which gives an optical rotation of [α]D22 27.1 and a melting point of 43–45 °C. These values are very close to the natural duryne (62)'s reported [α]D 29° and 44–45 °C, respectively [82].
Duryne (62) is similar in structure to adociacetylene (1), which we have successfully resolved using lipase from Pseudomonas sp. [16] For most secondary alcohols, the rate of acylation is faster for the (R)-configuration than for the (S)-configuration. However, for the acetylenic alcohol 62 the alcohol with (S,S)-configuration is acylated faster because the small acetylenic group has a higher priority in the nomenclature system. From these considerations and the data obtained, we surmise that the natural duryne (62) has a (Z)-central double bond and (S,S)-configurations at the chiral centers. However, in order to remove the doubt that the corresponding isomer with a central (E) double bond might have a similar optical rotation, we decided to synthesize the (E)-isomer and resolve the racemic mixture as well (Scheme 25).
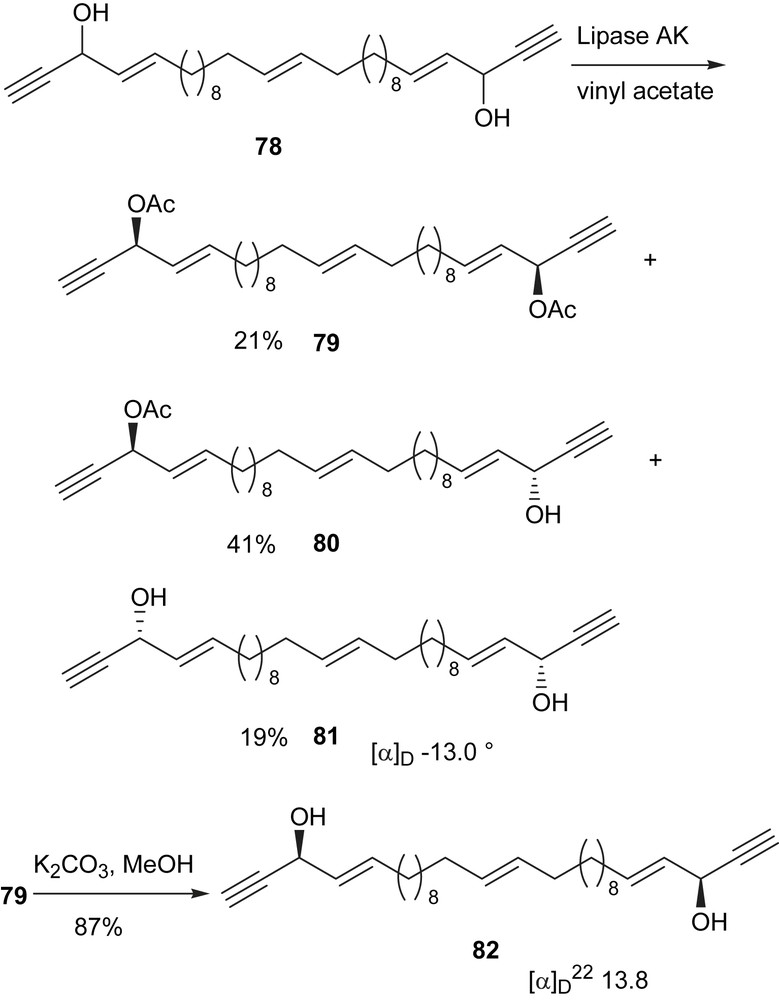
The (E)-geometry of the central double bond of the trans isomer was constructed with LiAlH4 reduction of an internal alkyne precursor [83]. The racemic mixture of the (E)-isomer 78 was prepared following the procedures by Deshpande [83]. After enzymatic resolution of 78 the optical activity obtained for the (15E), (3S, 28S)-diol 82 is [α]D22 13.8 and the (15E), (3R, 28R)-diol 81 is [α]D22 −13.0. Thus by synthesizing both geometric isomers of duryne (62) and the subsequent enzymatic resolution, we conclude that the natural (+)-duryne (62) reported by Wright et al. has a central double bond geometry of (Z) and absolute configuration of (S,S) as shown in structure 62. This conclusion not only is consistent with all experimental data reported in this study, but also consistent with several known C2 symmetric acetylenic alcohols isolated from marine sources [7,8,70,89].
4.3 Dideoxypetrosynol A (63)
Dideoxypetrosynol A (63) was found to have potent anti-cancer activity and exhibited an ED50 of 0.02 μg/mL against human ovarian cancer cells and of 0.01 μg/mL against human skin cancer cells [90]. It is noteworthy that the cytotoxic activities of compound 63 is one order of magnitude higher than those found for doxorubicin, which is one of the most commonly used chemotherapeutic drugs and exhibits a wide spectrum of activity against solid tumors, lymphomas, and leukemias [91]. Dideoxypetrosynol A (63) was also found to inhibit DNA replication at the initiation stage [92]. Our total synthesis of dideoxypetrosynol A (63) has just been accepted [93]. Here is a brief description of the synthesis of this C2 symmetric natural product (Scheme 26).
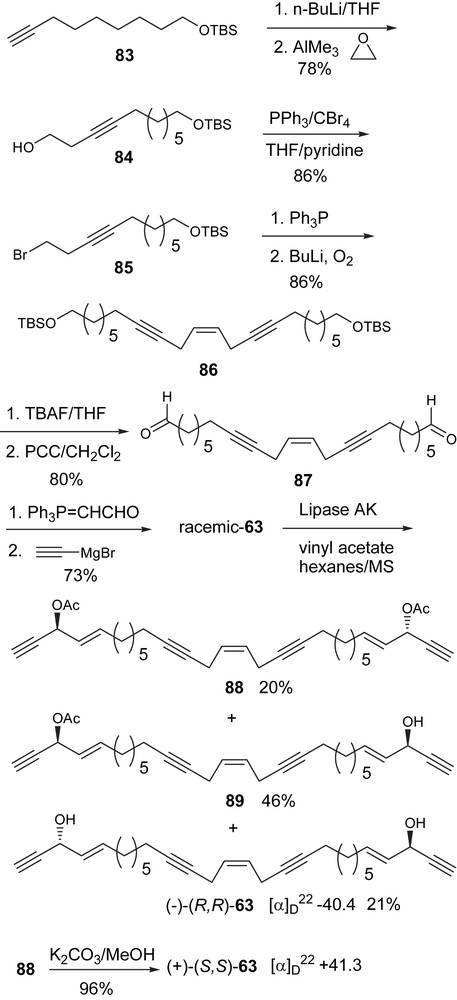
The synthesis of the target started with the known terminal acetylene 83 (Scheme 26) [16]. Homopropargyl alcohol 84 was prepared following a reported procedure [94]. This method employs Me3Al as an additive in the ethylene oxide opening reaction and gives a reproducible yield of the desired product. Compound 84 was converted to the homopropargyl bromide (85) by a modification of the Appel reaction [95]. The preparation of the phosphonium salt from 85 follows the reported procedure by Dawson [87]. n-BuLi treatment of the resulting phosphonium salt followed by saturating the reaction mixture with oxygen and reflux in THF produced the cis-enediyne 86 in 86% yield. With the key intermediate 86 in hand, dial 87 was obtained through a two step sequence: (1) removal of the TBS protecting group with TBAF in THF and (2) PCC oxidation of the resulting diol (Scheme 26). The reaction of the dialdehyde with Ph3PCHCHO yielded an α,β-unsaturated dial and the addition of acetylenic magnesium bromide to the dial produced the racemic natural product 63. Although the natural product dideoxypetrosynol A was reported as a possible racemic mixture, it was unclear whether racemization had occurred during the isolation–identification process [90]. We thought it was important to identify the absolute configurations for each enantiomer.
The general procedures using lipase AK from Pseudomonas sp. for the enzymatic resolution of the acetylenic alcohols were followed [57,75]. The separation of the diacetate 88, the meso monoacetate 89, and the diol (−)-63 was done by column chromatography. (R,R)-Diol 63 has a negative optical rotation of [α]D22 −40.4°. The removal of the acetate group from 88 yields (S,S)-diol 63, which gives a positive optical rotation of [α]D22 + 41.3°. The assignment of the absolute configurations is based on Burgess active site model for the lipases from Pseudomonas sp. [75] and was corroborated by a comparison to several known C2 symmetric acetylenic alcohols isolated from marine sources [7,8,70,89].
5 Concluding remarks
Through the total synthesis of polyyne natural products, new and fascinating polyyne chemistry has been advanced. It is interesting to note that many of the polyyne natural products are relatively stable once in their completed natural form. The same cannot be said about the intermediates leading to them, especially the terminal uncapped polyynes. Another interesting observation involving polyynes is their dual properties. When thinking of polyynes, one might think of the conjugated π electrons to dominate in their chemical property. Polyynes are indeed easily polymerized under light and/or air. However, polyynes are also electrophilic as shown in the addition of nucleophiles such as amines to tetraynes. The successive sp-hybridized carbons make them more electronegative. This is shown by the stronger than usual polarity of compound 35, which has a methyl group that, caps three conjugated triple bonds. New chemistry is expected to be further developed in this area. More efficient synthesis will be possible by the development of new reagents and new methodologies.
Acknowledgment
Financial support from the National Institutes of Health (GM069441) is gratefully acknowledged.