

1 Introduction
The chemistry of alkyne, metalla-alkyne and coordination behavior of alkyne towards transition metals has been explored thoroughly for a large number of systems. Tailor-made conjugated molecules, oligomers, and supramolecules when equipped with one or more key functionalities, are able to self-assemble into organized arrays. Tremendous advances have been made in the past decades, aided by major improvements in both synthetic and analytical protocols, and numerous new molecular architectures have been discovered. We can anticipate that such molecular or supramolecular systems will soon begin to play critical roles in established fields, such as analysis and catalysis, whilst providing the nucleation point for new subjects, like molecular-scale electronics, electroluminescent devices and chemical sensors [1]. It is our contention, and the focal point of this account, that the special properties inherent to ethynylene-grafted oligopyridines facilitate the construction of novel structures which retain highly desirable properties difficult to attain by conventional routes. Indeed, the combination of a metal center or a boron center and ethynylene-substituted fragments leads to unexpected electronic effects that can be exploited for practical purposes and it has to be realized that the presence of an acetylene group can cause a dramatic change in the electrochemical and photophysical properties of the supramolecule with respect to its parent metal complex or boron derivatives. This opens up the possibility to convert standard reagents into more valuable building blocks simply by the judicious insertion of an acetylene group and to conceive unusual materials. For example, ruthenium(II) bis(2,2′:6′,6″-terpyridine) is essentially non-luminescent in fluid solution at room temperature but highly luminescent derivatives are attained by functionalization with a single ethynylene group at the 4′-position [2]. Much of the spectroscopic investigation that has exposed these special properties has been reviewed recently, although new developments continue to arise on a regular basis, but less attention has been given to the synthesis of highly sophisticated ligands and their corresponding transition metal or boron complexes. Here, we describe the advanced synthetic methodologies that have permitted the establishment of new families of complexes or dyes and illustrate the inordinately wide range of modern scientific problems that these new molecules can address.
Transition metal complexes are known to participate in a variety of processes involved in, for example, (i) solar light energy conversion [3], (ii) photocatalysis [4], (iii) artificial photosynthesis [5], (iv) chemosensors [6], (v) molecular electronics [7] and (vi) ferromagnets [8]. To be effective, it is necessary to optimize the choice of both the metal center and the coordinated organic framework and, because of their versatility and stability, oligopyridines are often the preferred chelating function. It is not sufficient, however, to use a simple metal oligopyridine complex for advanced applications. The organic backbone has to be designed to allow assembly of an organized array of metal centers that acts cooperatively. This realization provides a formidable challenge to the synthetic chemist, who seeks to control the chemical nature, complexation ability and intrinsic properties of each individual building block as well as their precise positioning within the array. For example, the construction of a photoactive molecular triad [9] requires the exact placement of sensitizer, donor, and acceptor units within the superstructure in order to engineer a system capable of vectorial transfer of information along the molecular axis. Even so, elegant examples of donor/sensitizer/acceptor triads that span liposomal membranes have been described and such systems have been used to transport protons across the insulating membrane [10]. This proton-motive force, driven by a simple photochemical reaction, has been used to trigger the synthesis of ATP, via an ATP synthase incorporated inside the liposomal bilayer [11]. In this way, a crude artificial system mimics the process by which photosynthetic bacteria convert light energy into chemical potential.
Nature is a source of tremendous inspiration for scientists and it is clear that the efficient and unidirectional processes that characterize most natural organisms have evolved by careful optimization of each and every step in the overall mechanism. It is necessary to perfect each stage by balancing the energy requirements and by precise positioning of all the reactants [12]. The same type of critical evolution has to be applied to the engineering of totally artificial systems and, for this reason, it is essential that the synthetic protocol is readily adaptable for preparation of molecules of different size and shape. Simple ways have to be found for fine-tuning the basic electronic properties of the supramolecules and for favouring selective interactions with the environment. Stability and integrity of structure are mandatory, especially in those systems designed for ultrafast information transfer. A choice has to be made between direct (i.e., long-range) and sequential (i.e., short-range) transfer and, as with natural systems, a great amount of screening and testing goes into identifying the correct building blocks. We can illustrate this point by specific reference to the design of photonic wires [13] and photoactive molecular-scale wires [14].
We now concentrate on the design principles which have been established in our laboratory to create novel multi-metallic scaffoldings, and highly luminescent dyes and complexes possessing valuable properties. The emphasis will be on the versatility of the synthetic procedures and the more straightforward results will be highlighted in order to further stimulate the development of the next-generation of supramolecules. Information relating to the applications of these new materials can be found elsewhere [15,16] while synthetic details and purification procedures may be found in the cited literature.
2 Diethynyl-thiophene as spacer in the synthesis of polytopic ligands: design and principle
From a general point of view ligands bearing heteroaromatic moieties have attracted great attention because complexation with ruthenium, osmium, iridium and rhenium, in particular, provides complexes which are luminescent in fluid solution at room temperature [17,18]. Such complexes have been extensively studied in recent decades due not only to their fascinating optical but also their unusual electronic and redox properties, and have been long considered likely to have many different applications such as in chemical sensing [19], electron [20] or photon [21] donation, light harvesting [22] and electro-generated luminescence [23]. In many cases, the electronic properties of the metal complexes are sensitive to the nature, substituent patterns and shape of the surrounding ligands. Grafting unsaturated fragments close to the ligands provides means to easily construct sophisticated units and also to tune the intrinsic properties of the complexes by prolongation of the excited-state lifetimes and by modification of the absorption of the charge-transfer state, as well as the redox properties, of one of the peripheral ligands.
Particularly attractive are molecular systems where photoinduced energy or electron transfer processes can be realized over large distances and in a preferred direction [24]. Earlier discoveries have shown that the nature of the spacing units separating photoactive terminals plays a crucial role in the efficiency and mechanism of information transfer [25]. Here, unsaturated systems have been explored and appear to be the most attractive. Amongst these systems, p-phenylenevinylene oligomers [26], polyenes [27,28], polyalkynes [29–33], polyphenylenes [34–36], polyphenyl/alkyne [37,38] and polythiophenes [39–41] units have been extensively studied due to their chemical stability and synthetic accessibility.
The uses of oligothiophenethynylene [42] for the construction of cyclic nanoarrays [43–45] and quasi-linear molecules capped by various stoppers have been reported [45,46]. Furthermore, thiophene-based functional polymers have also attracted significant attention due to their application as conductive and active layers in OLEDs [47]. Oligothiophenes show high conductivity and electroluminescence, and have excellent characteristics as organic thin film transistors [48,49]. Based on a thorough understanding of structure–property relationships and the ingenuity of synthetic chemists, ambipolarity, tunability, high quantum efficiencies and stability have all been achieved [50].
Ethynyl grafted thiophenes are among the most promising units to connect chromophoric centers and to provide interesting optical properties [51,52]. Although there are many manuscripts pertaining to closely-coupled binuclear and trinuclear metal complexes, little is known about the more extended systems in which energy hopping processes might be a critical feature. This situation is in marked contrast to that in analogous organic systems, where it is well known that the optical properties depend on the degree of oligomerization [53,54].
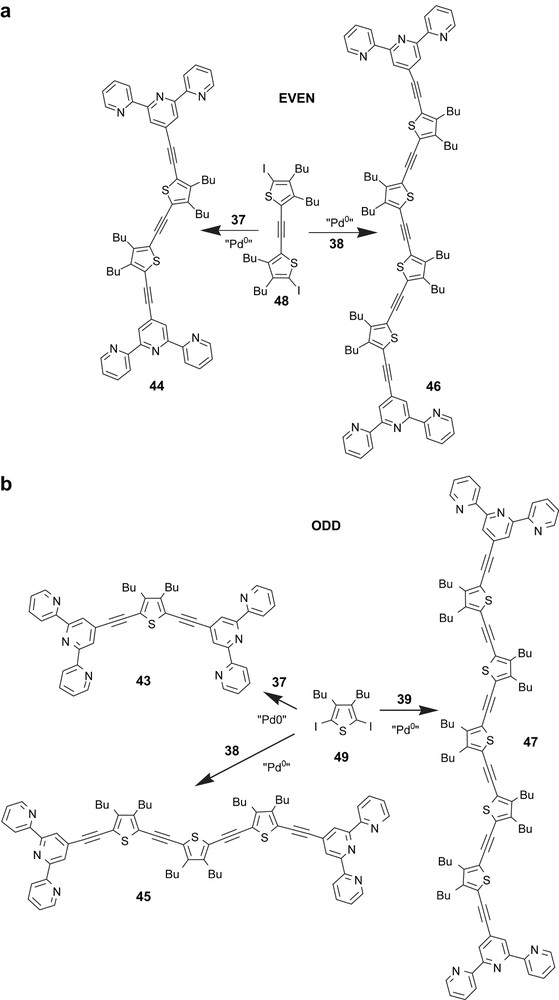
We succeed in the preparation of a series of soluble polybipyridine ligands comprising one to five bipyridine units sandwiched between rigid carbon–carbon triple bonds substituted by 3,4-dibutylthiophene repeating units. Scheme 1 defines a single strategy applicable to the synthesis of multitopic ligand [55]. Two main pathways can be distinguished depending on the presence of an odd or even number of bipyridine units. These require two pivotal building blocks constructed around a 5,5′-disubstituted-2,2′-bipyridine core and a thiophene moiety. In the top case, coupling between a synthon carrying two terminal alkynes and a mono-iodo-substituted hybrid bpy/thiophene molecule leads to the progressive increase of two bpy links providing the ligand precursors.
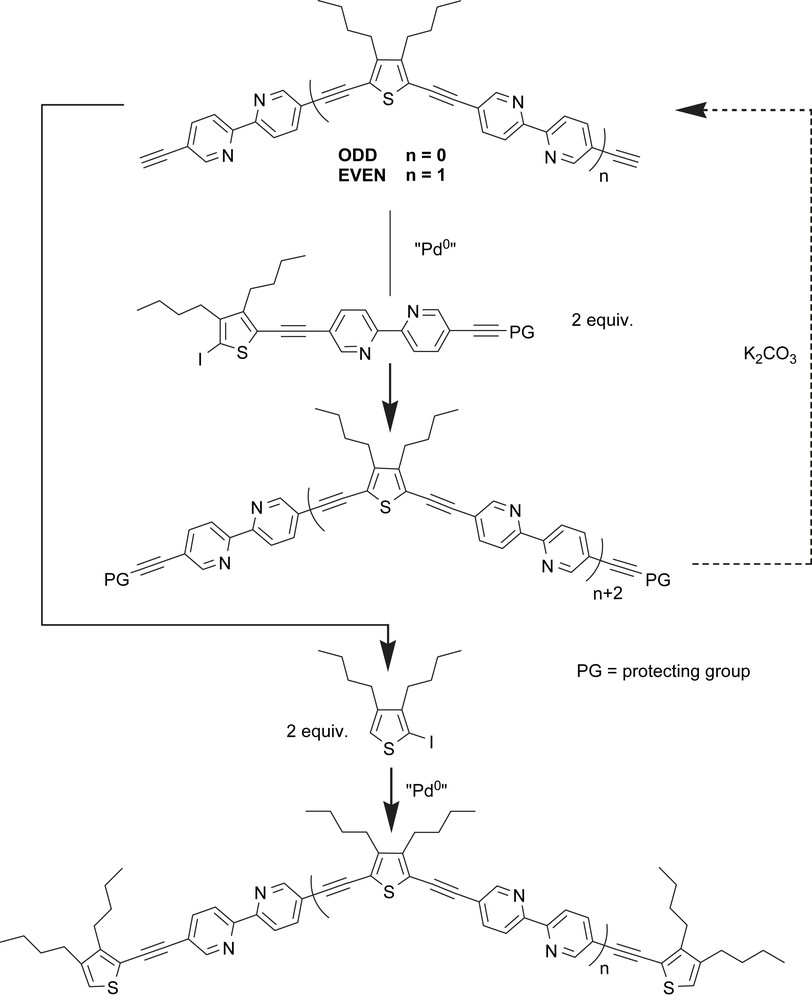
The final ligands are obtained by cross-linking of these deprotected intermediates with 3,4-dibutyl-2-iodothiophene. The key concept is the provision of the target ligands by an iterative sequence of reactions involving a step-by-step introduction of each extending unit after a facile deprotection reaction. The presence of one protecting group (PG) and one reactive iodo function on the second starting material is critical for the success of the methodology. This approach allows an increase in the number of bpy units in every two steps. The idea was to start from a 5,5′-dibromo-2,2′-bipyridine moiety which in our hands is easy to produce on the gram scale, and has been previously proved to provide transition metal complexes with exceptional photophysical properties.
The general protocol is based on Pd(0) promoted cross-coupling reaction between selected building blocks. Various combinations allow the interconnection of the terminal alkyne to 3,4-dibutyl-2,5-diiodothiophene or 3,4-dibutyl-2-iodothiophene leading to bipyridine frameworks bearing two acetylene protected groups or one acetylenethiophene/one acetylene protected function. It is possible thereby to construct dimeric to pentameric bipyridine ligands 2–5 (Scheme 2), where the chelating subunit is bridged by a 3,4-dibutyl-2,5-diethynyl thiophene spacer and end-capped by a 3,4-dibutyl-2-ethynyl thiophene stopper, the size of the fully extended ligands lies between 20 and 74 Å.
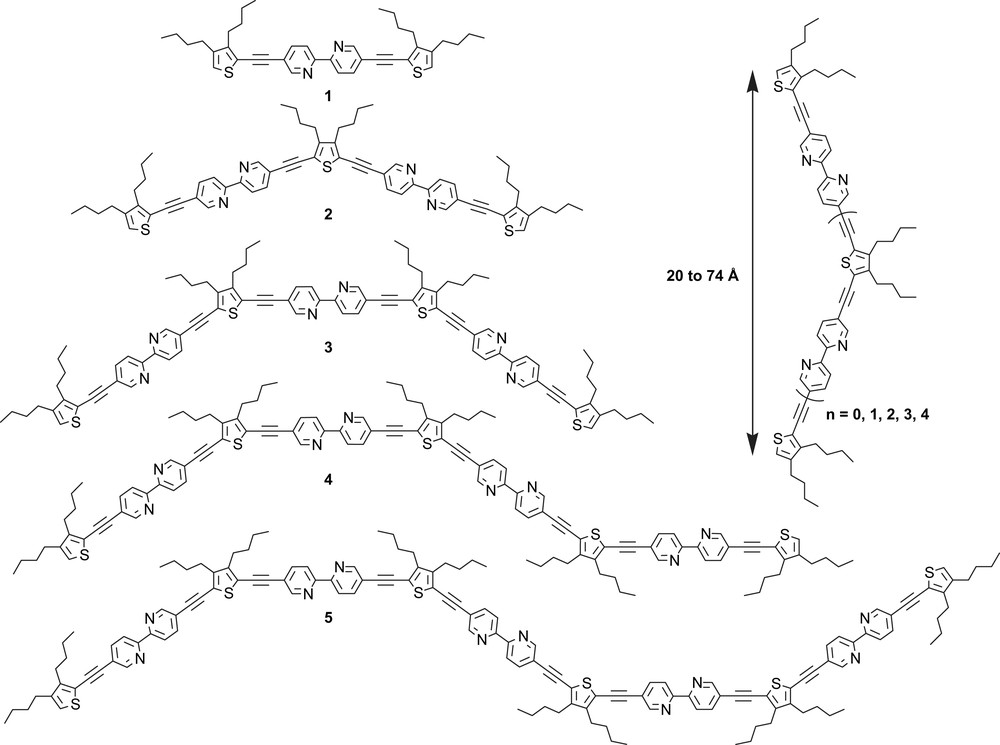
The good yields obtained for the syntheses of these extended ligands underline the versatility of the synthetic protocol. It is anticipated that this procedure will be useful for the synthesis of larger systems because only slightly decreasing yields have been observed as a function of chain length for the various compounds obtained so far.
Spectrophotometric studies have revealed that by increasing the number of π-electrons from 24 to 104, a significant bathochromic shift occurs for absorption and emission maxima [55]. This effect is attenuated upon increasing the number of chelating sites. All ligands fluoresce strongly in solution at room temperature when excited in the weak energy absorption band, with the fluorescence quantum yield increasing from 31 to 43% with increased molecular length. The first reduction potential (LUMO level) of the oligomers does not change significantly along the series of oligomers and lies in the −3.06 to −3.18 eV range as determined experimentally by cyclic voltammetry. The thermal and photo-stabilities of these rigid-rod platforms make them attractive for the engineering of luminophoric d8-transition metal centers where each metallic dot might act as a relay station in a photon shuttling process. The whole array might be expected to operate as an artificial photon-harvesting system (Scheme 3) [55].

3 Controlled synthesis of multi-nuclear metal arrays using diethynyl-thiophene as spacer
On the basis of the present knowledge, a bridge providing both a conduit for strong interactions and an entity conveniently adapted to end-capped the molecular scaffolding is likely to be an ethynyl thiophene unit [55,56]. A plethora of polytopic ligands have been synthesized over the past decade but little is known on selective complexation of one chelating site vs the other. It appears that without resort to complicated protection procedures and the use of less readily accessible metal reactants, prospects for a direct preparation of mixed-metal species would seem to be particularly poor using the existing molecular technologies.
Recently, we have shown that employment of synthesis at preformed scaffolds provides an easy entry into sophisticated complexes in which the centers are linked by alkyne spacers. The methodology encompasses a wide variety of functional groups, and it is worthwhile noting that the advanced protocols employ both Ru and Os building blocks with reactive C-iodo or terminal alkyne groups. Some examples are highlighted in Schemes 4 and 5.
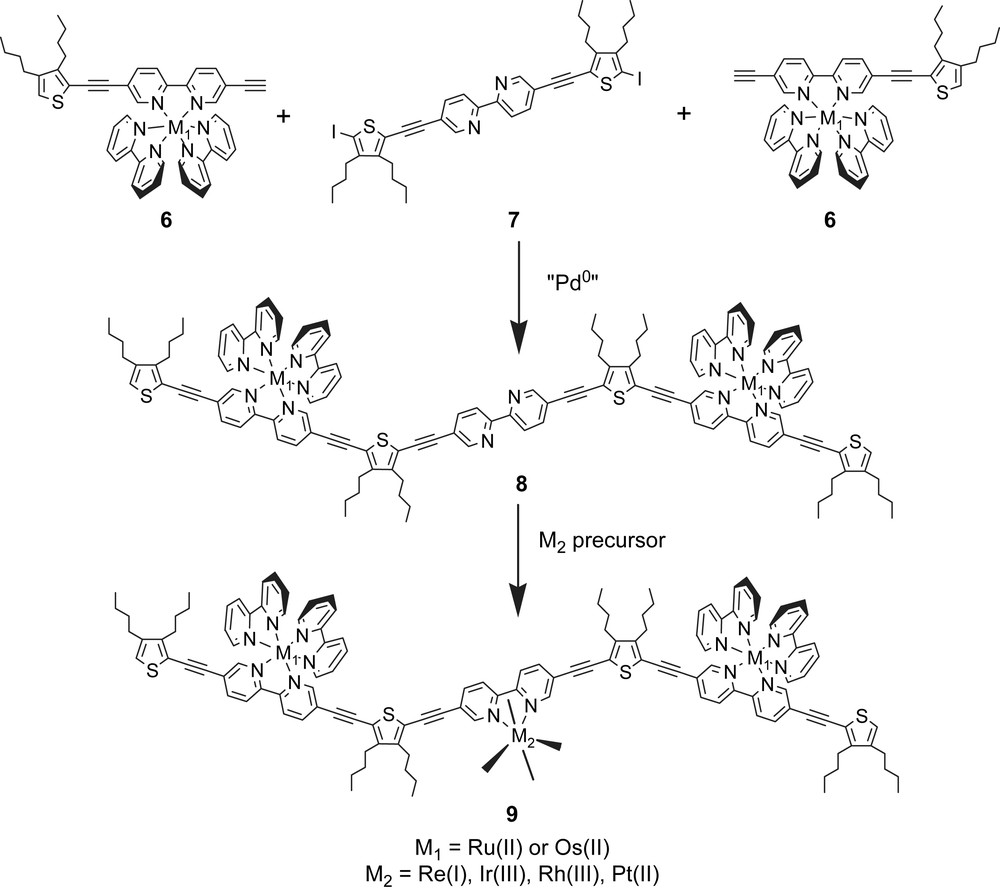

Numerous variations in the nature of transition metal and side chains may be envisaged, giving in principle access to a large variety of novel multichromophoric transition metals. One may note that no scrambling of the metal centers during the synthesis is proved by electro-spray mass spectroscopy [56]. This is a direct proof that synthesis based on the reactions of the coordinated ligands is a convenient and versatile method because of the mild conditions, the tolerance of various functions such as terminal alkyne and C-iodo. The overall process may then be described as a regiochemically controlled synthesis of polynuclear complexes [56].
4 Photophysical properties of Ru(II) based polypyridine bearing conjugated diethynyl thiophene appendages
A large number of ruthenium poly(pyridine) complexes have been synthesized in which an aromatic polycycle is appended close to the metal complex [1]. Such derivatives have been highly successful as a strategy for prolonging the lifetime of the lowest-energy, triplet-excited state associated with the metal complex [57–59]. Indeed, triplet lifetimes have been increased from a few microseconds to more than 100 μs in certain cases [57,58]. The most popular polycycle has been pyrene and a wide variety of such derivatives are now known. Prolongation of the triplet lifetime arises from the rapid establishment of a thermally equilibrated mixture of triplet states and requires that triplet states localized on the metal complex and the polycycle are almost isoenergetic [60]. A similar approach has been used with osmium(II) poly(pyridine) complexes [61] and employed for the construction of multi-nuclear metal complexes.
Recently, a new application for this same synthetic strategy has evolved [62]. Thus, considerable attention has focused on the design of metal poly(pyridine)-based systems capable of transferring triplet energy over unusually long distances [2,63]. This work has involved attaching different metal complexes to either end of a molecular-scale bridge constructed by the stepwise oligomerization of individual modules into a linear structure. The rate of triplet energy transfer between the terminals depends on the length and composition of the bridge. At first, interest was directed to those bridges where the triplet energy of the donor was well below that of the bridge – the so-called super-exchange mechanism – and several effective bridges have been identified [64]. Notable among the putative building blocks have been ethynylated aromatic groups [65].
The alternative design principle – the so-called hopping mechanism – uses a donor whose triplet energy is slightly above that of the bridge [66]. Here, illumination of the donor results in injection of triplet energy into the bridge, followed by energy migration and subsequent trapping by the acceptor. This approach can provide very fast rates of triplet energy transfer regardless of the length of the bridge [67]. It is necessary, however, to identify suitable molecular-scale connectors.
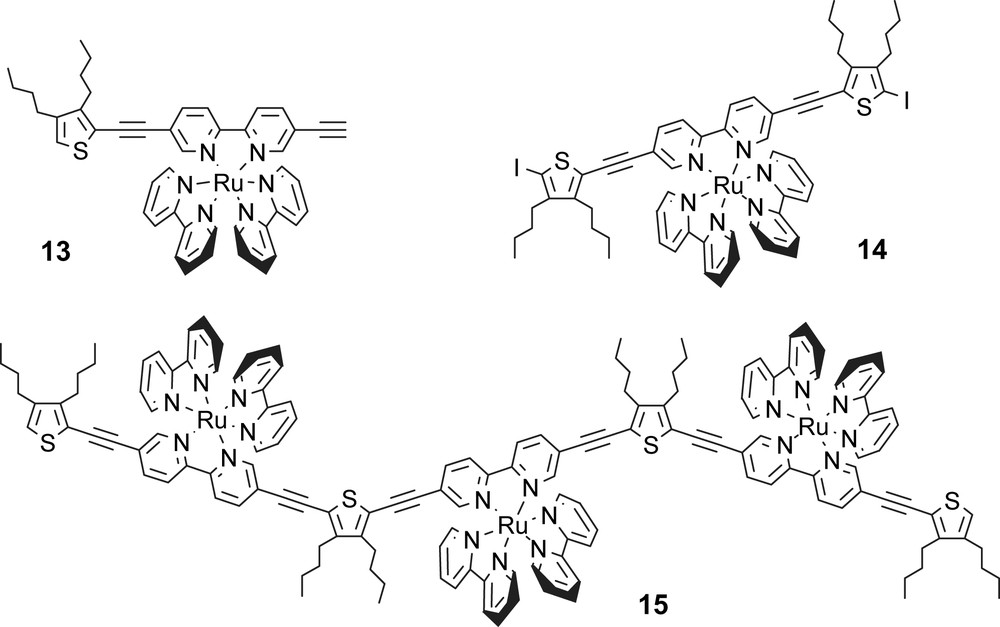
For the series of ruthenium(II) tris(2,2′-bipyridine) complexes depicted in Scheme 6, the photophysical properties depend on the conjugation length of the thiophene-based ligand and well defined absorption spectra and emission spectra recorded for the three complexes in acetonitrile solution are shown in Fig. 1.

Left: absorption spectra recorded for the three complexes in dilute acetonitrile solution; 13 (black), 14 (blue) and 15 (red). Right: luminescence spectra recorded for the three metal complexes in deoxygenated acetonitrile solution following excitation at 420 nm; 13 (black), 14 (blue) and 15 (red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The luminescence spectra recorded for the thiophene-based metal complexes at ambient temperature are relatively broad and contain features clearly apparent on the high-energy side of the main transition; this is especially evident for 13. Cooling to 77 K in butyronitrile resulted in a marked sharpening of the spectrum (Fig. 2). The features seen on the high-energy side disappear on cooling. There was also a significant blue shift for each emission maximum; in a butyronitrile glass at 77 K λLUM was found at 640, 660 and 680 nm, respectively, for 13–15. Such spectral shifts are well known for metal complexes [68] and can be attributed to destabilization of the MLCT triplet state on moving from a polar solvent to a rigid glassy matrix [69]. This finding suggests that the observed emission arises from an MLCT triplet state and the broad emission can be resolved by emission spectral curve-fitting routines (Fig. 3).

Luminescence spectra recorded for the three metal complexes in a deoxygenated butyronitrile glass at 77 K following excitation at 420 nm; 13 (black), 14 (blue) and 15 (red).
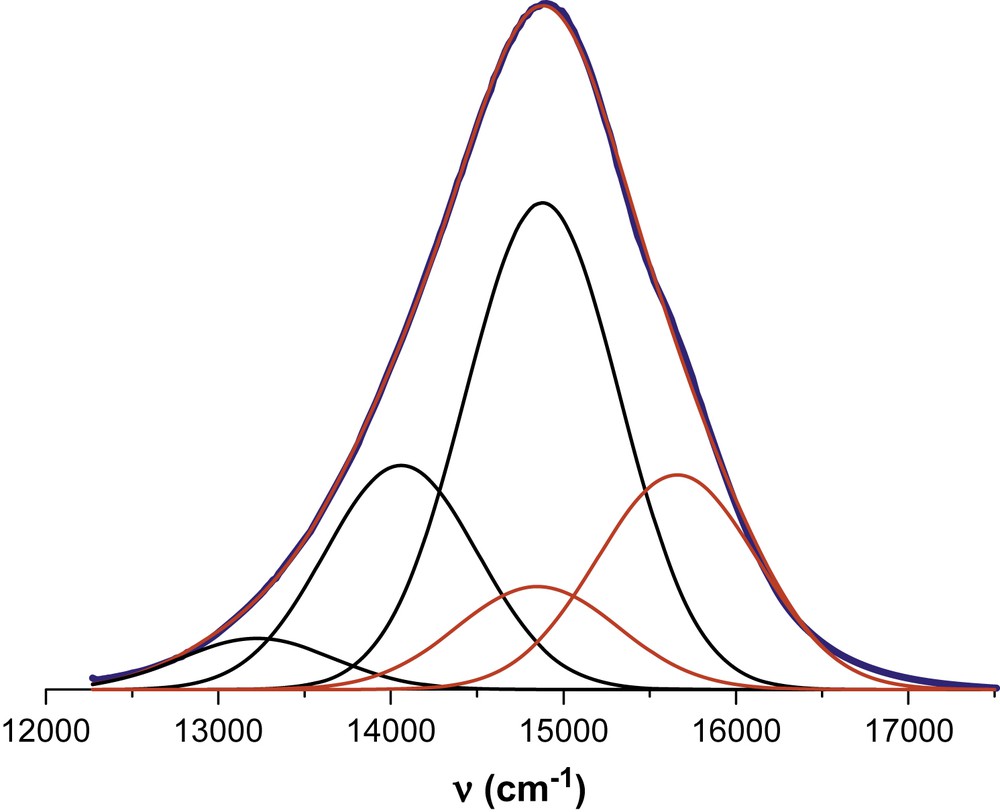
Deconvolution of the emission spectra recorded for 13 at room temperature in butyronitrile solution into the minimum number of Gaussian-shaped components. The ‘normal’ vibrational progression is indicated in black while the ‘hot’ emission is represented in red. The fit to the entire spectrum is overlaid with the observed spectrum.
In each case, dual emission is observed and best explained as the two emitting states reside in thermal equilibrium at ambient temperature. This allows the properties of the two states to be evaluated in both fluid butyronitrile solution and a transparent KBr disc. It is concluded that both emitting states are of metal-to-ligand, charge-transfer (MLCT) character and, despite the presence of conjugated thiophene residues, there is no indication for a low-lying π,π∗ triplet state that promotes nonradiative decay of the excited-state manifold. A key feature of these systems is that the conjugation length imposed by the thiophene-based ligand helps to control the rate constants for both radiative and nonradiative decay from the two MLCT triplet states.
There are a few other reports of dual emission from close-lying, triplet-excited states for metal poly(pyridine) complexes but these mostly refer to cases where MLCT and π,π∗ triplet states lie at comparable energies [58d,70]. The spectroscopic properties of the complexes described in Scheme 6 for the two emitting species do not correspond to a mixture of MLCT and π,π∗ triplet states but appear to match the properties expected for MLCT triplet states. It is, in fact, well known that many ruthenium(II) poly(pyridine) complexes possess a higher-lying MLCT triplet that can reside in thermal equilibrium with the lowest-energy MLCT triplet [71]. Furthermore, there are at least two cases of osmium(II) bis(2,2′:6′,2″-terpyridine) complexes [72] that display dual emission from two MLCT triplets that differ in terms of their spin-orbit coupling properties, with the higher-lying MLCT state retaining increased singlet character. The behavior reported here is entirely consistent with equilibration between two such MLCT triplets. Apart from their obvious difference in energy, these two MLCT states differ with respect to the extent of their interaction with the conjugated ligand. The experimental results are strongly indicative of the upper-lying state being more strongly coupled to the conjugated substituent.
The relative ordering of π,π∗ and MLCT triplet states reported here for 13 and 14 agrees with that described earlier [20] for mononuclear RuII tris(2,2′-bipyridine) complexes bearing one or two ethynylatated thiophene residues. Indeed, the photophysical properties, with the exception of the noted dual emission, of these various mononuclear complexes are reasonably comparable, given the somewhat different experimental conditions. We might expect that the π,π∗ triplet level for 15 should lie below the corresponding MLCT triplet state, but this is not the case. The most likely reason for the π,π∗ triplet remaining at relatively high energy is that this state is highly localized over a small fragment of the ligand. Presumably, this situation arises because internal motions around the aryl units, most notably the vacant 2,2′-bipyridine groups, restrict extended π-electron conjugation along the molecular backbone. In this respect, it is interesting to note that coordinating zinc(II) cations to the vacant 2,2′-bipyridine groups, thereby forcing the two pyridine rings into a coplanar arrangement, does not affect the emission spectral properties despite the obvious introduction of charge-transfer absorption bands in the near-UV region.
This work clarifies the nature of the lowest-energy, excited triplet state in such systems, with particular reference to the relative energies of ligand-centered and metal complex localized excited states [52b,73], and to evaluate the temperature dependence for any luminescence issue from the equilibrated system. This latter feature is an important tool for examining energy gaps and injection rates. A long-term goal of the work is to devise improved pathways for very long-range triplet energy transfer.
5 Dimeric ruthenium(II)-bipyridine units linked by ethynylene–oligothiophene or ethynylene bridges
Hybrid conjugated materials that contain oligothiophene wires are opening interesting perspectives for the development of technologies for electronics and optoelectronics [74]. The combination of ruthenium(II)–polypyridine complexes and thiophene-based oligomers offers a series of advantages and is actively pursued [52c,75]. These include the fact that mononuclear and polynuclear ruthenium(II)–polypyridine compounds are one of the most studied family of compounds [76]. This family of complexes is also used in optoelectronic applications [77]. On the other hand, poly- and oligothiophene are also attractive compounds in the absence of metal, particularly promising for the development of molecular wires, and their optical and electrochemical properties are likewise extensively studied, particularly for the smaller oligomers [78].
Inclusion of ruthenium(II)–polypyridine complexes, and other metal centers, as side or end units of linearly arranged oligothiophene frameworks, may result in a unique combination of photophysical and electrochemical properties. This is particularly appealing bearing in mind that the oligothiophene backbone can transport electrons or favour energy transfer [79]. Therefore, for these hybrid materials it is of importance to investigate and understand the nature, energetic, and spatial location of the various excited states, with particular emphasis on the lowest-lying ones, originated by the integration of ruthenium(II)–polypyridine and oligothiophene units.
The syntheses of the ligand and relevant metal complexes are depicted in Scheme 7. In particular, our studies are aimed at understanding (i) the role of the various excited states, including those originated from possible charge separation (CS) processes, that in our cases may be present at the interface between the terminals and the oligothiophene bridge, and (ii) what is the electronic layout that establishes within this type of molecular wires.
The dimeric 18 and 21 species examined are hybrid systems integrating two types of electroactive and photoactive centers, the [Ru(bpy)3]2+ chromophore and the conjugated oligothiophene wire of variable length. These complexes nicely illustrated the possible interplay of excited levels of diverse electronic nature and spatial origin. Light absorption can take place at physically separated subunits and generates accordingly both 1MLCT levels (at the ends of the wire) and 1π,π∗ levels (at the thiophene backbone). The former correspond to metal–ligand charge-transfer separation at the terminals of the wire while the latter levels spread over the oligothiophene backbone because of excitonic delocalization (Fig. 4) [80].

Room temperature energies of the excited states for the compounds studied and for the reference complex Ru(bpyDBT), see Scheme 8; at 77 K, the CS levels (of T+–bpy− origin) are expected to be strongly destabilized, see text. BpyDBT accounts for 2,2′-bipyridine-5-(3,4-dibutylthiophene).
In principle, charge-separated states (CS) can also come into play, corresponding to thiophene-localized oxidation and reduction at a connected bpy ligand from the coordination environment of the Ru(II) center. This rich diversity in the nature of the excited states originated upon light absorption turns in a simpler picture when examining the layout of the thermally equilibrated levels. Our results indicate that for our species, which contain relatively large oligothiophene backbones, the typical 3MLCT luminescence of [Ru(bpy)3]2+ and its analogues is quenched by the lowest-lying levels, of 3π,π∗ nature, delocalized over the oligothiophene backbone.
These triplet levels are related to the π,π∗ excitonic levels, and have the potential to be “conductive” of excitation energy, as confirmed by recent studies wherein Ru- and Os-based 3MLCT level, located at the terminals of complex 22, species are respectively higher in energy and lower in energy (2.1 and 1.6 eV, respectively) than the 3π,π∗ triplet level (1.83 eV), see Scheme 8. The absorption and luminescence properties observed for 21 and some reference complexes indicate that the Ru- and Os-based metal units (at an intermetal distance of 1.9 nm) are connected via electronic levels that allow physical (i.e., not virtual) flow of excitonic energy to the Os-based trap (Fig. 5) [81].


The 3ππ excitonic band at the bridge allows Ru-to-Os in complex 22 excitation injection by which the bridge plays a role as a real transmission path.
6 Critical comparison of energy transfer between ethenyl-thiophene and ethynyl-thiophene substituted bipyridine dimers decorated with phosphorescent subunits
The basic strategies employed for synthesizing the ligands with double bonds are based upon Horner–Wadsworth–Emmons preparative methodologies. The synthetic protocol required first the synthesis of the corresponding phosphonate and aldehyde precursors. Scheme 9 illustrates the synthetic sequence for the synthesis of ligands 23 and 25. Like typical Wittig-type reactions, 24 was obtained as a mixture of E and Z vinylene isomers. Only one diastereoisomer, ZZ, was isolated in the case of ligand 23. Again only the all-Z diasteroisomer 25 was obtained with 84% yield. The stereochemistry of the two ligands is likely explained by the photoisomerization Z/E of the double bonds. As the conjugation increases with the size of the ligands, the absorption spectra show the expected red shift assigned to a low-lying π,π∗ absorption [82]. We assumed that this weakly energetic transition facilitates the isomerization of the E alkene to the thermodynamically more stable Z isomer.

The Ru and Os complexes (Scheme 10) were prepared with the procedure that we used previously for the synthesis of d6-transition metal bipyridine complexes [83]. Complexes 26 and 27 were obtained by reaction between ligands 23 or 25 and cis-Cl-[Ru(bipy)2Cl2]·2H2O [84] or cis-Cl-[Os(bipy)2Cl2] [85]. For the ditopic ligand, unavoidable mixtures of mono- and binuclear complexes could be separated by chromatography on alumina. In all the cases, the complexation with Ru gave better results than with Os. So the preparation of the heteronuclear complex 30 was carried out from the mononuclear Os complex of ligand 25. The basic strategies employed for synthesizing the ligands with triple bonds are based upon Sonogashira cross-coupling reactions highlighted in Scheme 1. Regiospecific complexation with Ru or Os precursor was realized as described above for the double bonded ligands.
The luminescent behavior of the dinuclear complexes 28–30 is consistent with the ligand-centered triplet levels (3Th) lying at an intermediate energy between 3(Ru → L CT) and 3(Os → L CT) levels, the latter being located at ca. 1.5 eV for the examined Os-containing species (according to estimates from the emission maxima of 26, 29, and 30). In fact, (i) 28 is not emissive, and (ii) 29 and 30 exhibit a quite similar emission efficiency, ϕem = 3.4 × 10−4 and 3.9 × 10−4, respectively. The latter emission intensity is lower than that exhibited by [Os(bpy)3]2+, ϕem = 3.2 × 10−3 and by 26, ϕem = 2.5 × 10−3. This suggests that, for 29 and 30, additional deactivation processes are affecting the luminescence properties, even if we have no simple explanation to offer in this regard.
It is of interest to notice that this series of complexes shows a close structural resemblance with a series of mononuclear and dinuclear complexes for which the various subunits are connected by triple bonds, instead of double bonds. The systems can apparently to be viewed as more rigid, with the various subunits subject to a tighter electronic connection than for the cases with triple bond linkage [83]. For the 31 and 32-based complexes, the emission properties were ascribed to excited levels of predominant MLCT nature, while for the 26-based complexes a predominant ligand-centered nature is proposed for the lowest-lying level, which explains the lack of luminescence. This outcome might be understood based on comparison of the electrochemical properties of both the mononuclear and the dinuclear cases for the two series. It is to be noticed that for the complexes with the triple bonds, the “redox energies”, Δ = e(Eox − Ered) eV, are systematically lower than for those of series with double bonds. For instance, for the 33 dinuclear complex, first oxidation is at 1.32 V, first reduction is at −0.99 V vs SCE [83]; and Δ = 2.31 eV. For the 28 dinuclear complex, first oxidation is at 1.30 V, first reduction is at −1.04 V, and Δ = 2.34 eV. Based on the well-known correlation between redox properties and MLCT levels [86], this might indicate that the MLCT levels for the complexes bridged by double bonds are slightly higher in energy (by ∼150 to 250 cm−1) than the corresponding complexes bridged by triple bonds. Thus, it is possible that the different electronic properties of the triple and double bonds regulate the switching between closely lying 3Ru → L CT and 3LC (3Th) levels for the complexes shown in Fig. 5.
Also worth noting here is the strong fluorescence measured in CH2Cl2 solvent, ϕem = 49 and 39%, for respectively the monobipy and the dibipy double bonded free ligands. Related triple bonded free ligands fluoresce with quantum yields of 33 and 38% under the same experimental conditions [55].
The nature of the energy transfer process in complexes 35 and 30 is best described by a double electron-exchange mechanism (whereby the constituent electron and hole transfers appear to exhibit some peculiar aspects), as mediated by the thiophene/bipy/thiophene/bipy/thiophene backbone (Fig. 6). These studies provide useful grounds in view of the exploitation of hybrid materials integrating metal units and oligothiophene backbones.
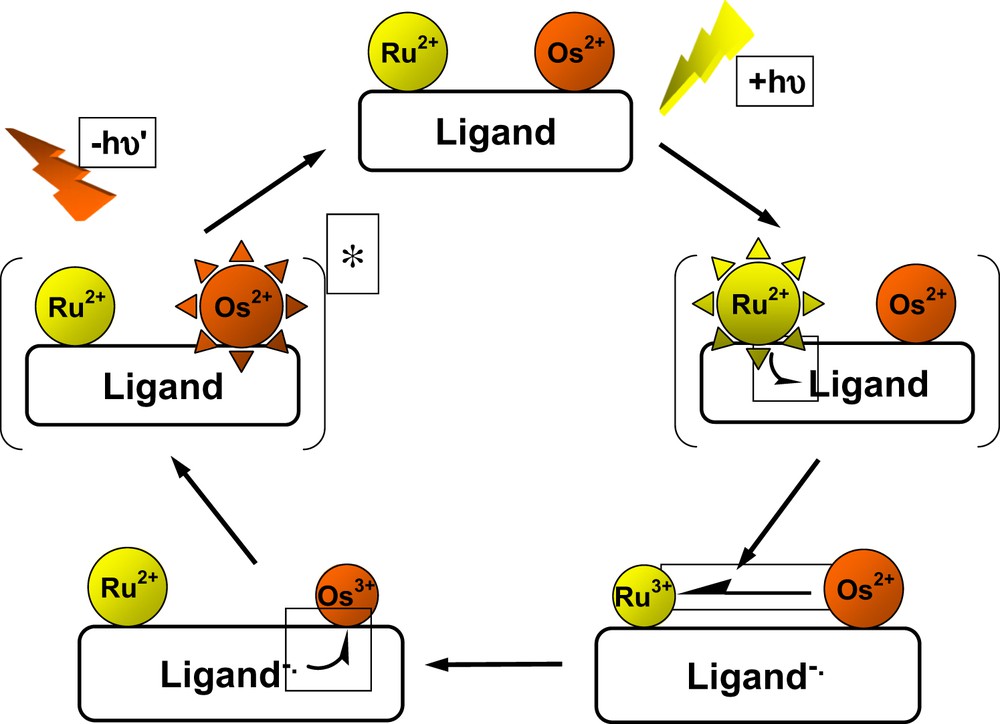
Schematic representation of energy transfer from a ruthenium to an osmium center by double exchange of an electron and a hole according to the Dexter mechanism.
7 Oligo (diethynyl thiophene) bridged back-to-back terpyridine ligands
A series of wire-like terpyridine-based ditopic ligands bearing one to five 2,5-diethynyl-3,4-dibutylthiophene [51,86] spacing modules have also been synthesized (Scheme 11).

Two synthetic strategies are envisaged. The first would be the construction of the target ligands by a step-by-step implementation of a terpy building block carrying a terminal acetylenic function with the key building block 36. The use of an iterative sequence of reactions involving cross-coupling/deprotection provides the molecules with the requested number of modules. In a final step, cross-coupling of these molecules with a triflate or a bromide substituted terpyridine would access to the ditopic ligands. The tedious and difficult purification of the molecules carrying three thiophene modules prompted us to change the synthetic strategy for the preparation of the higher oligomers. The second synthetic protocol clearly distinguishes two main pathways (Scheme 12) depending on the presence of an even or odd number of thiophene modules. Direct coupling reactions between the mono-terpy intermediates 37–39, prepared in the first strategy, with the diiodo compounds 48 and 49 give the back-to-back terpy ligands respectively with even modules (molecules 44 and 46 in Scheme 12a), and odd modules (molecules 43, 45 and 47 in Scheme 12b).
The mono- and dinuclear complexes [87] of corresponding ruthenium (Scheme 13) are prepared from these ligands and the precursor cis-[Ru(terpy)(DMSO)Cl2] [88].
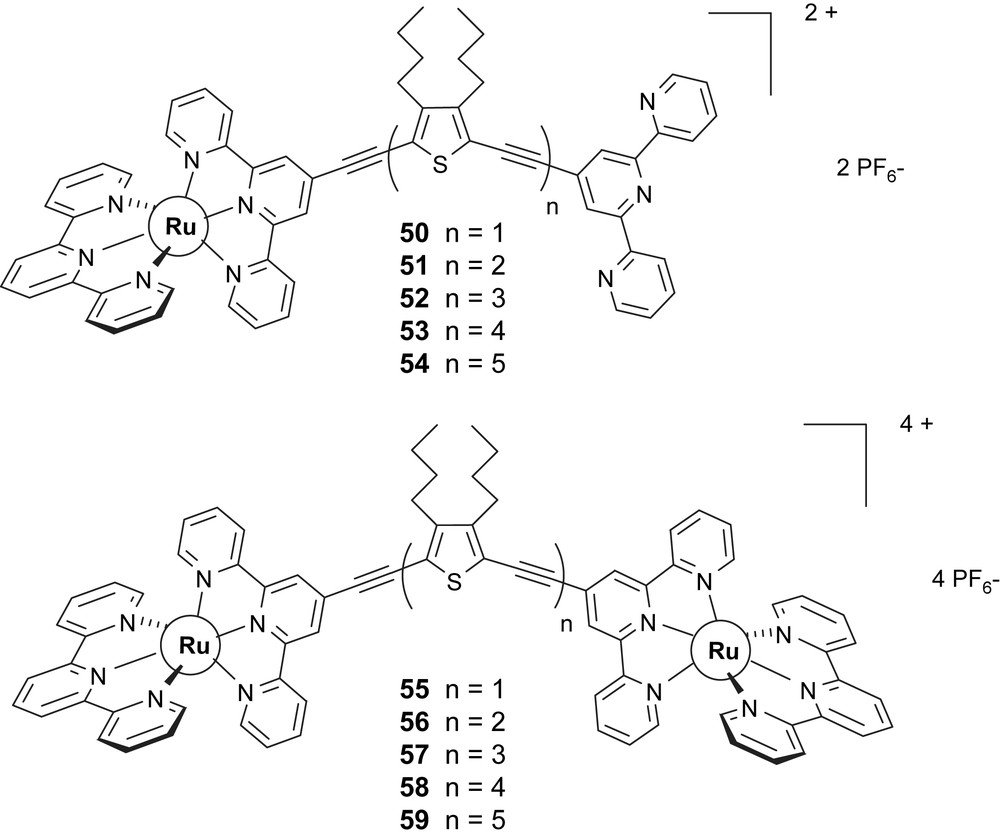
The complexes contain back-to-back terpyridine ligands connected to π-conjugated 2,5-diethynyl-3,4-dibutylthiophene oligomeric fragments, terpy–DEDBTn–terpy, with n = 1–5 [87]. In the binuclear complexes 55–59, the connecting ligands provide a structurally rigid linkage between the two chromophoric centers, with an estimated intermetal separation of 18.5, 24.9, 30.7, 36.1, and 41.6 Å for n = 1, 2, 3, 4 and 5, respectively as deduced by molecular mechanical calculations.
For all cases, the oxidation of the ruthenium center(s) occurs in the same potential range, without any significant splitting of the waves for the dinuclear complexes. In the mononuclear complexes an additional, irreversible thiophene-based oxidation is observed with the higher number of ethynyl thiophene modules. Well-resolved reduction potentials are found, corresponding to reduction steps at the various terpy segments. The absorption spectra of the complexes feature several types of transitions, either localized on the ligands (both on terpy and ethynyl thiophene subunits) or of MLCT (Ru → terpy) nature. The photophysical properties of the complexes are dominated by relatively long-lived, 3MLCT luminescent excited states (τ = 100–160 ns, at room temperature), which are ascribed of largely localized Ru → terpy character. For both mononuclear complexes 50–54 and dinuclear complexes 55–59 series, the electrochemical and spectroscopic results indicate that the electronic interaction between the terpyridine subunits of the ligands (as mediated by the interposed diethynyl thiophene oligomeric fragments) is weak, particularly for the larger molecules, n ≥ 3. The weak coupling between terpy and diethynyl thiophene subunits could explain why low-lying 3π,π∗ triplet states localized at the oligo ethynyl thiophene framework do not completely quench the 3MLCT luminescence. On the other hand, photoinduced intramolecular electron transfer (both for oxidative or reductive schemes), as driven by the energy content of the excited Ru-terpy levels, and involving the diethynyl thiophene fragments, are inhibited by an unfavorable energetic balance in all cases [87].
Given the interest of such wire-like complexes as molecular components of thiophene-based photonic devices, and in order to gain control over competitive energy disposal paths, ligand tailoring is clearly a powerful tool to adjust the energy levels of the excited states localized at the Ru-based and thiophene-based portions of the species.
8 Multiple ethynylpyrenyl based Ru(II)–terpyridine and Ru(II)–phenanthroline complexes
It is generally admitted that [Ru(terpy)2]2+ derivatives have much less attractive photophysical properties than [Ru(bpy)3]2+, most notably due to a short excited-state lifetime at room temperature [2,89]. However, much efforts have been devoted to design tridentate ligands with prolonged lifetime [58], including the use of cyclometalating ligands, electron-withdrawing and -donor substituents, and ligands with extended π∗ orbitals [1]. Within the last approach, species based on ethynyl substitution have given quite interesting results [14,87]. There has been recent interest in the study of bichromophores consisting of a Ru(II) tris-bidentate complex, [Ru(bipy)3]2+, covalently linked to pyrene (and others) [60] using a variety of tethers [73a,90,91]. For these compounds, the approach to generate long lifetime emission at room temperature is to select a ligand system whose lowest-lying ligand-centered (3ππ∗, 3LC) level is slightly lower in energy relative to the metal-to-ligand charge-transfer (3MLCT) emitting level of the complex core, with an energy gap of the order of kBT, about 200 cm−1. In such bichromophores at room temperature, visible light absorption results in a sequence of steps, (i) population of the 1MLCT level at the [Ru(bipy)3]2+ unit, (ii) rapid intersystem crossing to 3MLCT and 3LC states, (iii) establishment of thermal equilibrium between these two triplet levels, (iv) light emission from the level with the higher radiative rate constant, kr; for [Ru(bipy)3]2+–pyrene systems this usually implies predominant emission from the 3MLCT level (for the separate components, ϕemMLCT > ϕemLC, and krMLCT > krLC) [92], with the 3LC level playing as “energy reservoir” [79,93]. This scheme works well only if the two chromophores are weakly interacting so that “localized” excitations can be considered. In most cases, the excited-state lifetimes observed from these complexes were significantly enhanced relative to model compounds as a result of the interstate dynamics involving 3MLCT and 3LC levels [94] (Scheme 14).
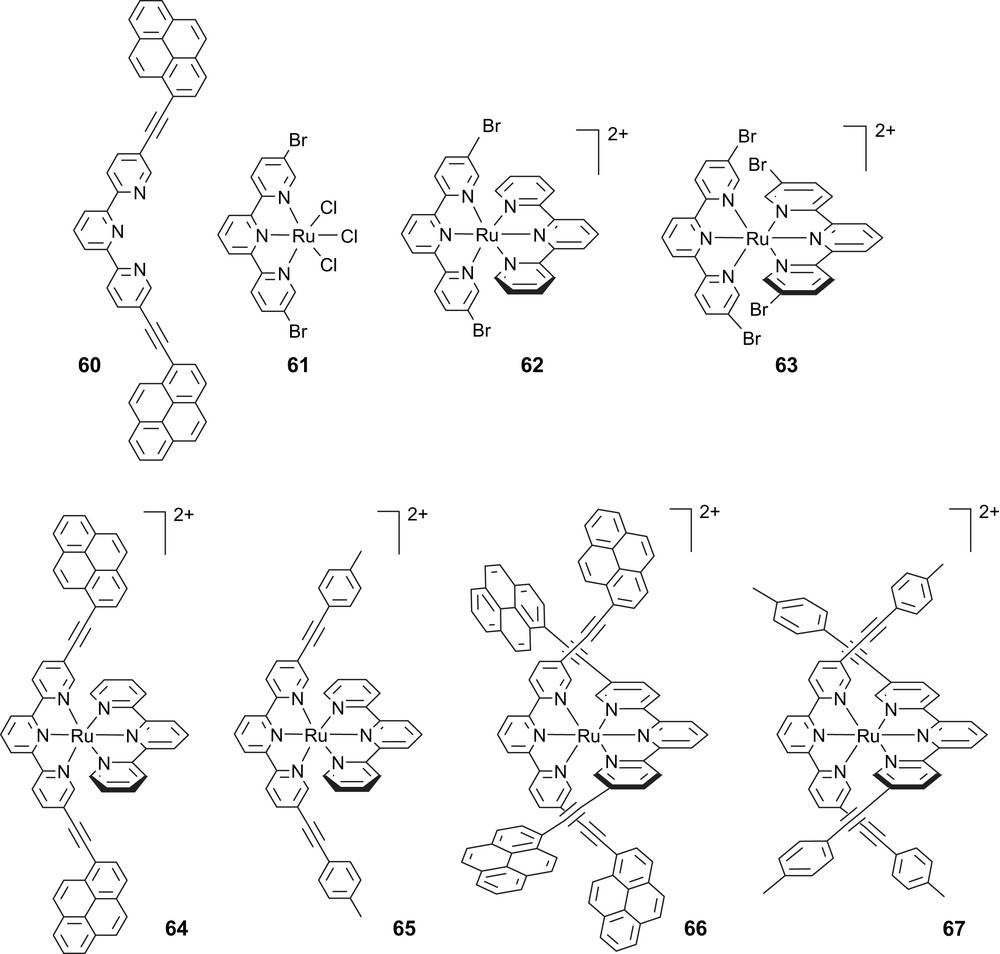
It is also interesting that Ru(II) bis-terdentate complexes, [Ru(terpy)2]2+, have been covalently linked to pyrene in the attempt to enhance the 3MLCT-based luminescence behavior. Thus, in [(pyr-terpy)Ru(terpy)]2+ the presence of the ethynylpyrene appendage results in an improved luminescence (by more than two orders of magnitude, for both the intensity and the lifetime, ϕ = 5 × 10−3 and τ = 580 ns, respectively), with respect to the reference [Ru(terpy)2]2+ species (pyr-terpy = 4-(1-ethynylpyrene)-2,2′:6′,2′′-(terpyridine)2,2′-bipyridine). Remarkably, the lowest-lying triplet in [(pyr-terpy)Ru(terpy)]2+ was attributed of 3MLCT nature. Therefore, 3LC (pyrene-centered) states, lying at higher energy, cannot apparently play as “energy reservoir” which leaves somewhat unexplained the reasons for the luminescence performance.
Several such heteroleptic and homoleptic ruthenium–terpyridine complexes bearing two and four ethynylpyrenyl or ethynytolyl residues have been prepared from complexes carrying reactive bromo functions. Again the chemistry at the complex is a viable pathway to produce all these complexes by cross-coupling promoted by low valent palladium(0) on preformed halogen complexes (Scheme 14) [95].
The new multichromophoric complexes have characteristic absorption spectra and are luminescent both in solution and in rigid matrix at 77 K, with room temperature lifetimes and quantum yields significantly larger than [Ru(terpy)2]2+ (Fig. 7). At room temperature, the tolyl-substituted complexes are triplet metal-to-ligand charge-transfer (3MLCT) emitters whereas for the pyrene-grafted complexes pyrene-centered emission is observed. For the latter complexes, the energy gap, ΔTT, between higher 3MLCT levels and lower ligand-centered (3ππ∗, 3LC) levels is in the 640–730 cm−1 range, which results in the interstate dynamics at the basis of the observed luminescent behavior. At 77 K, for the pyrene-grafted complexes, the emission reveals features that are tentatively ascribed to intraligand interactions involving the pyrene and terpyridine units.
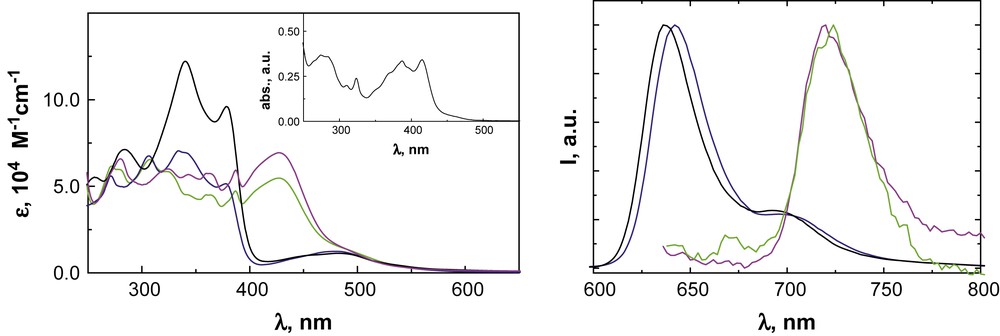
Left: absorption spectra of complexes 64 (green), 66 (magenta), 65 (blue), and 67 (black) in CH3CN, and of ligand 60 (inset) in CH2Cl2. Right: Normalized 77 K emission spectra of CH3CN solutions of complexes 64 (green), 66 (magenta), 65 (blue), and 67 (black); λexc = 490 nm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Similarly, the synthesis, electrochemical and optical spectroscopic properties of a Ru(II)–diphenylphenanthroline complex decorated with ethynylpyrene (EP) appendages were studied (Scheme 15).
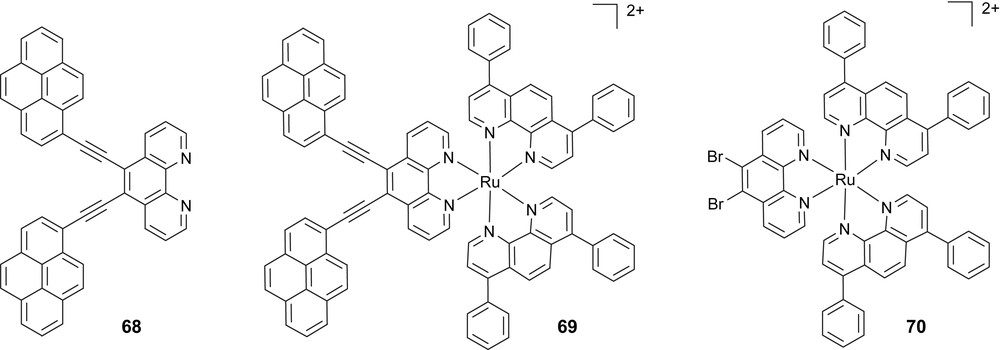
In CH3CN solvent, both the substituted complexes are redox active with metal-centered and ligand-centered processes assigned with reference to the analogous steps for [Ru(phen)3]2+ and to steps for the EP appendages. The complexes exhibit strong absorption bands in the visible and near-UV due to ligand-centered and MLCT transition, respectively; for 69, the lowest-lying absorption band is slightly red-shifted due to a better delocalization at the ligand bearing the pyrene subunits. At room temperature and in O2-free solvent, complex 69 appeared to be practically non-luminescent while 70 exhibited intense 3MLCT emission with ϕem 9 × 10−4 and 6.1 × 10−2, respectively; λexc = 462 nm in both cases. Transient absorption experiments allowed assigning the lowest-lying state of 69 to 3LC levels of EP localization, which are non-luminescent thus explaining the lack of room temperature luminescence for this complex. At 77 K, 70 is emissive, as expected for 3MLCT levels; however also for 69 a moderately intense 3MLCT emission is observed at this temperature. For the latter case, this outcome is tentatively explained in terms of local trapping of 3MLCT states at the 4,7-diphenyl-phenanthroline ligands free of EP appendages [96].
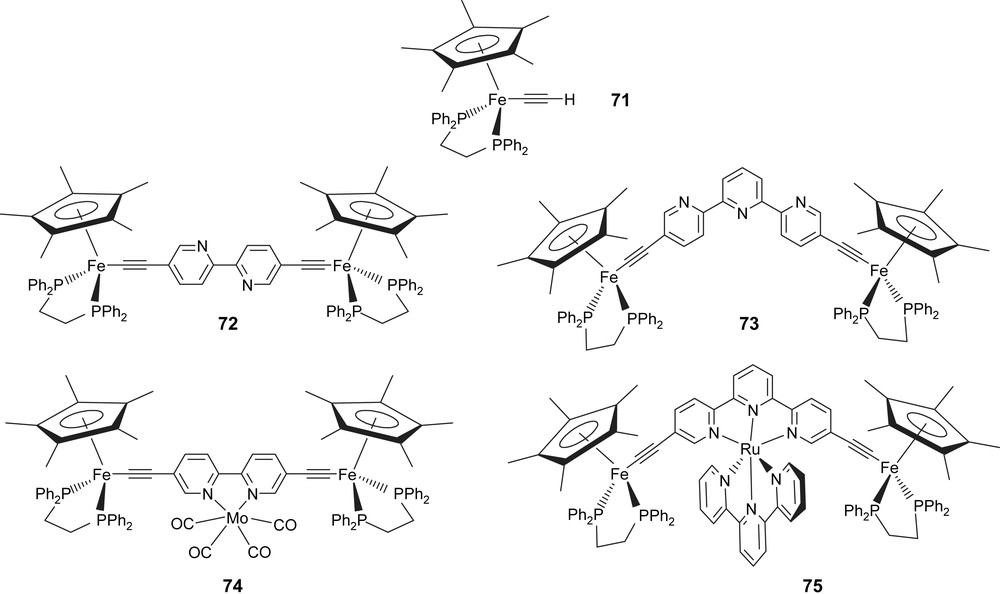
9 σ-Connection of alkynyl oligopyridines to iron and borodipyrromethene dyes
From a general viewpoint judicious spatial or topological arrangements of chromophores or redox-active endgroups can lead to molecular architectures presenting unique properties for information storage and processing at the molecular level [97].
In the open literature we can found several cases where the “(η2-dppe)(η5-C5Me5)FeCC″ fragment can import interesting electronic properties to various molecular assemblies [98,99]. Fluorescence switching or modulation might be envisaged by changing the redox state of the organoiron endgroup [100] or the nature of the central ion [65b]. Furthermore, when the ligand has more than one redox-active substituent appended, one should also be able to tune the electronic communication between the two redox-active endgroups by simply changing the complexed metal center. The use of the classical vinylidene route [101] to connect bipy or terpy chelating fragments to organoiron endgroups is not operating here. However, classical “metalla-Sonogashira” coupling conditions [102], between compound 71 and the adequate bromo-substituted terpyridine or bipyridine provide the desired bifunctionalized terpy 73 and bipy 72 complexes (Scheme 16) [103]. Interestingly, the central terpy or bipy fragment could be chelated respectively by Ru or Mo using either [(terpy)Ru(Cl)3]or [(norbornadiene)Mo(CO)4] as metal precursors leading respectively to complexes 75 and 74.
X-Ray molecular structures were obtained in most of the cases (Fig. 8). For complex 73 the terpy ligand adopts a planar conformation with the two iron atoms lying ca. 17.3 Å apart and for complex 72 the bipy ligand adopts a quasi-planar conformation with the two iron atoms lying ca. 16.0 Å apart. The complexation of the molybdenum center reinforces the planarity of the bipy fragment in complex 73, as would be expected from a strict electronic viewpoint [103].

ORTEP representation of complex 73 (Top), of complex 72 (Middle) and of complex 74 (Bottom).
Whereas in complexes 72 and 74 the two iron-centered oxidations were barely separated, suggesting weak electronic coupling between the two redox centers, complexation of molybdenum produced a new absorption at 442 nm due to a dMo → (π∗)bipy MLCT [104] and a bathochromic shift from 487 to 606 nm for the dFe → (π∗)CCAr MLCT band. Similarly, complexation of 73 with Ru generates a new absorption at 481 nm corresponding to the dRu → (π∗)terpy MLCT along with a significant shift of the dFe → (π∗)CCAr MLCT band from 464 nm in the metallo-ligand 73 to near 700 nm in complex 75. No evidence for the presence of emissive triplet states either at rt or at cryogenic temperatures has been obtained [103].
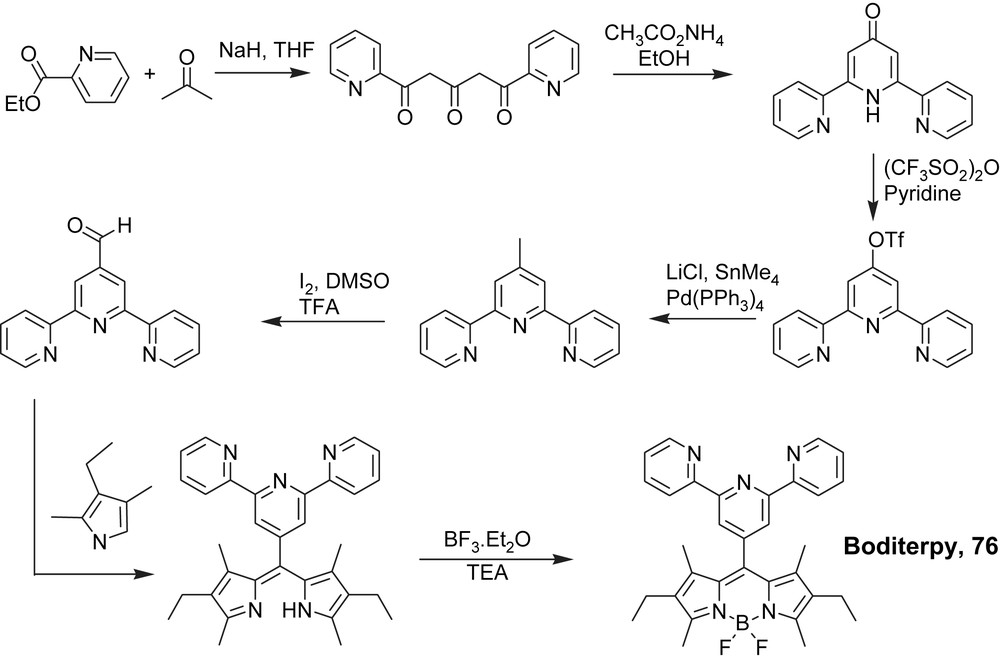
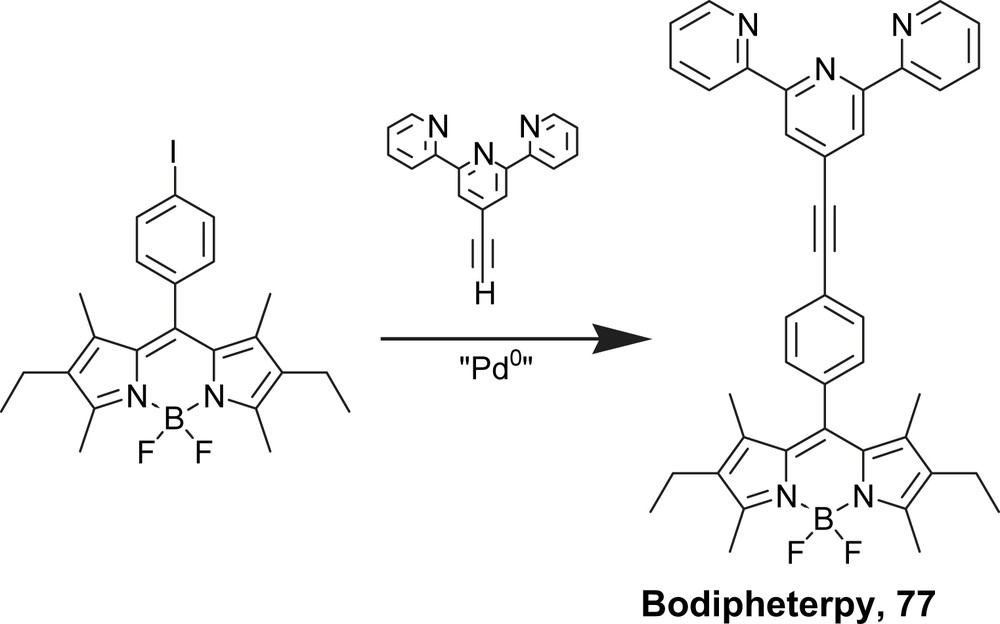
However, the linkage of the well-known singlet emitter boroazadipyrromethene (Bodipy) to chelated oligopyridine provides interesting insights into the formation of emissive triplet states localized on the organic fragment. Here the use of an alkyne tether is not useful to connect both subunits. The hybrid system (Boditerpy) 76 was constructed from the terpyridine-4-carboxaldehyde synthon and the Krypto pyrrole (Scheme 17), whereas Bodipheterpy 77 was prepared by a Pd(0) promoted cross-coupling reaction (Scheme 18) and from the phenyliodoBodipy [105] and 4′-ethynyl-2,2′:6′,6″-terpyridine [106].
Complexation of the Boditerpy and Bodipheterpy ligands [107] with Zn(II) provides the homoleptic complexes 78 and 79 respectively (Scheme 19). For ligand 76, absorption and fluorescence spectral profiles are dominated by contributions from the bodipy unit but at 77 K or in the presence of iodoethane at rt, the low energy emission band is assigned to the triplet emission of the Bodipy subunit (Fig. 9). The role of iodoethane is not to alkylate the free donor atoms but to favour intersystem crossing from the singlet to the triplet-excited state.
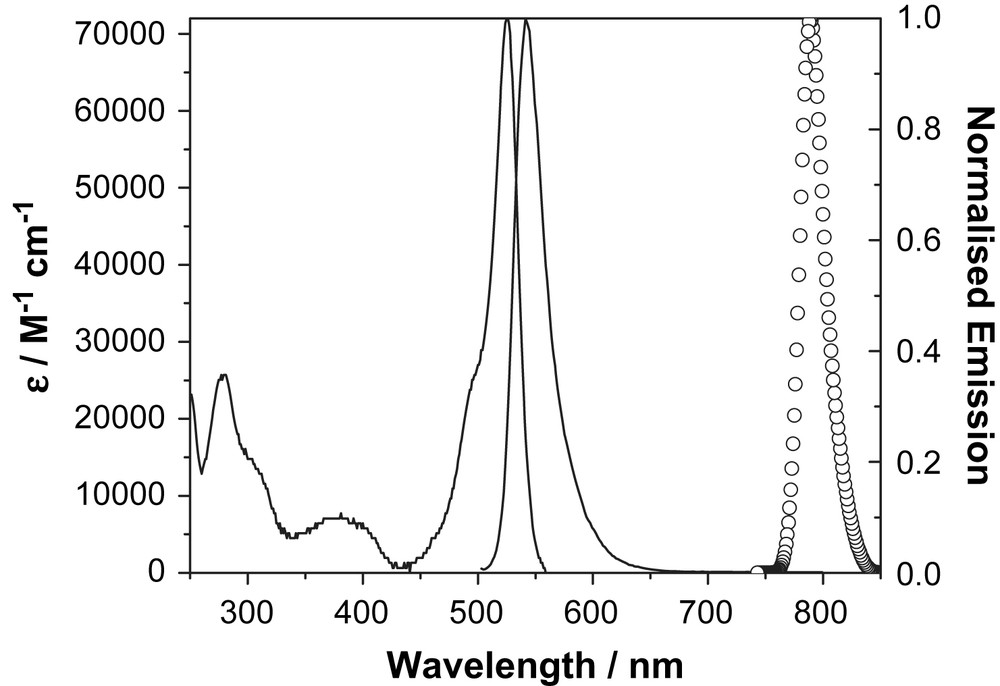
Absorption and fluorescence spectra recorded for Boditerpy 76 in acetonitrile at room temperature. The low-temperature phosphorescence spectrum recorded in ethanol containing 10% v/v iodoethane is shown as a circled curve.
For the two photoactive complexes 78 and 79 the fluorescence from the terminal bodipy dye is extensively quenched due to intramolecular electron transfer [108]. This latter process is activated at high temperature but becomes activationless in a glassy matrix when the reactants are closely coupled. For the shorter analogue, 78, electron transfer occurs along the connecting σ-bond and electronic coupling is relatively high. Electron transfer occurs under non-adiabatic conditions, despite the close proximity. Extending the molecular axis, as in 79, decreases the extent of electronic coupling but electron transfer is still the dominant means for deactivation of the excited singlet state. Although the triplet-excited state of the bodipy dye lies at lower energy than the charge-separated state, charge recombination occurs preferentially to reform the ground state. In both cases, the rate of charge recombination exceeds that of charge separation such that charge-separated products are not seen in the transient absorption spectral records.
Charge recombination might be enhanced by preferential localization of the positive charge at the bridgehead meso carbon at the bodipy π-radical cation level or by quantum mechanical effects. Although triplet state formation is relatively unimportant compared to deactivation of the charge-separated state, there is still an impressive increase in the rate constant for intersystem crossing (kISC) relative to the parent bodipy dye. Thus, kISC for 76 (Boditerpy) and 77 (Bodipheterpy) is less than ca. 5 × 105 s−1. For the corresponding Zn–terpy complexes, kISC increases to around 5 × 108 s−1; i.e., an increase of at least 1000-fold. It has been shown previously [109] that fast intersystem crossing is to be expected for conformations where the nodal planes of the donating and accepting molecular orbitals are approximately perpendicular to each other. Thus, the orthogonal geometries inherent to 78 and 79 should give rise to a large spin-orbit coupling matrix element. There is also the realization that the cation provides additional spin orbital coupling via the heavy-atom effect [110]. The fact that triplet formation does not dominate over charge recombination is considered to be a consequence of the poor spin orbital coupling properties of the parent dye.
An interesting feature of the present systems is that the Zn–terpy complex acts as a light harvester [111,112] and channels absorbed photons to the bodipy dye, as evidenced by fluorescence excitation spectroscopy. For both 78 and 79, the rate of singlet–singlet energy transfer from the metal complex to the bodipy dye exceeds 5 × 1010 s−1. The most likely mechanism involves Förster-type, dipole–dipole interactions since there is strong spectral overlap between fluorescence from Zn–terpy and absorption by bodipy and almost optimal orientation of the relevant transition dipoles. The Zn–terpy chromophore absorbs in the spectral region where bodipy is relatively transparent and can be illuminated with good selectivity [113].
10 Functionalized oligopyridine ligands bearing accessory dipyrromethene-BF2 fluorophores
Finally, the piecewise synthetic strategy approach, although tedious in operation, permits the engineering of a plethora of multicomponent molecular systems embodying preferential binding properties by the specific use of preorganized modules. Selected examples of stable and highly luminescent pyridine-, bipyridine-, phenanthroline-, bipyrimidine-, and terpyridine-based ligands bearing one or two 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (bodipy) modules are provided in Schemes 20 and 21. For the phenylethynyl-linked molecules, direct coupling between the bodipy-phenyliodo and the 4′-ethynyl-2,2′:6′,2″-terpyridine, 6,6″-diethynyl-2,2′:6′,2″-terpyridine, 5-ethynyl-2,2′-bipyridine, 5,5′-diethynyl-2,2′-bipyridine, 6,6′-diethynyl-2,2′-bipyridine and 5,5′-diethynyl-2,2′-bipyrimidine substrates is promoted by Pd catalysts and sonication. This procedure provides the advantages of efficiency, versatility and rapidity (Scheme 20) [114].
A second set of experimental conditions is required to produce the 3,8-disubstituted-1,10-phenanthroline and 5,5″-disubstituted-2,2′:6′,2″-terpyridine derivatives [114]. Due to the weak reactivity of the bromo-substituted partners and limited stability of the ethynyl substituted phenanthroline and terpyridine a one-pot reaction was envisaged. The deprotection of the alkyne and the cross-coupling were conducted in situ by a phase transfer process with aqueous NaOH and Et3BzN+Cl− as mediator, and the benzene phase contains [Pd(PPh3)4] and CuI as catalysts (Scheme 21).
Complexation of some ligands with Ru(terpy) fragments provides heteroleptic complexes (Scheme 22) for which absorption spectra, luminescence properties (both at room temperature in fluid solution and at 77 K in rigid matrix), and redox properties have been investigated [115]. Absorption spectra and redox behavior indicate the supramolecular nature of the multichromophoric assemblies, allowing to assign the various absorption features and the several redox processes to specific subunits. In spite of the good luminescence properties of the separated components, the hybrid Bodipy/Ru complexes do not exhibit any luminescence at room temperature. However, transient absorption spectroscopy evidences that for all of them a long-lived (microsecond timescale) excited state is formed, which is identified as the bodipy-based triplet state (Fig. 10). It is proposed that such a triplet state is formed in most cases by the intervening of a charge-separated level from the bodipy-based 1π, π∗ state. At 77 K, all complexes exhibit the bodipy-based fluorescence, although with a slightly shortened lifetime compared to the corresponding free ligand(s), and also exhibit a phosphorescence assigned to the bodipy subunits. To the best of our knowledge, this is the first time that phosphorescence of bodipy species has been observed and we propose that it can be an effective decay process thanks to the presence of the ruthenium heavy atom and of the closely lying 3MLCT state of the Ru(terpy)2-type subunits [116].

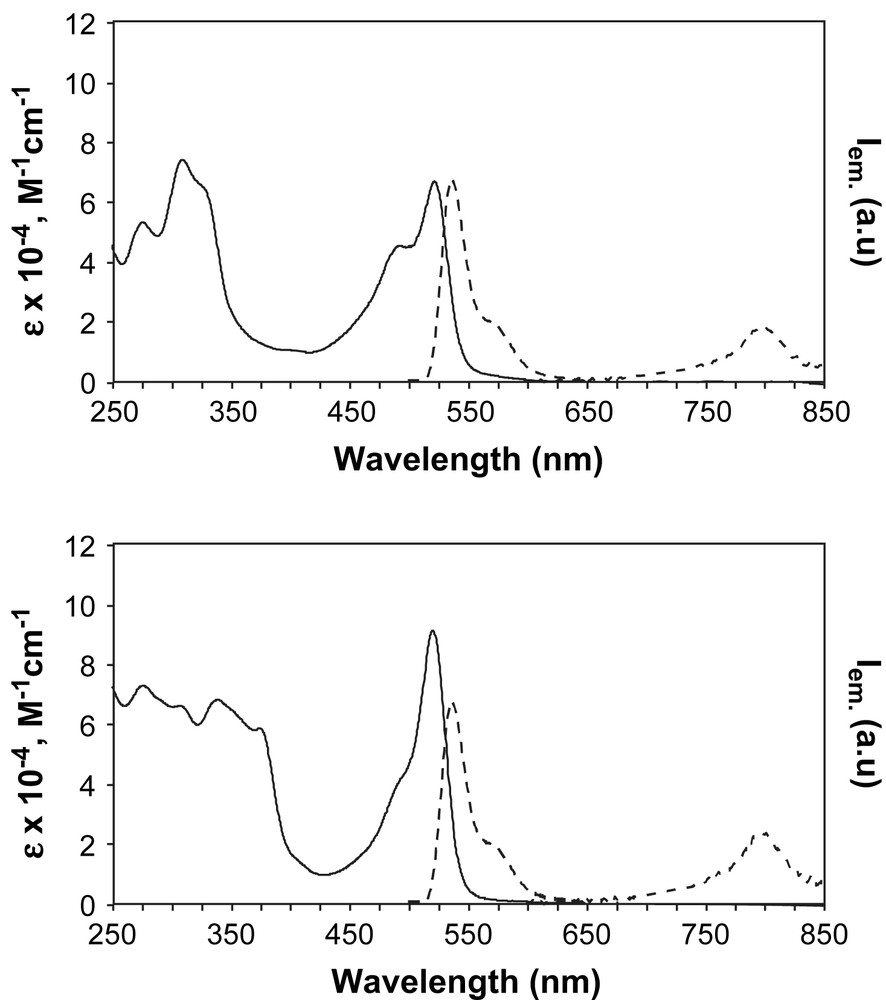
TOP: absorption (in acetonitrile, solid line) and 77 K emission (in butyronitrile, dashed line) spectra of 86. BOTTOM: absorption (in acetonitrile, solid line) and 77 K emission (in butyronitrile, dashed line) spectra of 87.
Finally, additional confirmation of the presence of emissive triplet state is coming from platinum(II) connection to Bodipy subunits. Cross-coupling of 89 with [(terpy)Pt(Cl)](BF4) is straightforward in the presence of CuI under anaerobic conditions [117] (Scheme 23).

Interestingly, in the hybrid system 91 the bodipy fluorescence is not quenched, although an oxidative photoinduced electron transfer is thermodynamically allowed, because of unfavorable electronic factors due to the structural arrangement of the hybrid species. On the contrary Pt-based 3MLCT luminescence is quenched by energy transfer to the bodipy triplet state both at 77 K and rt (Fig. 11). At 77 K, phosphorescence of Bodipy also occurs, as a consequence of the presence of the heavy metal center [118].
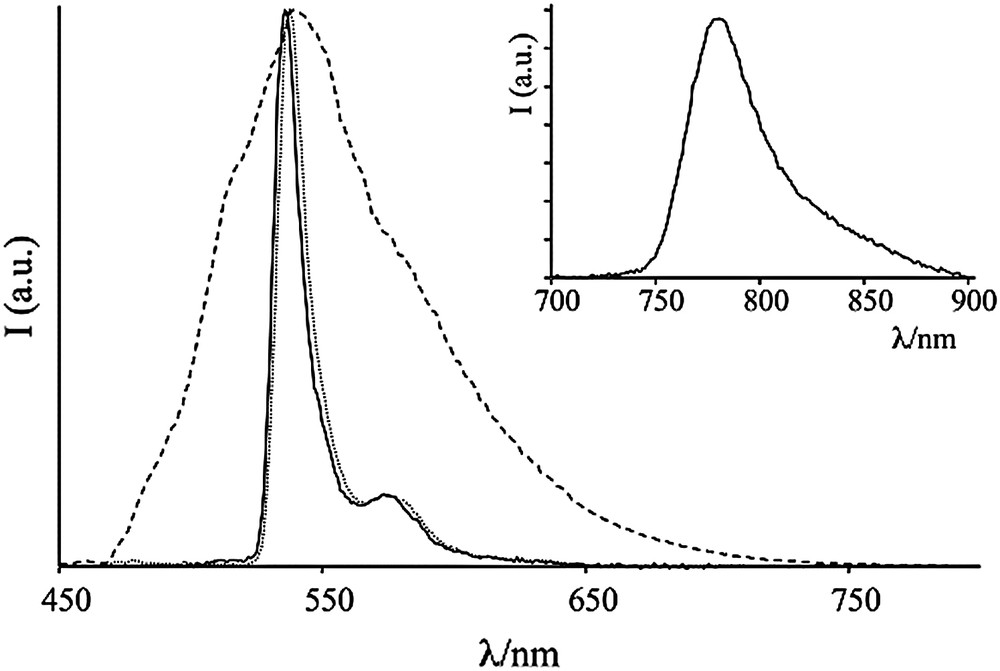
Emission spectra of 88 (thin line), 90 (dashed line) and 91 (thick line) in butyronitrile matrix at 77 K. Inset shows the emission spectrum of 82 recorded with a delay of 0.1 ms from excitation pulse.
The presence of residual phosphorescence from the Pt center in the hybrid complex 91 allows to determine the rate of the energy transfer process (Fig. 12).

Energy levels (not in scale) and decays of 91.
11 Conclusion and perspectives
The development of segmented multichromophoric species of nanometric dimension is still in its infancy. However, it has been shown that intramolecular triplet energy transfer by the electron-exchange mechanism offers a suitable analytical method to measure the efficacy of molecules at conducting electrons. A particularly appropriate system has RuII/OsII poly(pyridine) complexes at either end of the array. The best system produced to date displays triplet energy transfer over a distance >70 Å. This value, although impressive, is insufficient for the construction of viable molecular photonic wires. Considerably more success has been achieved with respect to the prolongation of the triplet lifetime of the RuII-based donor and it is now possible to prepare luminescent complexes with triplet lifetimes on the order of 150 μs in deoxygenated solution at room temperature. There still remains the problem, however, of engineering triplet energy transfer over distances in excess of 100 Å.
An alternative approach to the construction of photonic molecular devices is to incorporate numerous identical chromophores into a one-dimensional array. Several such systems have been built in recent years, using conjugated aromatic residues or fused porphyrins. Polyacetylenic complexes have been synthesized that contain metal atoms in the conjugation pathway. It has also proved possible doping carbon nano-wires with transition metal ions. Related systems are starting to appear that have many RuII poly(pyridine) complexes attached to a linear polytopic ligand. Such materials might operate as artificial light harvesters able to transport photons over long distances by way of a random walk. If so, it should be possible to cap the end of the wire with an OsII-based acceptor, thereby creating an efficient photonic wire in which each chromophore lies in the same chemical and electronic environment. Such ligands have recently been engineered with well-adapted protocol for which the position of the metal is controlled by the synthesis and the reactive functions present at the molecules.
An interesting strategy for constructing long, linear arrays has been developed. This approach involves the synthesis of ethynylated ditopic ligands equipped with a metal complex at one end. Addition of a suitable metallo-synthon results in the self-assembly of the corresponding trinuclear complexes. Such materials display an interesting variety of intramolecular electron and/or energy transfer processes whilst the ZnII complex exhibits extended electron delocalization at the triplet level. These systems could be adapted to produce arrays of higher nuclearity by careful selection of the bridging cation and reaction conditions. Indeed, it should be possible to use a bridging cation that introduces a metal bis(2,2′:6′,2″-terpyridine) complex with triplet energy between those of the primary donor and the ultimate acceptor. This would provide a photonic wire where reversible energy migration allows for unusually long-distance excitation energy transfer. The main advantage of this type of approach is that it avoids the synthesis of multiple ligands of limited solubility.
There is indeed a rich palette of hybrid structures and properties to be generated by blending transition metals phosphorescent scaffoldings with organic fluorophores allowing electronic π-networks to be expanded so that highly tuneable electronic and spectroscopic features can be envisaged.
Acknowledgements
We gratefully acknowledge generous funding from the CNRS and the Université Louis Pasteur de Strasbourg and Johnson-Matthey Ltd for the loan of precious metal salts. The research efforts of many talented postgraduate and postdoctoral associates are acknowledged with our most sincere gratitude. The contributions of colleagues are credited by reference to original literature citations. We also warmly thank the reviewer of the manuscript for improving the readability of this manuscript.