

1 Introduction
Long-range energy and electron transfer is a topic of considerable importance owing to its fundamental and practical implications with solar energy conversion. Our ability to rationally design molecular systems featuring long-distance electron or energy transfer processes represents a crucial step towards the development of systems that mimic the natural photosynthesis process [1,2]. Besides, the output of these researches can also have a strong impact in photovoltaics or in molecular electronics [3]. In this context, covalently linked donor–bridge–acceptor (D–B–A) is the archetypal molecular system used to perform and study photoinduced charge separation reactions. In these molecular systems, the importance of the linkage is quite significant because it has two fundamental roles. The first one is a geometrical feature, since a rigid bridge positions two neighbouring active centres at a fixed distance and with a well-defined geometry. Secondly, the bridge may also fulfil a functional feature if it promotes electronic communication to assist the electron and/or energy migration between the two bridged units. According to the current theory, the electronic coupling in a donor–bridge–acceptor system (D–B–A) is usually dominated by through-bond interactions, via the electronic properties of the bridge, which is known to decrease exponentially with the separation distance (d) between A and D (Eq. (1)) [4,5].
| (1) |
Our interest in the development of D–B–A systems derives from the aim of performing bridge-mediated electron transfer, that is, to use the electronic properties of the bridge to assist the charge separation between D and A over a long distance [8–11,39]. Extensive theoretical analyses have been devoted to this problem, but, surprisingly, there are relatively few molecular systems that are based on the implicit utilization of a long spacer to assist the electron transfer for photoinduced charge separation. Relevant examples are the para phenylenevinylene spacers used to connect a tetracene sensitizer to a pyromellitimide acceptor described by Wasielewski and coworkers [6,12]. More recently, the use of the same spacer was reported by Martin and coworkers to prepare fullerene based dyads [13]. Other systems, based on other π-conjugated wires, were also reported recently to promote long-range charge separations [14,15]. We have been particularly interested in the use of porphyrin chromophore to build photomolecular systems for single step long-range charge separations, because porphyrins are versatile dyes with tunable photo- and electro-chemical properties which make them well-suited to be used either as light collectors, electron donors or electron acceptors depending on the nature of the metal ions complexed inside the macrocycle and/or its peripheral substituents [16]. Until recently, porphyrin arrays, designed in view of photoinduced energy and electron transfers, generally featured very weak electronic interactions, mostly because the bridge was appended on the meso-aryl substituents of the porphyrin [2,17]. In that case, the electronic communication is weak because in meso-tetraaryl porphyrins, the aromatic substituent is forced out of the porphyrin macrocycle, interrupting the conjugation with this substituent [18]. Pioneering works of Arnold [19] and Anderson [20–22], followed by those of Therien [23,24], Osuka [22,25] and other groups [26], demonstrated that the oligoynyl bridge provides exceptional interporphyrin electronic communication if connected directly on the meso position of the porphyrin macrocycle. The strong electronic interactions brought by this molecular design was essentially exploited for the development of new materials for non-linear optical properties. For example, Therien and coworkers reported push–pull porphyrin systems exhibiting exceptionally large first hyperpolarizability [24]. In the same vein, Anderson and coworkers prepared very interesting multiporphyrin arrays linked by triple bond with large two-photon absorption coefficient [20]. Surprisingly, the attachment of the bridge directly on the porphyrin core via a triple bond has been rarely exploited for long-range photoinduced charge separation. This review focuses on recent porphyrin systems performing long-range photoinduced charge separation (Fig. 1).
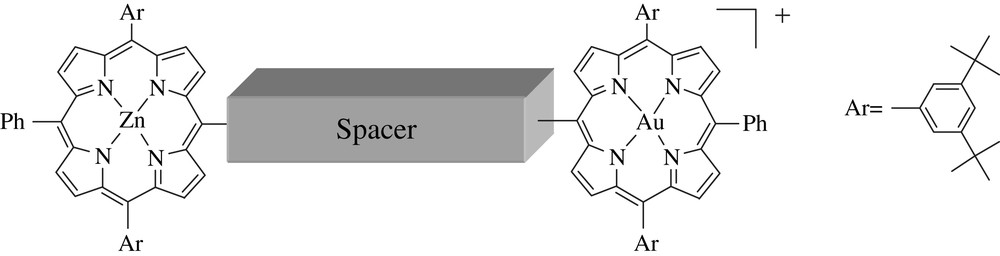
General structure of zinc/gold porphyrin dyads.
2 Weakly coupled zinc/gold porphyrin dyads
In this first section, we have summarized the results of the known weakly coupled systems composed of a zinc porphyrin and a gold porphyrin as respective photoactive electron donor and electron acceptor units. In the early 1990s, the Sauvage group in Strasbourg had the bright idea to use a gold porphyrin in association with a zinc porphyrin to perform photoinduced charge separation [27,28]. The idea, which led to this choice was certainly driven by the fact that the metal inside the porphyrin should render the complex a bad energy acceptor (high-lying excited state) and a good electron acceptor (low reduction potential) with respect to the zinc porphyrin. These latter features would favor the electron transfer over the energy transfer as it was usually observed in many bisporphyrin systems [29]. The oblique bisporphyrin bridged by a phenanthroline was at the origin of many beautiful and elaborated molecules thanks to the coordinating properties of the two nitrogens of the spacer, which open up the route to the construction of entwined catenanes or rotaxanes (Fig. 2) [30]. The exceptional properties of most of these zinc and gold porphyrin dyads is that light excitation of any chromophore (ZnP or AuP+) brings about a fast charge separation with quantum yields close to 100% [27]. Indeed, there are few multi-hetero-chromophoric arrays in which the light excitation of any of the chromophores induces a systematic electron transfer reaction.
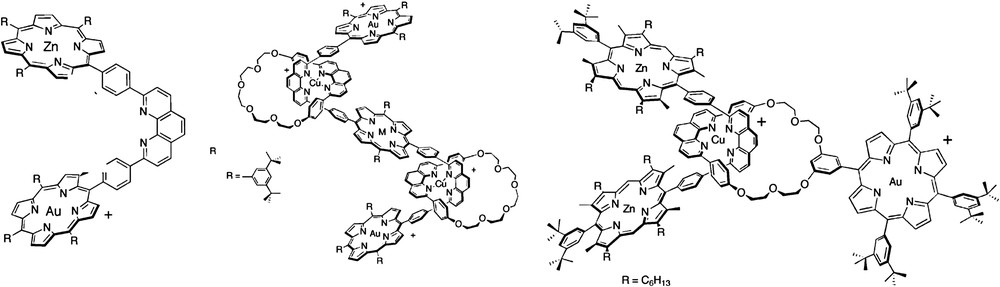
Structures of some of the ZnP/AuP+ systems prepared by Sauvage and coworkers. M = Zn(II). Reprinted with permission from Ref. [30a].
The suitability of the couple ZnP/AuP+ to perform photoinduced charge separation prompted several other groups, such as Mårtensson [15,31–33], then Crossley [34] and our group [8,35], to use it to prepare other dyads based on this successful combination (Fig. 3).
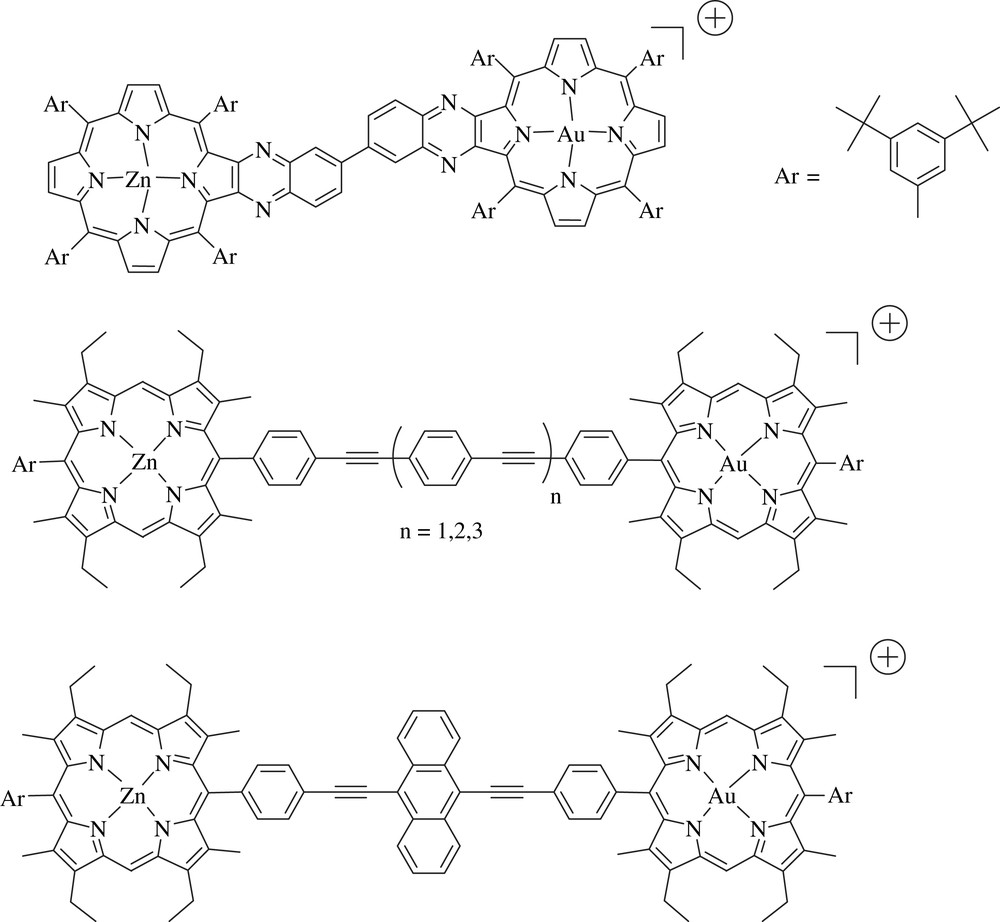
Dyad prepared by Crossley and coworkers (top) and those by Mårtensson and coworkers.
The specificities of the dyad of Crossley relies first on the use of quinoxaline porphyrins, in which the bridge is in interaction with the porphyrin via the fused ring across the β,β′ pyrrolic positions. This latter aspect represents an interesting characteristic of this type of connectivity since it opens a channel to propagate electronic communication. Quinoxaline porphyrins have been employed recently by Imahori and coworkers to design particularly efficient sensitizers for dye-sensitized solar cells [36]. The second unusual feature of the zinc/gold porphyrin prepared by Crossley and coworkers is the long-lived charge separated state that was reached with this dyad (τ = 10 μs at 298 K in cyclohexane), whereas most of the ZnP/AuP+ systems usually lead to relatively shorter charge separated states, hence to a relatively low kCS/kCR ratio.
Mårtensson and Albinsson report [15,31–33] a series of very interesting octaalkyldiaryl zinc and gold porphyrins bridged by various spacers (Fig. 3). The absorption spectra of all these dyads are essentially a linear combination of the spectra of their constituents, which indicate that there is a weak electronic interaction in the ground state. This is consistent with the attachment of the bridge on the meso phenyl substituent of the porphyrin and the presence of ethyl groups in the β pyrrolic positions, which limit the π-conjugation with the bridge. One valuable aspect of the studies on these dyads is the comprehensive investigation of the electron transfer mechanism as a function of the electronic properties of the bridge. For example, it has been demonstrated that some spacers promote bridge-mediated electron transfer by a superexchange mechanism, whose rates are nicely correlated with the energy gap between the zinc porphyrin excited-state and the bridge LUMO levels [32,33]. However, due to the weak electronic coupling, the efficiency of the electron transfer quantum yield becomes very low with the long bridge (14% in benzonitrile for the central dyad with n = 3 in Fig. 3).
3 Electronically coupled zinc/gold trisaryl porphyrins connected via an ethynyl linkage
This part is devoted to our most recent results collected with trisaryl porphyrins in which the bridge is directly connected in the porphyrin meso position via an ethynyl or ethenyl linkage to an oligophenyleneethynylene spacer (Fig. 4 and Scheme 4).
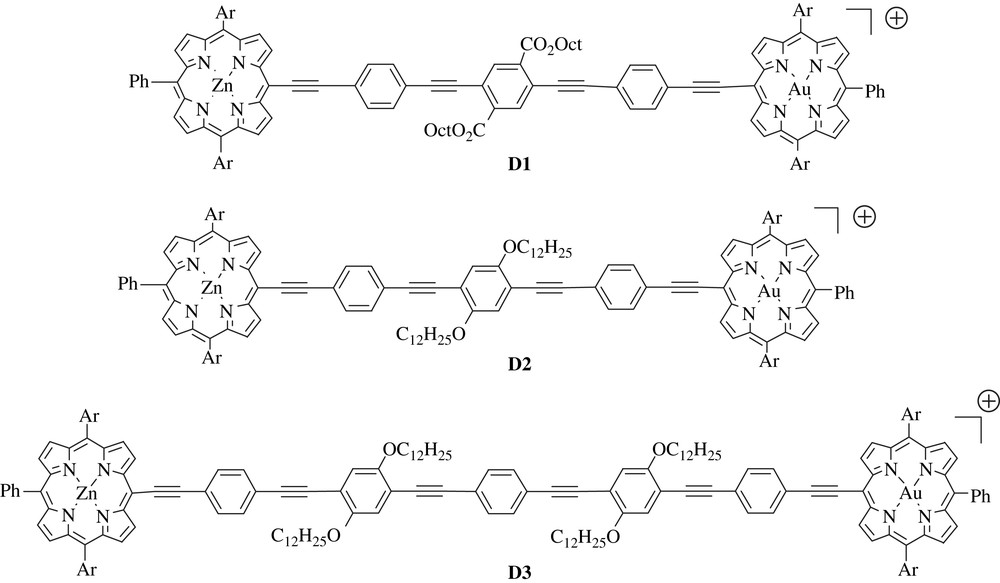
Schematic representation of electronically coupled porphyrin dyads D1–D3.
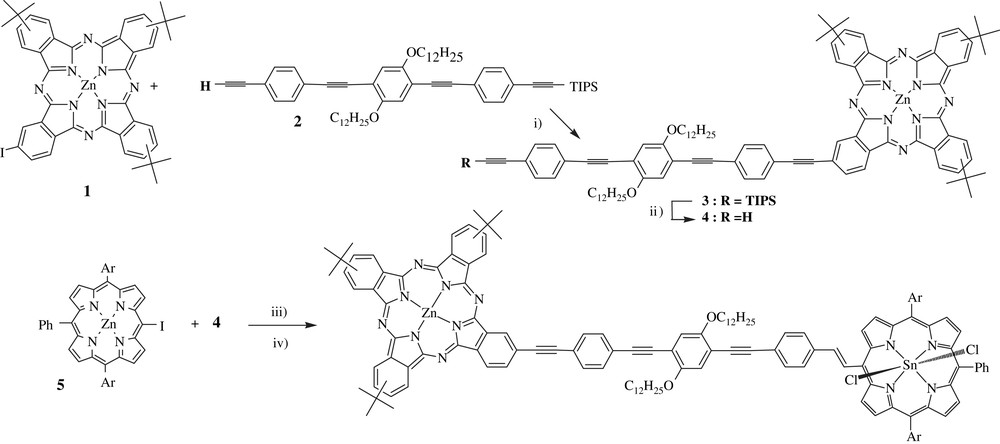
Preparation of the dyad D4. Reagents and conditions: (i) piperidine, Pd2dba3–CHCl3, AsPh3, 40 °C, 15 h (43%); (ii) Bu4NF, THF (100%); (iii) THF, Et3N, Pd2dba3–CHCl3, AsPh3, 60 °C, 15 h (33%); (iv) HCl, CH2Cl2 then pyridine, SnCl2–2H2O, 185 °C, 3 h (75%).
3.1 General synthetic strategy for the preparation of the dyads D1–D3
In this project, we set out to use trisaryl porphyrins instead of bisaryl porphyrins, because in the latter the remaining free meso position could lead to potential instability problems during the reactions and/or the purifications. For example, no gold bisaryl porphyrin lacking substituent on the β pyrrole positions is reported in the literature and in our hands the preparation of such compound has not been successful. These facts therefore pointed towards some difficulties in preparing the halogeno bisaryl gold porphyrin analogue of 7–8 (Scheme 1). Besides, we felt that octaalkyl porphyrins were not suitable building blocks for this project, since steric hindrance of the alkyl chains with the spacer would considerably reduce the π-conjugation, hence the electronic coupling.
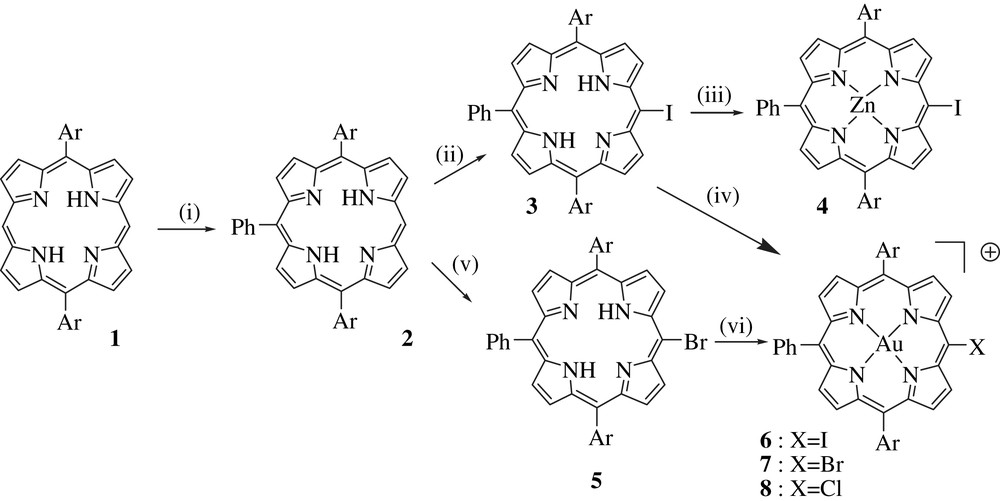
Synthesis of the porphyrin building blocks. Reagents and conditions: (i) 6 equiv. PhLi, THF, −78 °C, then H2O and DDQ (92%); (ii) I2, (CF3CO2)2IPh, CHCl3 (99%); (iii) Zn(OAc)2, CH2Cl2, MeOH (94%); (iv) Au(tht)2SbF6, 2,6-lutidine, CHCl3 (38%); (v) NBS, CH2Cl2, −20 °C at r.t. (90%); (vi) HAuCl4·3H2O, NaOAc, CH3CO2H, 120 °C (75%).
The preparation of the heterometallic porphyrin dyads was accomplished according to a stepwise modular approach, which implies the initial preparation of the three key building blocks; namely the iodo zinc porphyrin 9, the gold halogeno porphyrin 7–8 and the bridge functionalized at each of its extremity by an ethynyl group protected by a silyl group (orthogonal protection).
The synthesis of the porphyrin key building blocks is illustrated in Scheme 1. Briefly, the third aryl group was introduced on the diaryl porphyrin 1 by nucleophilic substitution with phenyl lithium, according to Senge's methodology [37]. This procedure constitutes an improvement of our previous synthetic routes [11,38,39] because it offers a higher yield and it is simpler. The resulting trisaryl porphyrin 2 is then iodinated using bis(trifluoroacetoxy)iodobenzene and metallated by zinc to afford the first building block 1. Iodo gold porphyrin 6 could be prepared from 2, using the mild conditions developed by Chambron and coworkers [30g], but in a modest yield (32%), which prompted us to use, instead, the more stable bromo trisaryl porphyrin 5. Porphyrin 2 was brominated with NBS and then metallated by gold using HAuCl4 in refluxing acetic acid with sodium acetate. During this reaction, the bromo group was partly substituted by chlorine leading to an equimolar mixture of porphyrins 7 and 8 with an overall 75% yield (Scheme 1). This side reaction was not a problem, because the two porphyrins were not separated and used as a mixture in the next step, since their reactivities proved to be high with respect to the next Sonogashira cross-coupling reaction. The syntheses of the spacers were accomplished according to well-described established strategies in the literature and they will not be presented in this review [8,39].
The two last steps of the synthesis consist in connecting porphyrins 4 and 7–8 to the bridge (Scheme 2). The zinc iodo porphyrin was first coupled, because we observed that the cleavage of TIPS group with fluoride anions in the presence of the gold porphyrin led to the formation of by-products, which was not the case with the zinc porphyrin. The zinc iodo porphyrin 4 was therefore reacted with the appropriate spacer using different catalytic systems (phosphine ligands such as (tertBu)3P, (oTol)3P, AsPh3), but the best conditions found were essentially those initially described by Lindsey and coworkers [40]. After deprotection of the triisopropyl silyl group with tetrabutylammonium fluoride, the halogeno gold porphyrins 7 and 8 was connected to the spacer. Classical Sonogashira conditions (Pd(PPh3)4, CuI with an amine as a base) were not suitable for this coupling since they did not afford the expected coupled product [40]. Such a phenomenon was previously observed with a gold porphyrin substrate by Mårtensson and coworkers [33]. We found that Pd(dppf)Cl2 catalyst (dppf = diphenylphosphinoferrocene) with copper iodide in a mixture of triethylamine and dimethylformamide proved to be particularly efficient for this coupling since the two porphyrins 7 and 8 were completely consumed after 5 h at 50 °C and the expected dyads were formed with 50% yields. During the course of our work, Mårtensson and coworkers reported that the classical catalyst Pd2dba3–CHCl3 with AsPh3 is active for this type of coupling provided that the polarity of the medium is sufficiently high to ease the deprotonation of the terminal alkyne proton [41]. However, these new conditions were not tested, since they appeared in the literature after the completion of the synthesis of all the dyads described herein.
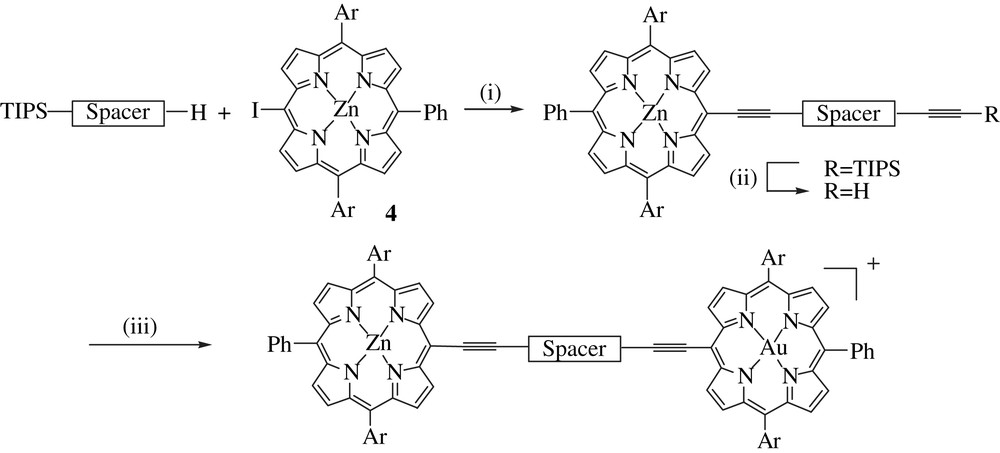
Synthetic route for the stepwise synthesis of the dyads D1–D3. Reagents and conditions: (i) AsPh3, Pd2(dba)3.CHCl3, toluene, Et3N, 60 °C (19–36%); (ii) Bu4NF, THF, r.t. (95–99%); (iii) equimolar 7/8, CuI, Pd(dppf)Cl2, Et3N, DMF, 40–60 °C (45–50%).
3.2 Zinc porphyrin linked to gold porphyrin via an oligophenyleneethynylene spacer
Among the π-conjugated molecules potentially suitable for being used as spacers, oligophenyleneethynylenes appear as good candidates because they are linear, relatively rigid π-conjugated molecules, and they exhibit relatively high-lying electronic excited-states, even when they contain a large number of repeating units [42]. This latter property will prevent deleterious energy transfer reaction of the photoexcited porphyrin to the bridge. In the dyads D1–D3, the bridges are substituted by electron-withdrawing groups such as esters (D1) or by electron-donating groups such as alkoxy chains (D2, D3). These substituents were used not only to bring about the solubility but also to tune the electronic properties of the bridge.
The significant electronic interactions in the dyads are clearly evidenced by the inspection of their electronic spectra. The spectra of the porphyrin substituted on the meso position by a π-conjugated bridge via an ethynyl linkage differ significantly from those of regular tetraaryl zinc or gold porphyrins (Fig. 5).
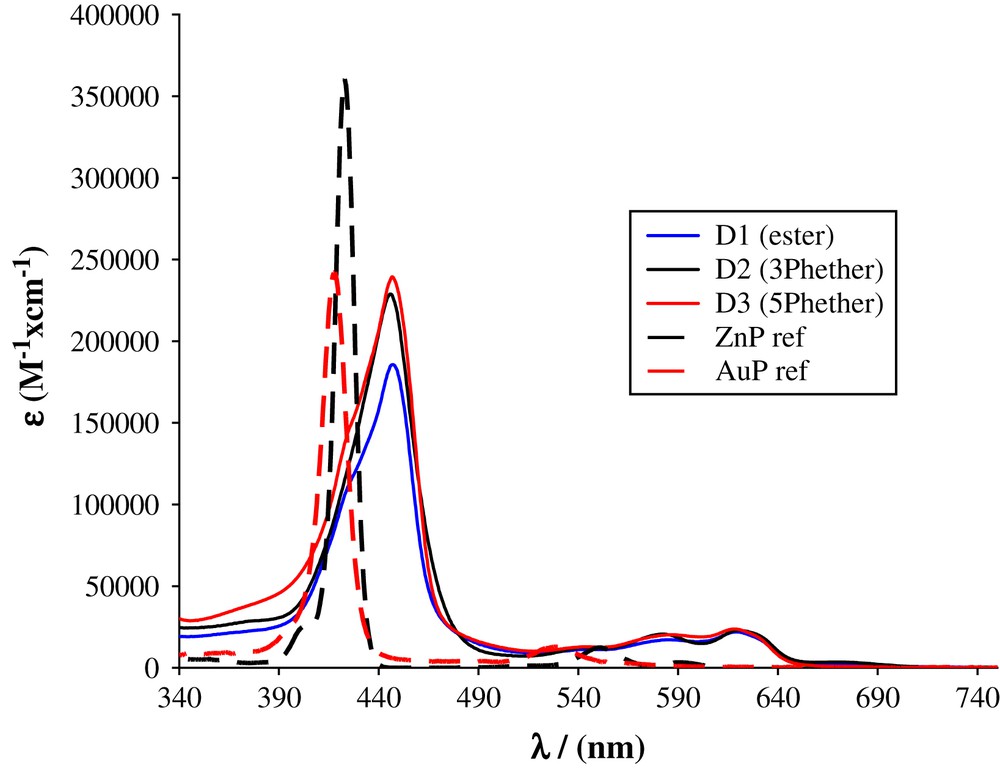
UV–vis absorption of the dyads D1–D3 along with those of tetra(3,5-ditertbutylphenyl) zinc(II) (ZnP reference) and tetra(3,5-ditertbutylphenyl) porphyrin gold(III) (AuP+ reference) recorded in dichloromethane.
As already reported in ethynyl substituted porphyrins, the spectra of the dyads D1–D3 show a bathochromic shift of all the visible absorption transitions, a broadening of the Soret band and an inversion of the intensities of the Q-bands (Fig. 5) [8,39]. These features suggest the existence of noticeable ground-state electronic interactions owing to the effective π-conjugation between the porphyrin ring and the bridge.
The fluorescence study showed that the zinc porphyrin singlet excited-state in these dyads is strongly quenched both in dichloromethane and in toluene (more than 75% in all the dyads). The femtosecond transient absorption spectroscopy permits to elucidate the deactivation processes occurring after the excitation of the zinc or the gold porphyrin and a summary of the photophysical behaviour of these dyads is given in Scheme 3. Summarily, light excitation of the zinc porphyrin of any of the dyads D1–D3 either in dichloromethane or in toluene leads to a fast electron transfer to the nearby gold porphyrin to form the charge separated state +ZnP–B–AuP, whose lifetime depends on the bridge and on the polarity of the solvent (toluene or dichloromethane). Naturally, the longer the bridge is, the longer is the charge separated state lifetime and the longest lifetime was obtained in the apolar solvent (toluene) in agreement with a charge recombination reaction which is pushed up further in the inverted region as the energy of the charge separated state is raised. It is noticeable that an efficient photoinduced electron transfer takes place in dyad D3 with an interporphyrin separation distance of 4.5 nm. This result underscores the pertinence of the molecular design presented here if one desires to perform long-range charge separation. On the other hand, light excitation of the gold porphyrin leads almost exclusively to the energy transfer forming the triplet excited state of the zinc porphyrin in dyads D2–D3. Conversely, in dyad D1, charge separation by hole transfer is the main deactivation pathway when gold porphyrin is photoexcited in dichloromethane.
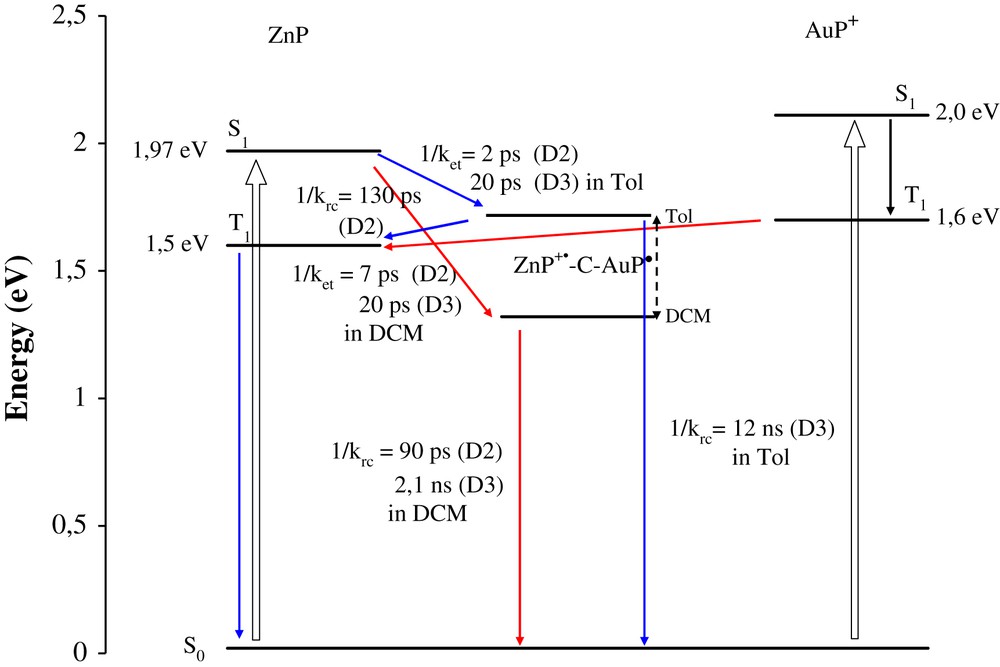
Summary of the photophysical behaviour of the dyads D2–D3. Arrows in blue, photophysical processes occurring in toluene and arrows in red, those occurring in dichloromethane.
3.3 Zinc phthalocyanine connected to a tin porphyrin via an oligophenyleneethynylene spacer
The design of the last dyad D4 and particularly the choice of the replacement of the zinc porphyrin by a zinc phthalocyanine (ZnPc) and the gold porphyrin by a tin porphyrin (SnP) was motivated by three reasons. First, the association of zinc phthalocyanine to a porphyrin permits one to cover a larger spectral window of the solar spectrum thanks to the intense and near infra-red Q-bands of ZnPc. This latter property increases the light harvesting efficiency of the system as photons of different colours (energy) will be collected (Fig. 6). Second, the distinct absorption bands of porphyrin and phthalocyanine enables selective excitation of each pigment, which facilitates the photophysical study. Thirdly, tin porphyrins display a more negative reduction potential than gold porphyrins (ERed(AuP+/AuP) ≈ −0.6 V vs SCE; ERed(Cl2SnP/Cl2SnP−) ≈ −1 V vs SCE), therefore upon charge separation a stronger reducer will be produced. This feature is a real advantage in view of using the charge separated state to perform useful redox transformations (artificial photosynthesis).
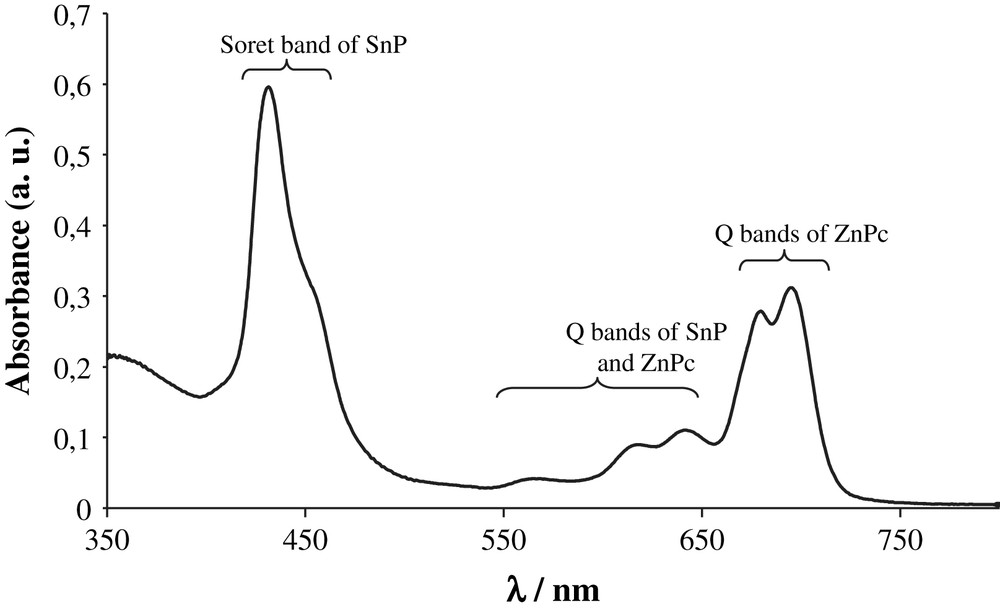
Absorption spectrum of the dyad D4 recorded in benzonitrile.
The synthesis of the dyad D4 is depicted in Scheme 4 and it relies on a similar strategy as that used for dyads D1–D3, namely a stepwise coupling of the chromophores on the spacer 4 via Sonogashira cross-coupling. The presence of a double bond between the bridge and the tin porphyrin, instead of the triple bond introduced in the preceeding dyads D1–D3, is the consequence of the unexpected reduction that occurred during the metallation of the porphyrin by tin dichloride (Scheme 4) [9]. However, in this particular case the ethenyl linkage has not ruined the photophysical properties of the dyad. Indeed, the photophysical study revealed that light excitation of the dyad D4 in any of its chromophore (ZnPc or SnP) is accompanied by a very fast charge separation leading to +ZnPc–B–SnP− with an almost quantitative quantum yield. This charge separated state has a lifetime of 85 ps and it decays by a charge recombination reaction directly to the ground state (Scheme 5).
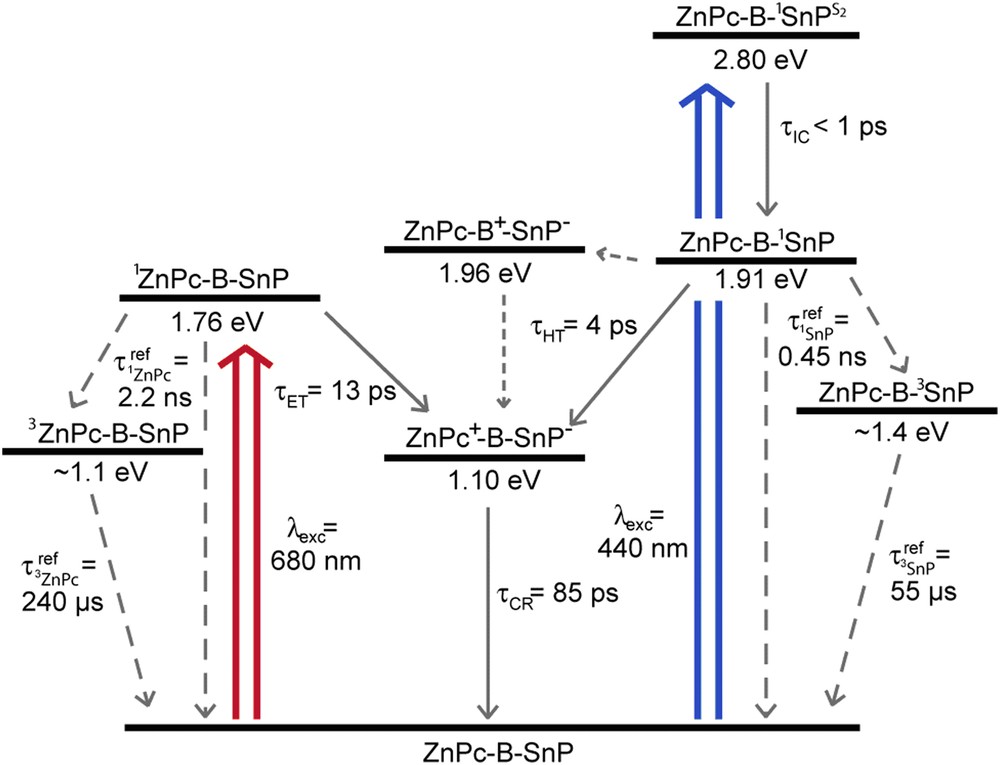
Summary of the relevant states and kinetics recorded in benzonitrile at room temperature in dyad D4. Reprinted with permission from Ref. [9].
The photophysical property of this dyad is particularly interesting because it can transform all the absorbed photons into a charge separated state storing a large amount of the initial energy introduced in the form of light. Besides, the charge separated state recombines into the ground-state in contrast with several phthalocyanine based systems, whose charge separated state usually decays to the phthalocyanine triplet excited state, which is of relatively low energy [43]. This last dyad D4 is another example that illustrates the suitability of OPE bridge to construct porphyrin and phthalocyanine based systems for charge separation over long distances.
4 Conclusion
We developed a synthetic strategy to prepare heterometallic bisporphyrin systems performing fast single step photoinduced electron transfers over very long distances via a bridge-mediated superexchange mechanism. The main difference of these bisporphyrins systems lies in the direct link of the bridge on the meso position of a triaryl porphyrin via an ethynyl linkage (Fig. 4). The dyads reported here exhibit absorption spectra and redox potentials that are significantly altered relative to the regular tetraaryl porphyrin and therefore can be considered as true supermolecules, because their electronic properties differ from the isolated pigments as a consequence of the strong electronic coupling between the porphyrin and the spacer. However, it is important to note that the excited-states of each porphyrin module remain individual separate states.
The photophysical properties of these dyads show that the attachment directly on the meso position of a π-conjugated bridge via ethynyl or ethenyl linker is a well-suited strategy to perform efficient and long-range photoinduced charge separation between two distant porphyrins. The ethynyl or the ethenyl linkage directly attached in the meso position of the porphyrin is certainly the key design feature that promotes the interchromophore electronic communication, because it ensures an effective π-conjugation between the porphyrin or the phthalocyanine with the adjacent bridge. In the superexchange model, the electronic coupling between D and A arises from the mixing of the wave functions of the donor and the acceptor with those of the bridge and depends on their respective energy proximity, but also on the effective overlap of the frontier molecular orbital of each unit (D with B and B with A) [5]. Besides, we showed that oligophenyleneethynylenes represent attractive bridging units since they exhibit wire-like behaviour when they are attached directly to the meso position of the porphyrins. Lastly, the comparison of the dyads D1–D3 with those of Mårtensson and coworkers highlights the determining role of the connectivity on the electronic coupling for a given bridge. As convincingly presented by Albinsson, the beta value is not only a bridge specific property, as it is often inferred in many papers [44]. It is important to realize that β also depends on the couple donor/acceptor units that are linked, on the mode of connectivity used to anchor the bridge to A and D and also on its anchoring position to A and D.
Acknowledgements
The French Research Ministry is gratefully acknowledged for the financial support of this research through the programs “ACI Jeune Chercheur” and the ANR Blanc “PhotoCumElec”. These studies were also supported by the European program COST D35.