1 Introduction
Thiophene-based polycyclic aromatic compounds, linearly fused systems in particular, have been attracting a great deal of interest in view of application to organic materials for electronic devices such as organic field-effect transistors (OFETs) and light emitting diodes (OLEDs) [1]. For instance, anthradithiophene, which is an isostructural analogue of pentacene, displayed improved solubility, robustness toward oxidative degradation, and solid-state ordering in favor of π stacking compared to pentacene [2]. Furthermore, Takimiya and coworkers reported that the extended benzothiophene derivatives showed good characteristics of OFETs [1b,3]. Compared to the linearly fused thiophene-based aromatics, little is known for angularly fused systems with regard to application to organic materials [4], presumably because they would exhibit larger HOMO–LUMO gaps than the linear systems.
For tetracyclic naphthodithiophenes, for example, of the 12 possible isomers shown in Fig. 1, which are classified according to the molecular shapes as S-, U-, J-, and I-types, only three of them, naphtho[2,1-b:6,5-b′]dithiophene (1a), naphtho[2,1-b:7,8-b′]dithiophene (2a), and naphtho[1,2-b:8,7-b′]dithiophene (2b), are known [5]. However, although their synthesis was reported, their physical data are not cited. We report herein the synthesis of some of the naphthodithiophenes using flash vacuum pyrolysis (FVP) of readily available dithienyldienynes [6].
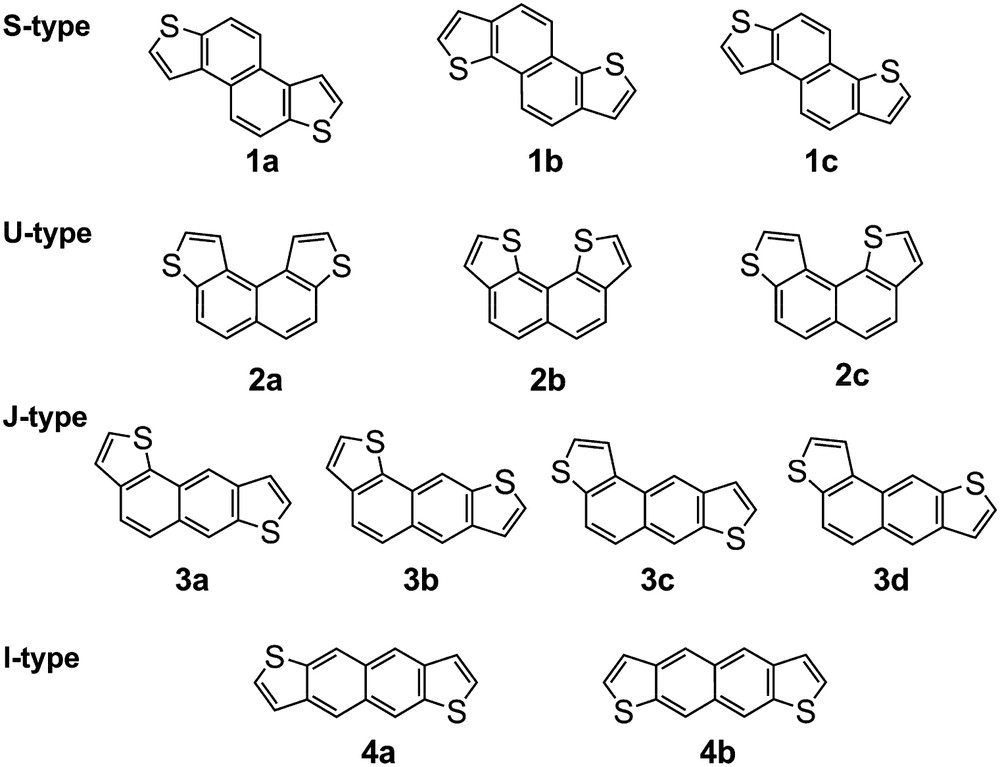
Structures of naphthodithiophene isomers.
As a part of our work on the synthesis of new polycyclic aromatic compounds based on tandem cyclization including alkyne linkages, we reported that flash vacuum pyrolysis (FVP) of diphenylhexadienyne derivative having a bromine atom 5 (Ar = phenyl) gave chrysene as a major product by 6-endo cyclization at high temperatures (Scheme 1, left) [7]. In contrast, we also found that palladium-catalyzed cyclization of 5 (Ar = phenyl) gave the biindenylidene derivatives as a sole product by 5-exo cyclization (Scheme 1, right) [8a]. Similarly, Pd-catalyzed cyclization worked well with the thiophene derivatives 5a and 5b (Fig. 2), producing cyclopentene-fused dithienylethenes [8b]. It is therefore usual to extend our work to FVP of 5a and 5b with an expectation of the formation of naphtho[2,1-b:6,5-b′]dithiophene (1a) and naphtho[1,2-b:5,6-b′]dithiophene (1b) as the major products, respectively. We found, however, the results were more complicated because of not only the two different reaction centers, α and β positions, in the thiophene ring but also the less selective 6-endo cyclization mode, leading to the formation of mixtures of isomers. From the mixtures, new compounds 1c and 2c were isolated in pure forms and fractions containing 1a and 1b as each major component were also obtained (Fig. 1).
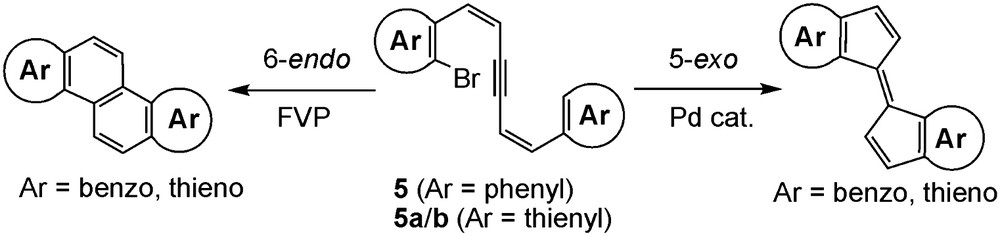
5-Exo and 6-endo tandem cyclizations of 5.
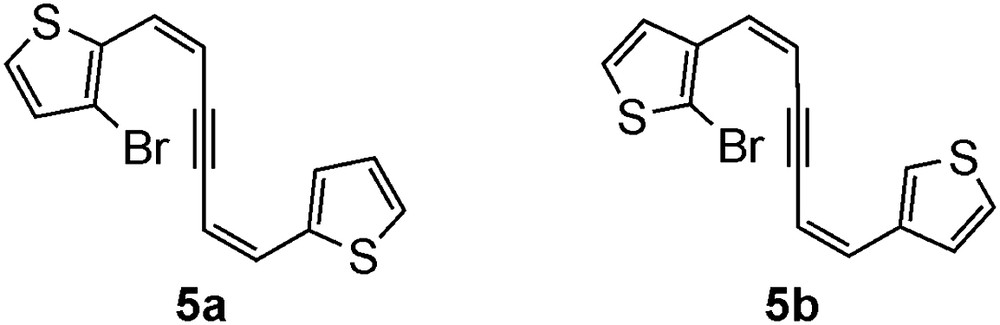
Precursors 5a and 5b for FVP.
2 Results and discussion
The substrates of FVP 5a and 5b were prepared by the previous method developed by our group [8b]. First, we carried out FVP of 5a at 850 °C (Scheme 2). In contrast to the FVP of 5 (Ar = phenyl) [7], the reaction of 5a afforded a complex mixture of products as indicated by the 1H NMR spectrum of the crude product (Fig. S2). The crude product was subjected to GPC separation, giving two fractions A and B. From the first fraction A, one of major products was isolated, of which structure assignment is outlined below. The 1H NMR spectrum (Fig. S3) exhibits eight doublets and the 13C NMR spectrum shows 14 signals. These results indicate the product has an unsymmetrical structure such as 1c or 2c (Fig. 1). The most characteristic feature of the 1H NMR spectrum of this product is the remarkable low field shift of two naphthalene protons which appear at 8.27 (d, J = 8.7 Hz, 1H) and 8.07 (d, J = 8.8 Hz, 1H) ppm and one thiophene proton which appears at 8.03 (d, J = 5.4 Hz, 1H) ppm owing to the anisotropic deshielding effect, indicating that these protons are located in the bay region. Among the possible products shown in Fig. 1, only 1c fits the observed NMR spectra. Moreover, the deshielded chemical shifts agree well with the calculated values (8.28, 8.12, and 8.03 ppm, respectively) obtained by the GIAO method at the B3LYP/6-31g(d,p)//B3LYP/6-31g(d,p) level (Scheme 2 and Fig. S4). The second GPC fraction B contained at least five products as indicated by the 1H NMR spectrum (Fig. S5) and GC analysis. Moreover, GC mass spectrum of the major peaks of this fraction exhibited molecular ions corresponding to the naphthodithiophenes (m/z 240), indicating the absence of fragmentation to small fragments. The fraction B was separated by column chromatography on aluminum oxide, giving a fraction (B1) consisting of a sole product and a fraction (B2) containing another product as a major component. The 1H NMR spectrum of fraction B1 (Fig. S6) also shows eight doublets, indicating that this compound possesses an unsymmetric structure. In this case, however, only one low field-shifted signal for a thiophene proton was observed at 8.49 (dd, J = 0.7, 5.4 Hz, 1H), indicating that this compound contains one thiophene proton in the bay region. Accordingly, we assign this compound as compound 2c. This assignment is also supported by the calculated NMR shift (8.49 ppm) for the thiophene proton (Scheme 2 and Fig. S4). Analysis of the 1H NMR spectrum of the other fraction B2 (Fig. S7) allowed us the assignment of its major component to 1a; it shows four doublet signals, consistent with a symmetrical structure, with two of which (for naphthalene protons at 8.29 (d, J = 8.5 Hz, 2H) ppm and for thiophene protons at 8.03 (d, J = 5.5 Hz, 2H) ppm) being observed at low field due to the anisotropic deshielding effect. The calculated NMR chemical shifts of the above protons of 1a (8.29 and 8.05 ppm, respectively) agree well with the observed ones (Scheme 2 and Fig. S4).
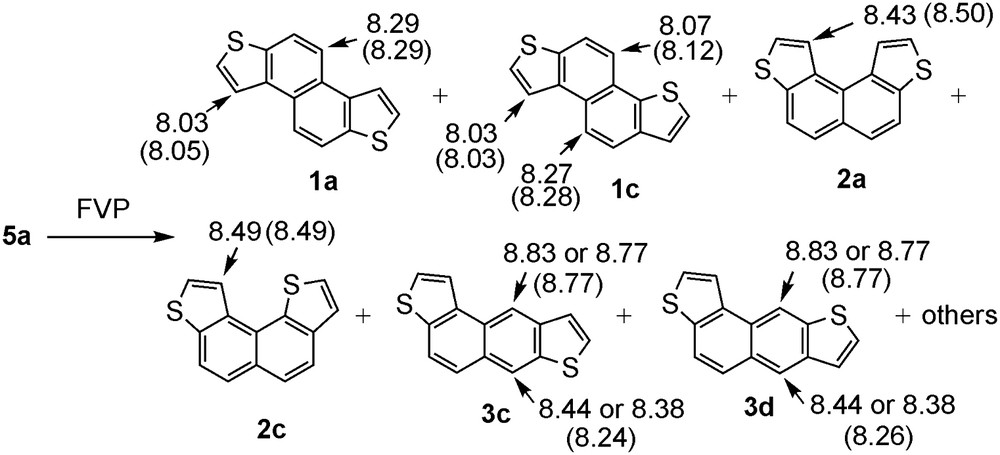
The structures of the products of FVP of 5a and the observed 1H NMR chemical shifts. Values in parentheses are the calculated chemical shifts by GIAO-B3LYP/6-31g(d,p)//B3LYP/6-31g(d,p) method.
Although we could not isolate other products or obtain fractions enriched with the others, the structures of the other minor products were assumed on the basis of the 1H NMR spectrum of the fraction B before subjection to column chromatography (Fig. S5). It exhibits the characteristic two pair of singlet signals at relative low field (8.38 and 8.77 ppm, 8.44 and 8.83 ppm). One of the signal in the each pair is subject to strong anisotropic deshielding effect. In addition to the spectroscopic data, the fact that in the case of 5 (Ar = phenyl), J-shaped product, benz[a]anthracene, was formed as a minor product led us to assume that the structures of the minor products to be 3c and 3d (Scheme 2 and Fig. S4). Also, the formation of compound 2a was suggested from the 1H NMR spectrum of the crude product, which exhibited the thiophene proton at 8.43 ppm located at the bay region (Scheme 2 and Fig. S4).
The product distribution was determined by the integral ratio of characteristic signals in the 1H NMR spectrum of the crude product of FVP of 5a (Fig. S2). The results at higher temperature (1050 °C) are also listed in Table 1. As shown, the distribution of products was not affected by changing the temperature from 850 °C to 1050 °C, indicating the absence of equilibrium between the products.
Product distribution by FVP of 5a and 5ba
| Temperature [°C] | Balanceb [%] | Product distribution (%)c | ||||||
| 1a | 1c | 2a | 2c | 3c | 3d | |||
| 5a | 850 | 55 | 27 | 27 | 14 | 22 | 5 | 5 |
| 1050 | 45 | 30 | 25 | 12 | 21 | 6 | 6 | |
| 1b | 1c | 2b | 2c | 3a | 3b | |||
| 5b | 850 | 54 | 33 | 22 | 15 | 15 | 7.5 | 7.5 |
| 1050 | 48 | 33 | 23 | 17 | 13 | 7 | 7 |
a The reactions were carried out at ca. 1 mmHg under a slow stream of Argon which was introduced through a capillary into the pyrolysis tube (see Supplementary material for the detail).
b Percent ratio of the weight of products relative to the starting material which passed through the hot zone of the pyrolysis tube assuming that all products have the same molecular formula C14H8S2.
c Based on the integral ratio of characteristic signals in the 1H NMR spectrum of crude reaction product after FVP.
Next, we carried out FVP of 5b. In this case, several products were formed too (Scheme 3 and Fig. S8). In the same manner as the purification of the products of 5a, the mixtures were separated by GPC and the structures were assigned on the basis of 1H NMR spectra. First, by separation of the crude products by GPC, three fractions (fractions C–E) were obtained. One of them, fraction C, was identified as compound 1c, because its 1H NMR data (Fig. S9) were identical with the one obtained from pyrolysis of 5a. The 1H NMR spectrum of the second GPC fraction D (Fig. S10) shows four doublets indicating 1b or 2b as the candidate among the possible structures (Fig. 1). In addition, since the signal of a naphthalene proton is observed at relatively low field of 8.06 (d, J = 8.4 Hz, 1H) ppm, we assume this compound to be 1b. The third GPC fraction E is assumed to contain two major compounds on the basis of the 1H NMR spectrum (Fig. S11). One of them was identified to be 2c by comparison of the 1H NMR data with that observed in the pyrolysate of 5a. The other component shows four doublet signals which are not subject to the anisotropic deshielding, indicating that this compound has a symmetrical structure and does not possess any proton in the bay region. These observations suggest that this compound is 2b. This is supported by the calculated NMR chemical shifts (Fig. S12). In addition, the 1H NMR spectra of all fractions C–E exhibit the characteristic pair of singlet signals (8.38 and 8.66 ppm, 8.44 and 8.61 ppm) of low intensities at relatively low field, indicating the presence of J-type compounds such as 3a and 3b (Figs. S9–S11).
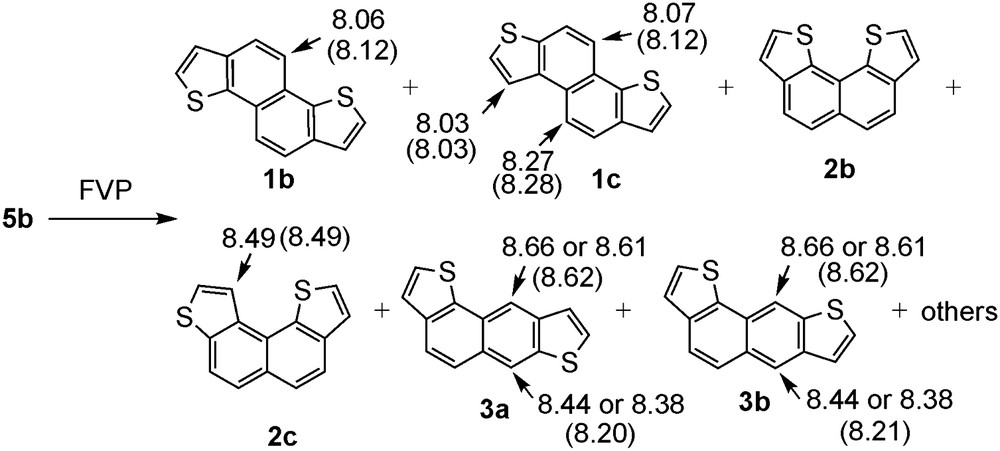
The structures of the products of FVP of 5b and the observed 1H NMR chemical shifts. Values in parentheses are the calculated chemical shifts by GIAO-B3LYP/6-31g(d,p)//B3LYP/6-31g(d,p) method.
The results of FVP of 5b at both 850 °C and 1050 °C are listed in Table 1. Similarly to the case of 5a, the distribution of the products produced from 5b was not affected by temperature, indicating that the products are formed under kinetic control.
The relative energy of the equilibrium geometry of naphthodithiophenes 1–4 calculated by using B3LYP method with 6-31G(d,p) basis set is shown in Table 2. Within the individual type isomers 1, 3, and 4, the relative energies are not much different. However, the case of U-type isomers 2a–c, the relative enthalpies are dependent on the position of sulfur atoms due to steric and electronic reasons. Moreover, the relative energies are also affected by the positions of fusion. The S-type products 1a–c, which were obtained as major products from 5a and 5b, were shown to possess relatively high energies. These results also implied that the formation of the naphthodithiophene products was conducted under kinetic control.
Comparison of the relative energies of the equilibrium geometries of naphthodithiophenes 1–4 calculated by using the B3LYP method with the 6-31G(d,p) basis set
| Type | Compound | E(RB + HF − LYP) (hartree) | E (kcal/mol)a | ΔE (kcal/mol)b |
| S | 1a | −1334.71720644 | −837547.7269 | −0.405 |
| 1b | −1334.71785138 | −837548.1316 | 0 | |
| 1c | −1334.71748486 | −837547.9016 | −0.230 | |
| U | 2a | −1334.71276953 | −837544.9427 | −3.19 |
| 2b | −1334.71654431 | −837547.3114 | −0.820 | |
| 2c | −1334.71583929 | −837546.869 | −1.26 | |
| J | 3a | −1334.71475090 | −837546.186 | −1.95 |
| 3b | −1334.71441062 | −837545.9725 | −2.16 | |
| 3c | −1334.71434996 | −837545.9344 | −2.20 | |
| 3d | −1334.71428595 | −837545.8942 | −2.24 | |
| I | 4a | −1334.70941672 | −837542.8387 | −5.29 |
| 4b | −1334.70932978 | −837542.7842 | −5.35 |
a 1 hartree = 627.5095 kcal/mol.
b Relative energy based on compound 1b.
The trend of product distributions is consistent with that of FVP of 5 (Ar = phenyl), though two products such as 1c and 2c were formed as S-type products by FVP of 5a and 5b because of the unsymmetrical structure of thiophene ring. For example, in the case of the FVP of 5 (Ar = phenyl) at 800 °C, the products were obtained in the following ratio, S-type (73%), U-type (21%), and J-type (6%) [7]. The selectivity to S-type products from 5a and 5b was slightly low as compared to that of 5 (Ar = phenyl), the reason of which is described later. In both cases with 5a and 5b, the material balance at 1050 °C was slightly lower than that at 850 °C presumably because of the greater possibility of degradation of the intermediate species of cyclization.
3 Plausible mechanism of the formation of the naphthodithiophene derivatives
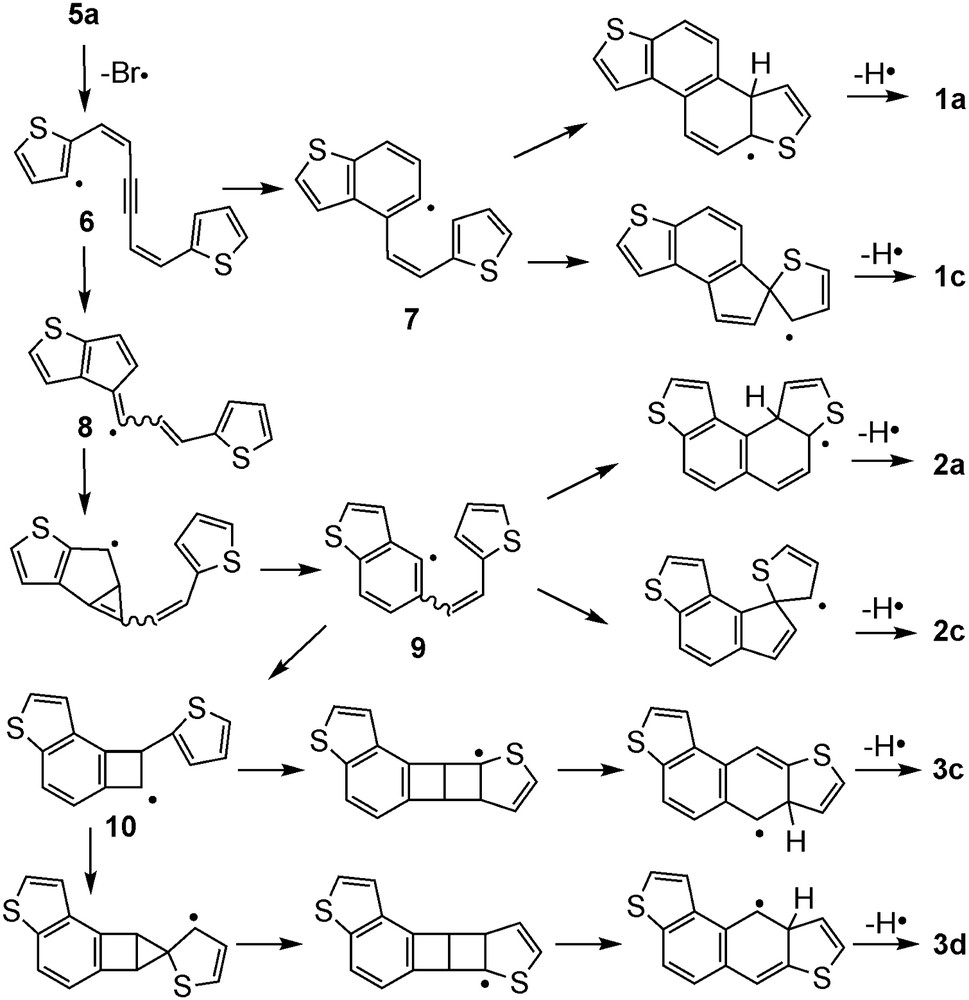
Plausible mechanisms of the formation of the naphthodithiophene derivatives 1a, 1c, 2a, 2c, 3c, and 3d are shown in Scheme 4. Obviously, at the high temperature,4 the homolytic cleavage of the C–Br bond should be the initial event [9], leading to the formation of radical species 6. Then, the generated radical 6 would form the benzothiophene radical species 7 by 6-endo cyclization. Subsequent 6-endo cyclization at β-position of the thiophene ring followed by elimination of hydrogen would afford compound 1a. On the contrary, 6-endo cyclization at α-position of the thiophene ring followed by the ring expansion would form compound 2c. On the other hand, 5-exo cyclization of the initially formed radical 6 to form 8 and subsequent three-membered cyclization and ring expansion would give another benzothiophene radical species 9. The formation of compounds 2a and 2c can be explained in terms of the same cyclization assumed for 1a and 1c from 7. In order to explain the formation of 3c and 3d, however, one should assume the formation of cyclobutyl radical intermediate 10 from 9. Thus compounds 3c and 3d might be formed from 10 through intermediates having highly strained bicyclo[2.1.0]pentane and bicyclo[2.2.0]hexane structures. Though they look peculiar in view of the strain, a similar intermediate has been proposed in the literature for the FVP of 3-(2-thienonyl)cinnoline [6d]. The mechanism is based on conjecture: it would be confirmed by DFT calculation of the intermediates, which is the topic of our future research. The relatively low product distribution of S-type products 1a and 1c from 5a compared to that of chrysene from 5 (Ar = phenyl) can be ascribed to the slower rate of the initial 6-endo cyclization relative to 5-exo mode owing to the smaller bond angle of the thiophene ring in intermediate 6. The formation of 1b, 1c, 2b, 2c, 3a, and 3b produced by FVP of 5b could be explained in the same manner (see Supplementary material).
4 Conclusion
In conclusion, FVP of 5a and 5b gave a mixture of naphthodithiophene isomers 1a–c, 2a–c, and 3a–d, from which 1b, 1c, and 2c were isolated in almost pure state. Their structures were elucidated on the basis of the observed and calculated 1H NMR spectra. The formation of the other products is assumed on the basis of 1H NMR spectra of the crude and partly purified products.
5 Experimental details
5.1 General
For FVP apparatus a ceramic electric furnace (Asahi Rika-Seisakusyo, ARF-30K) and a temperature control unit (Asahi Rika-Seisakusyo, AMF-N) were used. 1H NMR spectra were recorded on a Varian Mercury 300 spectrometer in CDCl3 and with Me4Si or residual solvent as an internal standard at 30 °C. IR spectra were recorded as KBr disks with a JASCO FTIR-410 spectrometer. Mass spectral analyses were performed on a JEOL JMS-700 spectrometer for EI ionization. GC/MS analysis was performed on a JEOL JMS-700 spectrometer coupled to a Agilent 6890 series (J&W Scientific HP-5 30 m × 0.320 mm). Column chromatography was performed with MERCK Aluminiumoxid 60 F254. Preparative GPC separation was undertaken with a JAI LC-908 chromatograph using 600-mm × 20-mm JAIGEL-1H and 2H GPC columns with CHCl3 as an eluent. DFT calculations were performed with Gaussian 03 program package [10]. The geometries of the compounds were optimized by using the B3LYP method with the 6-31G(d,p) basis set. The nature of the stationary points was assessed by means of vibration frequency analysis. The chemical shifts were calculated by the GIAO-B3LYP/6-31G(d,p) method.
5.2 Experimental procedure of FVP
The schematic apparatus of FVP is shown in Fig. S1. The hot zone consisting of a quartz tube was heated by an electric furnace to temperatures of 850 °C or 1050 °C. Under a slow stream of argon at reduced pressure (about 1 mmHg), a sample 5a or 5b (30 mg) was sublimed by heating with a heat-gun. The products passed through the hot zone were collected in a cold trap (liq. N2). After most of the sample was passed through the hot zone, heating was stopped. After cooling to room temperature, the products were collected by washing the quartz tube and the trap with CHCl3. This procedure was repeated three times to obtain enough amount of products for analysis. The combined crude product was separated by GPC and column chromatography. The relative ratio of the products was estimated on the basis of the 1H NMR integration for the characteristic signals.
5.2.1 FVP of 5a
GPC separation gave fractions A (compound 1c, 6 mg) and B (18 mg). Fraction B was subjected to column chromatography to give fraction B1 (compound 2c, 4 mg) and B2 (mainly compound 1a, 7 mg). 1H NMR spectrum of crude products before separation by GPC was shown in Fig. S2. Partial 1H NMR spectrum of fraction A (compound 1c) is shown in Fig. S3. 1H NMR spectrum of fraction B before subjection to column chromatography is shown in Fig. S5. Partial 1H NMR spectra of fraction B1 (compound 2c) and fraction B2 (mainly compound 1a) are shown in Figs. S6 and S7, respectively. The calculated 1H NMR chemical shifts of 1a, 1c, 2a, 2c, 3c, and 3d are described in Fig. S4.
Compound 1c: m.p. 139.0–140.0 °C, 1H NMR (400 MHz, CDCl3, 30 °C) δ 8.27 (d, J = 8.7 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 5.4 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.64 (d, J = 5.4 Hz, 1H), 7.53 (d, J = 5.3 Hz, 1H), 7.50 (d, J = 5.3 Hz, 1H); 13C NMR (75 MHz, CDCl3, 30 °C) δ 138.2, 136.9, 136.8, 136.7, 126.4, 126.3, 126.2, 125.0, 124.8, 122.3, 122.2, 121.2, 120.9, 120.8; IR (KBr) 3098, 2923, 1651, 1556, 1514, 1465, 1415, 1365, 1340, 1293, 1187, 1161, 1101, 1086, 907, 882, 808, 732, 698, 606 cm−1; MS (EI) m/z 240 (M+).
Compound 2c: m.p. 101.5–102.5 °C, 1H NMR (400 MHz, CDCl3, 30 °C) δ 8.49 (dd, J = 5.7, 0.9 Hz, 1H), 8.01 (dd, J = 8.7, 0.7 Hz, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.92 (d, J = 8.7 Hz, 1H), 7.91 (d, J = 8.7 Hz, 1H), 7.77 (d, J = 5.7, 0.9 Hz, 1H), 7.67 (d, J = 5.4 Hz, 1H), 7.60 (d, J = 5.4 Hz, 1H); MS (EI) m/z 240 (M+).
Compound 1a: 1H NMR (400 MHz, CDCl3, 30 °C) δ 8.29 (d, J = 9.0 Hz, 2H), 8.04 (d, J = 9.0 Hz, 2H), 8.03 (d, J = 5.4 Hz, 2H), 7.64 (d, J = 5.4 Hz, 1H); MS (EI) m/z 240 (M+).
5.2.2 FVP of 5b
GPC separation gave fractions C (compound 1c, 7 mg), D (compound 1a, 7 mg), and E (13 mg). Partial 1H NMR spectrum of fraction C (mainly 1c) is shown in Fig. S6. 1H NMR spectrum of crude products before separation by GPC is shown in Fig. S8. Partial 1H NMR spectrum of C (mainly compound 1c), fraction D (mainly compound 1b), and fraction E (mainly compounds 2b and 2c) are shown in Figs. S9–S11, respectively. The calculated 1H NMR chemical shifts of 1b, 2b, 3a, and 3b are described in Fig. S12.
Compound 1b: 1H NMR (300 MHz, CDCl3, 30 °C) δ 8.06 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 5.1 Hz, 1H), 7.49 (d, J = 5.1 Hz, 1H); MS (EI) m/z 240 (M+).
Compound 2b: 1H NMR (300 MHz, CDCl3, 30 °C) δ 8.02 (d, J = 8.1 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 5.4 Hz, 1H), 7.65 (d, J = 5.4 Hz, 1H).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas “Advanced Molecular Transformation of Carbon Resources” from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Appendix Supplementary material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.crci.2008.09.020.
4 At lower pyrolysis temperature (650 °C), the isomers of the substrate 5a such as (E,E), (E,Z), and (Z,E) isomers were also obtained together with recovered 5a and the pyrolysis products, which were identified in the 1H NMR spectrum of the crude product.