

1 Introduction
The creation of reactive metal-carbene intermediates has led to useful synthetic methods, with tremendous recent developments in catalysis. Well-defined metal-carbenes first constitute the basis of alkene and enyne metathesis that has brought revolutions in synthetic methodology to create CC and CC bonds [1]. The reaction of alkene metathesis molybdenum- and ruthenium-alkylidene catalysts with alkynes constitutes a way to initiate alkenylcarbene-metal intermediate formation, further leading to the building of complex molecular architectures [2].
In situ generated metal-carbenes, especially from diazoalkanes with copper and rhodium catalysts, play a key role on interaction with alkenes and alkynes [3] in the synthesis of cyclopropanes [4] and cyclopropenes [5] and their derivatives. More recently, in situ generated metal-carbene intermediates have been shown to result from the activation of alkynes and enynes with electrophilic metal catalysts, derivatives of Pd, Ru, Pt, Au metals [6]. They lead to skeleton rearrangement of unsaturated substrates and the synthesis of functional cyclopropanes and polycyclic molecules [6,7].
In parallel, attempts have been made to generate RuCl(CHR)Cp* moieties, on reaction of RuClCp* containing complexes with diazoalkanes, to study their action on the transformation of alkynes and enynes [8]. Such 16 electron species are isoelectronic with alkene- and enyne- metathesis Grubbs type catalysts RuCl2(CHR)(PCy3)L1 (L1PCy3, NHC), but their geometry and ligand lability are so different that the behaviour of RuXCp* systems is expected to lead to innovation with respect to metathesis reactions.
It is the objective of this review to show that the catalytic system arising from RuCl(COD)Cp*/N2CHR interaction, as the cyclo-1,5-octadiene (COD) ligand is very labile, plays a unique role in the catalytic modifications of alkyne and enyne derivatives, and leads to catalytic reactions that were not observed before with other metal/diazoalkane or metal-carbene systems. It will be also shown that the nature of the alkyne is crucial to create a new reaction type and that a variety of molecular architectures as shown in Scheme 1 can be obtained.
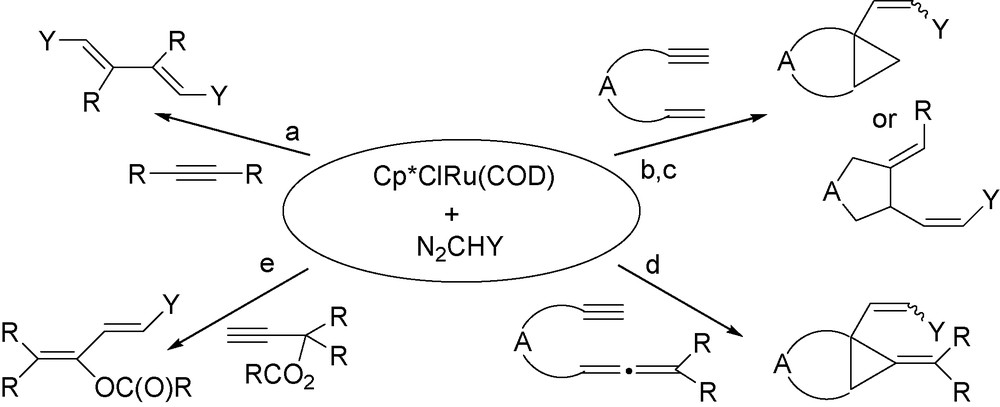
.
It will be shown that the catalytic system RuCl(COD)Cp*/N2CHY allows:
- • the selective double addition of carbene to the triple bond;
- • the bicyclisation of enynes into bicyclo[x.1.0]alkanes;
- • the transformation of enynes into alkylidene, akenylidene-cyclopentane derivatives;
- • the access to alkylidene cyclopropane derivatives;
- • the transformation of propargylic carboxylates into dienes, featuring a cross-coupling CC bond formation.
These different aspects will be successively presented and attempts to rationalize these catalytic reactions will be offered by discussing possible mechanisms and catalytic cycles.
2 Double catalytic addition of diazo compounds to alkynes : an easy synthesis of functional dienes
The reaction of disubstituted alkynes 1 with two equivalents of trimethylsilyldiazomethane (2 M in hexane) in the presence of 5 mol% of RuCl(COD)Cp* [9], catalyst A, in dioxane at 60 °C for 5–6 h, affords the 1,3-dienes, the (E,E)-1,4-bis(trimethylsilyl)butadienes 2a–d (Eq. (1)).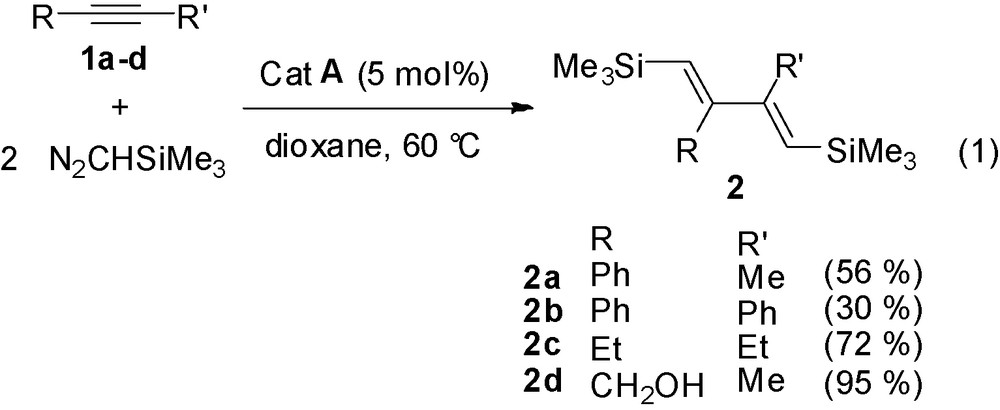
The reaction is slowed down when arylacetylenes are used (2b, 30%), but it tolerates propargylic alcohol functionality (2d, 95%). Only one (E,E)-stereoisomer was observed in the formation of these tetrasubstituted dienes, as shown by NMR and NOE experiments.
The same reaction performed with terminal acetylenes such as 3e–f, also leads to the complete transformation into dienes 4e–f in good yields (Eq. (2)). However, two diastereoisomers are obtained as the disubstituted double bond is formed with a E/Z stereoselectivity: 70/30 (4e), 89/11 (4f).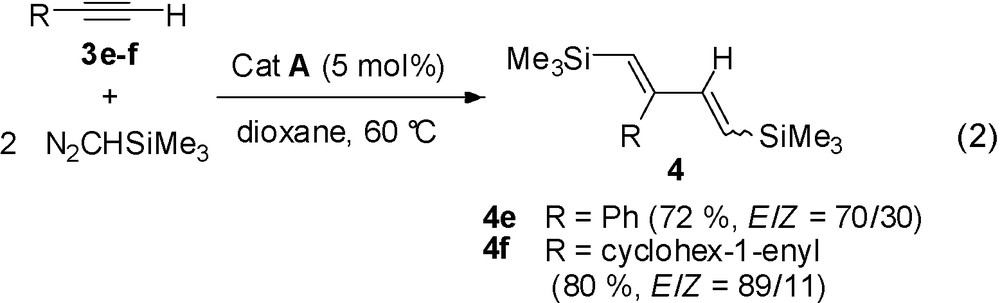
It is noteworthy that only RuCl(COD)Cp* type catalyst performs this catalytic reaction as the ruthenium(IV) complex RuBr2(η3-allyl)Cp* or ruthenium(II) complexes RuCl2(L)(arene) are inactive.
The addition of four equivalents of N2CHSiMe3 has been attempted on the conjugated diyne derivative Me3SiCC—CCSiMe3 5. This diyne 5 behaves as a monodisubstituted alkyne as the double addition takes place only to one triple bond with high stereoselectivity and the unsaturated carbon-rich dienylalkyne 6 was obtained in 48% yield (Eq. (3)). No double addition of the carbene to carbon atoms C1 and C4 was observed.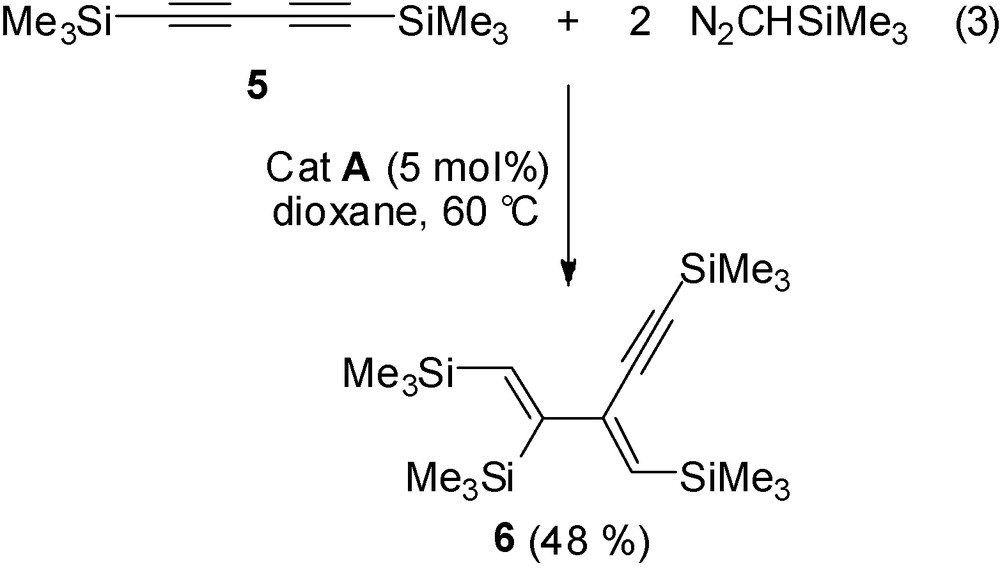
Ethyldiazoacetate is much less reactive in this reaction than trimethylsilyldiazomethane and requires more drastic conditions (100 °C, 20 h) to transform phenylacetylene 3e into 64% yield of 7, isolated as a mixture of stereoisomers (Eq. (4)).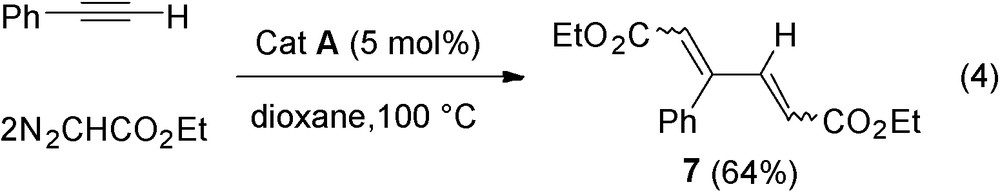
A possible mechanism for this reaction is illustrated in Scheme 2. Although an intermediate of the reaction of trimethylsilyldiazomethane with RuCl(COD)Cp* even at low temperature could not be observed, it is likely that the ruthenium-carbene intermediate is first formed Cp*RuCl(CHSiMe3) B. N2CHSiMe3 is known to interact with RuCl2(PCy3)(arene) complex to generate an NMR observable coordinatively unsaturated RuCHSiMe3 species [10]. In addition, phenylacetylene alone reacts very easily with RuCl(COD)Cp* to give the cis-biscarbene-ruthenium complex 8 via the head-to-head oxidative coupling of alkyne [11] (Eq. 5). Such a biscarbene-ruthenium complex was discovered in 1986 on reaction of RuBr(COD)Cp with phenylacetylene [11a]. It is noteworthy that the product 8 is not formed in the reaction of phenylacetylene 3e with N2CHSiMe3 and catalyst A (Eq. (2)), indicating that interaction of complex A with N2CHSiMe3 is a fast process with respect to alkyne oxidative coupling leading to 8.
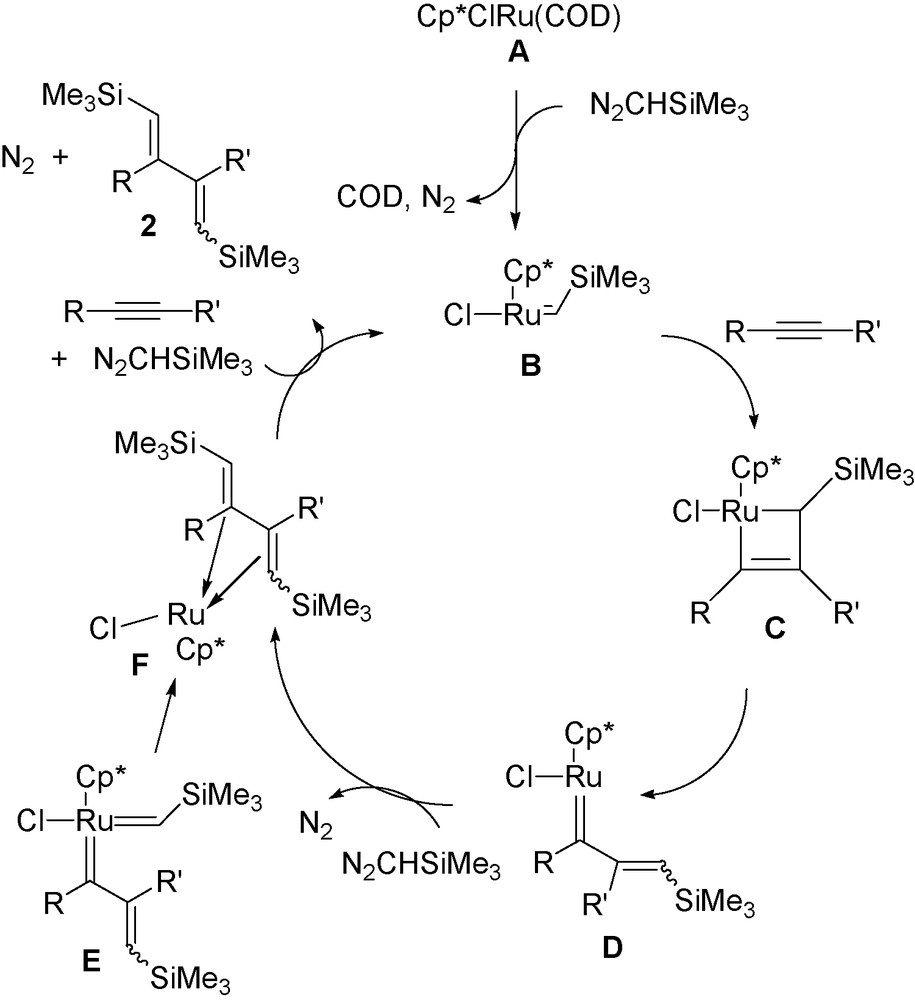
.
The coordination of the alkyne to the ruthenium carbene species B is expected to lead to the 2 + 2 cycloaddition intermediate C and then to the vinylcarbene D as for an alkyne metathesis process [2], as proposed for the Grubbs type catalysts for enyne metathesis [12] and observed in some stoichiometric reactions of RuCHR bonds with alkynes [13].
The vinyl carbene ligand in D can interact directly with N2CHSiMe3 to give the diene 2 [14] or its complex F, or alternatively to give a cis biscarbene ruthenium complex E analogous to the biscarbene 8 (Eq. 5). It is noteworthy that stoichiometric double addition of diazoalkane carbenes to alkyne-cobalt complex to give a diene-cobalt complex was observed by O’Connor et al. [15].
This reaction has some analogy with the addition of ethylene to alkynes catalysed by Grubbs type catalysts to form dienes via intermolecular ene-yne metathesis process as shown by Mori et al. [16] (Scheme 3).
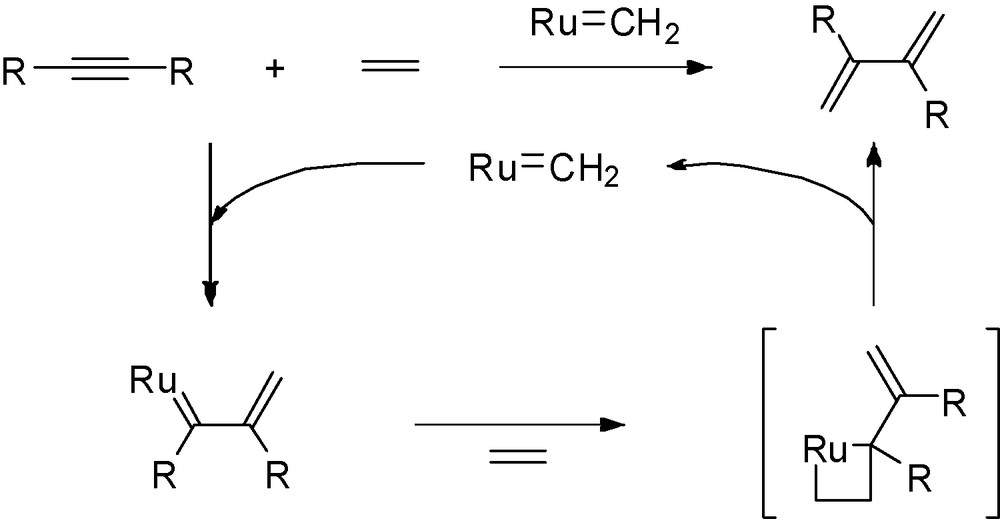
.
3 Catalytic addition of diazoalkane carbene to enynes: a novel way to generate alkenylbicyclo[x.1.0]alkanes
3.1 Evidence and scope of the reaction
Non conjugated enynes have become key starting substrates for the creation of very complex rearranged skeletons [17], including cyclic and bicyclic molecules, promoted by ruthenium (II) [18], gallium (III) [19], platinum (II) [20] and gold (I,III) [21] catalysts.
In a more classical way, the alkene metathesis of enynes performed with Grubbs type catalysts [22,23] has been shown very useful for the access to conjugated dienes, the alkenyl cycloalkenes (Eq. (6)).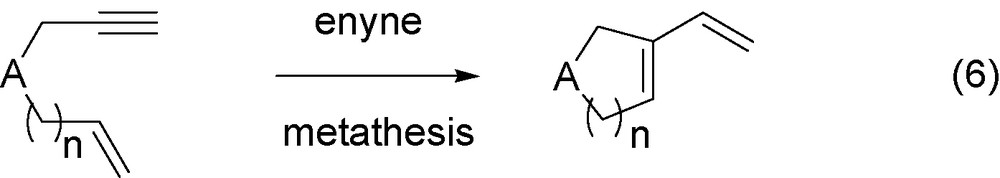
When the enyne triple bond is terminal, the proposed mechanism is shown in Scheme 4.

.
The RuCHR moiety reacts first with the CC bond to give the metathesis intermediates H and I, successively. Then the alkenylcarbene moiety in I traps the distant CHCHR bond to generate the alkenylcyclometallacyclobutane intermediate J, the latter via a classical metathesis process, generates the alkenylcycloalkene and a RuCHR intermediate allowing to initiate another catalytic cycle. When the triple bond is internal such as in bis(alkenyl)alkyne, then the Grubbs catalyst interacts first with a terminal double bond [23c].
The question was then asked, as the 16 electron Cp*(X)RuCHR moiety is isoelectronic of the Grubbs catalyst but has no labile ligand: does Cp*(X)RuCHR, arising from the reaction of RuCl(COD)Cp* with diazoalkane, transforms the non conjugated enyne as the Grubbs catalyst (Scheme 4)? or does it differently proceeds, such as for the double carbene addition to the CC bond or for cyclopropanation of double CC bonds?
The reaction of enynes with diazoalkanes in the presence of RuCl(COD)Cp* catalyst actually leads to the general formation of alkenylbicyclo[x.1.0] alkanes (Eq. (7)).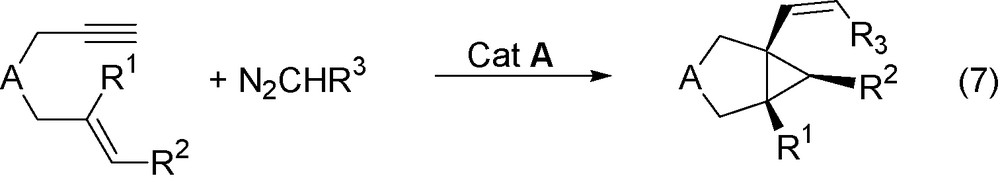
The initial reaction was performed with the enyne 9a and N2CHSiMe3 (2 M in hexane) and 5 mol% of RuCl(COD)Cp* A to afford after 1 h at 60 °C, 95% of 10a with a Z/E ratio = 4/1 (conditions A) (Eq. (8)). The reaction strongly depends on the reaction conditions as with 1.1 equiv. of N2CHSiMe3 (2 M in Et2O), 9a was completely transformed with 5 mol% of A in 5 min to produce 86% of 10a (conditions B).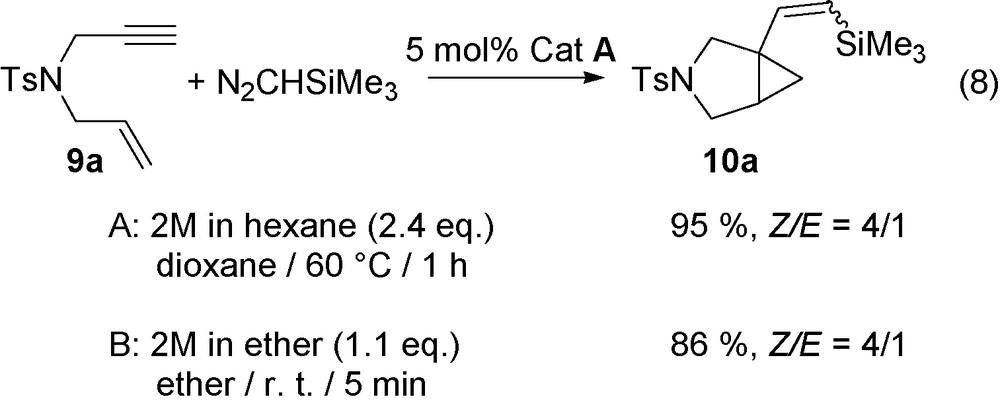
Under B conditions, a variety of enynes have been transformed into bicyclic derivatives 10 in good yields and sometimes with very high Z selectivity (Table 1). Only the Z-isomer was obtained with enynes 9 containing a substituent on the CC bond terminal carbon atom (Table 1, entries 4–6). The X-ray structure of the major diastereoisomer 10d (85/15) established the cis positions of the alkenyl and methyl groups.
Catalytic transformation of enynes 9 with N2CHSiMe3 in bicyclic compounds 10.
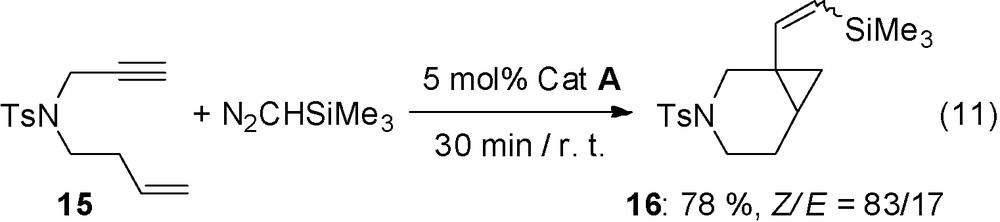
The above reaction applied to enynes arising from propargylic ethers are more difficult to performed (Eq. (9)). The reaction applied to enynes 13 with only a carbon chain give the corresponding bicyclic compounds 14 with good Z selectivity for the alkenyl chain (Eq. (10)). The bicyclisation reaction can also be efficiently applied to 1,7-enynes as shown by the transformation of 15 (Eq. (11)).
The reaction mechanism will be discussed later in Scheme 5.

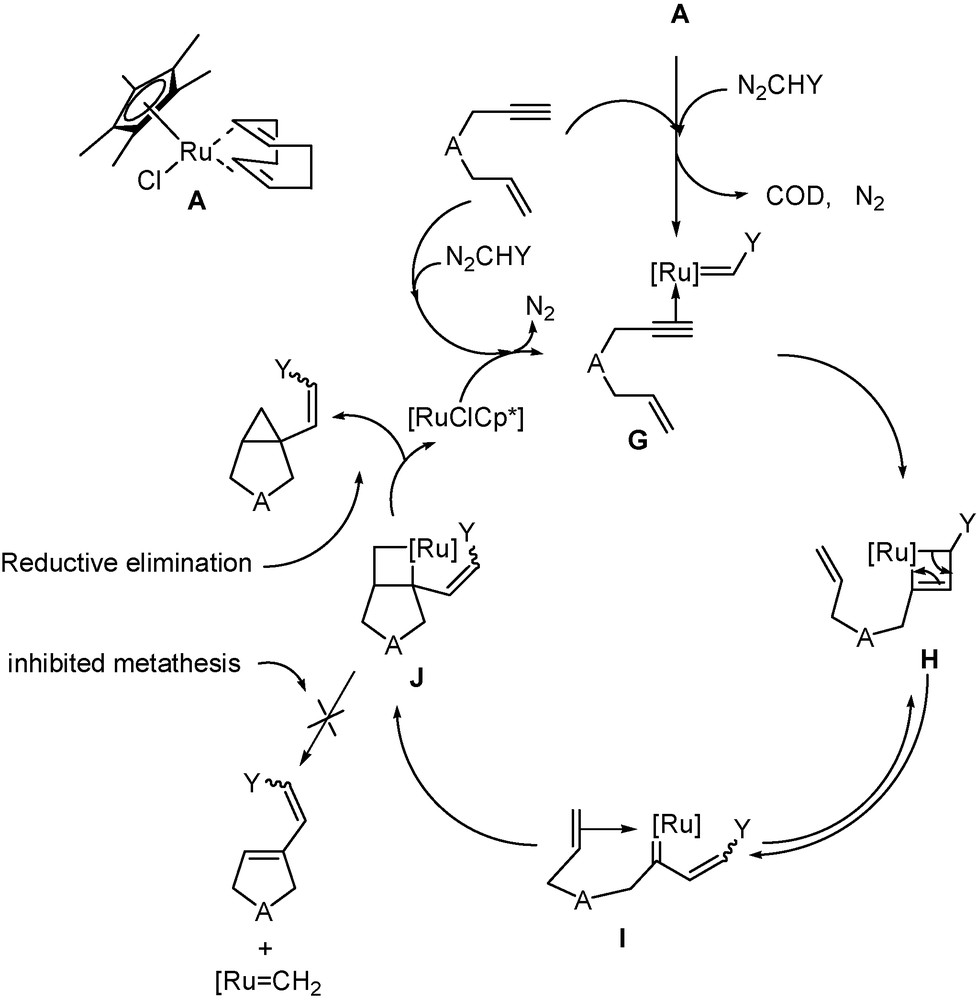
Proposed mechanism for the transformation of enynes into alkenylbicyclo[3.1.0]hexanes.
The diazoalkane carbene addition to enynes offers access to other compounds when the triple bond is disubstituted or when the propargylic carbon is disubstituted. In that case, the reaction leads, besides the formation to the expected bicyclic compound of type 10, to derivative 17, the cyclopropanation product of only the CC double bond, and/or to the alkenyl cyclopentane derivative 18 (Eq. (12), Table 2).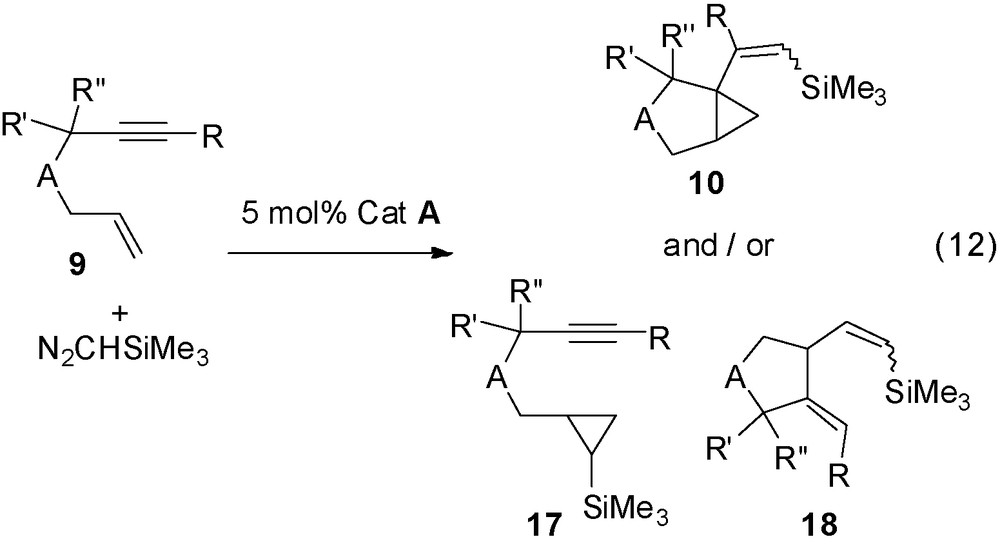
Catalytic transformation of enynes with disubstituted triple bonds or disubstituted propargylic carbon.
Table 2 gathers some representative examples:
- • with disubstituted triple bonds, cyclopropanation of the double bond only takes place as for a classical olefin and compounds 17 are obtained (entries 1, 2);
- • the functional enyne 9j with disubstituted triple bond gives the classical derivatives 10j in small amount and the 5-membered cyclic compound 18j was obtained as a major product (entry 3);
- • with the disubstituted propargylic carbon derivatives 9k and 9l, the analogous alkylidene derivatives 18k and 18l were obtained, and in the later case with the parallel formation of 19l (entries 4, 5).
Theses results will be rationalized later in Scheme 6.
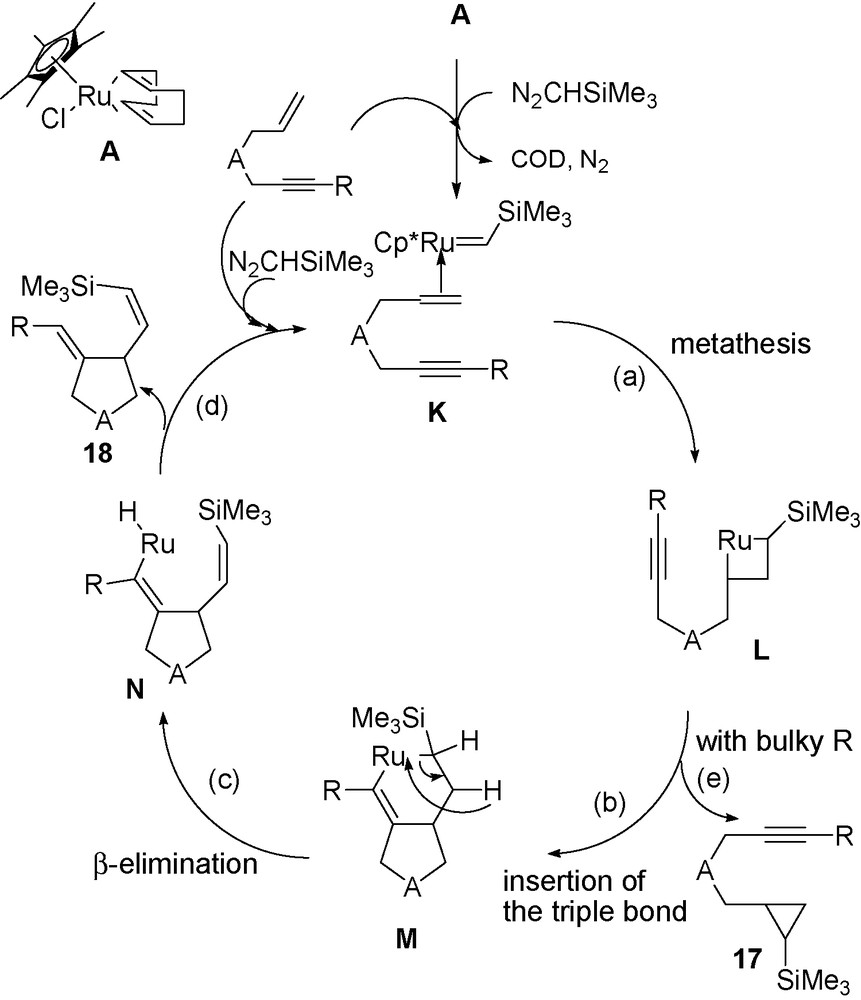
Proposed mechanism for the transformation of enynes into alkenyl alkylidene cyclopentanes.
The direct desilylation of bicyclic derivatives 10 does not take place under mild classical conditions (KOH, fluoride). Thus, the diazomethane was in situ generated on reaction of N2CHSiMe3 in methanol and a variety of desilylated products 20 were obtained in 60–80% yields (Eq. (13)).
The reaction can also take place in water without desilylation but it especially shows that the RuCl(COD)Cp*/N2CHSiMe3 system surprisingly tolerates water (Eq. (14))
The catalytic bicyclisation of enynes under the action of N2CHCO2Et and N2CHPh can also be performed but under more drastic conditions.
With N2CHCO2Et, the reaction is quite general and requires 100 °C for 1–24 hours and the resulting compounds analogous to derivatives 10 possess the E configuration of the double bond (Table 3).
Catalytic transformation of enynes with diazoacetate.
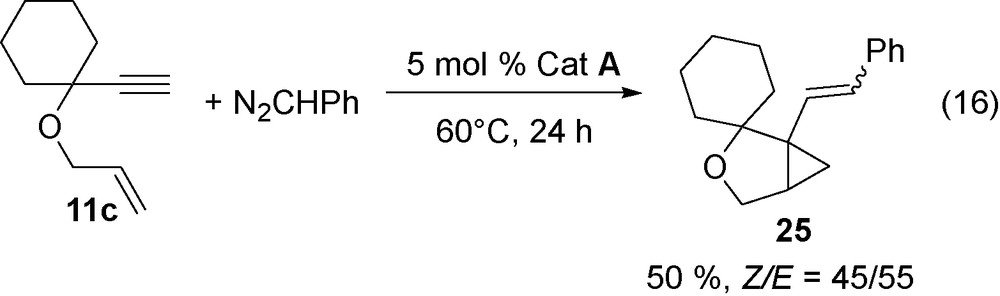
With N2CHPh, the reaction takes place at room temperature to 100 °C and leads to a mixture of Z and E isomers (Eqs. (15), (16)).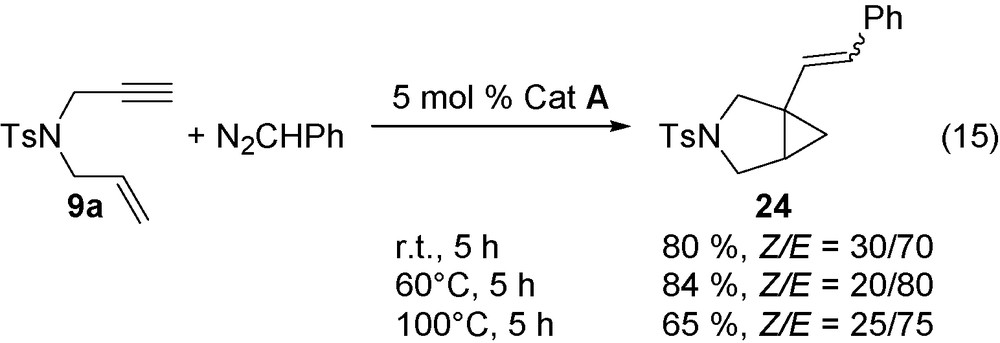
3.2 Proposed mechanisms according to enyne nature
For the catalytic cycles two situations have to be considered:
- • for terminal triple bond enynes;
- • for enynes with disubstituted triple bonds and disubstituted propargylic carbon enynes.
3.2.1 Mechanism with terminal triple bond enynes (Scheme 5)
The mechanism should account for the transformation of the D-labelled enyne 9a which shows that the triple bond deuterium is completely recovered on the vinylcarbon atom linked to the bicyclic carbon (Eq. (17)).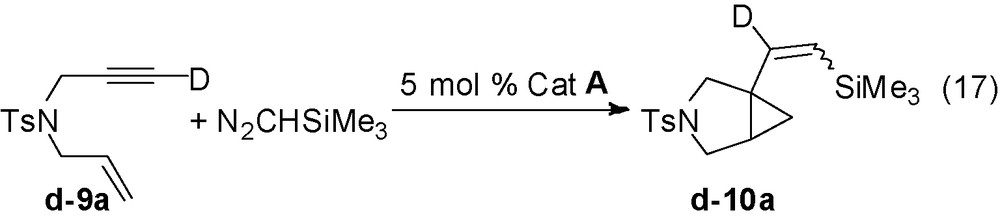
The mechanism likely involves the initial interaction of the RuCH carbene with the triple bond (Scheme 5), that successively leads to intermediates G→H→I→J via transformation as already observed in enyne metathesis (Scheme 4). With the RuClCp* moiety, the metathesis process appears inhibited to the profit of reductive elimination to give the formation of the cyclopropane ring and the RuClCp* moiety which regenerates the ruthenium carbene in the presence of diazoalkane.
DFT computational studies support this mechanism [8c]. They show that:
- • the RuC/CC 2 + 2 addition is favoured by the shift of η5-C5Me5 to η3-C5Me5 ligand to form intermediate J;
- • the coordination of the alkenyl group single double bond of J strongly stabilizes the metallacyclobutane intermediate and favours reductive elimination;
- • the metathesis process from J requires a much higher transition state energy than that leading to reductive elimination.
This catalytic cycle corresponds to that proposed for the stoichiometric reaction of Fischer type Tungsten-carbene, containing an alkene chain, with alkynes [24].
3.2.2 Mechanism from enynes containing disubstituted alkyne bond and/or disubstituted propargylic carbon atom (Scheme 6)
For sterically hindered triple bond enynes likely the initial RuCHY carbene interaction with the CC double bond takes place to give intermediate L (Scheme 6), is faster than the interaction with the sterically hindered triple bond. This intermediate L either gives the reductive elimination when the triple bond is disubstituted to give cyclopropane derivatives 17 or the triple bond insertion into the RuC bond of L takes place to give intermediate M, which on β-elimination, leads to intermediate N the precursor of 5-membered cyclic derivatives 18, via reductive elimination.
The catalytic diazoalkane carbene addition to enynes generating bicyclic compounds presented here, actually shows that the in situ generated carbene RuCl(CHY)Cp* behaves as an alkene/enyne metathesis inhibitor and rather favours reductive eliminations.
This reaction of enyne with diazoalkanes has recently been observed by Li and Montgomery but with Nickel(0) catalysts [25]. This new straightforward catalytic synthesis of alkenylbicyclo[3.1.0]hexanes directly from enynes offers further skeleton rearrangements. Indeed, vinylcyclopropanes can be transformed into cyclopentene derivatives with Ni(0) catalysts [26] and alkynes can insert into metal–carbon bond of the intermediate when rhodium [27] or ruthenium [28] catalysts are used.
4 Application of tandem catalytic carbene addition/bicyclisation of enynes
4.1 Synthesis of fluorinated bicyclic aminoacid derivatives
Cyclic aminoacids introduce a conformational restriction into peptides that are useful for drug discovery [29]. In addition, a fluorinated group at the α position of aminoacid moiety is expected to slow down related peptide hydrolysis. As the fluorinated enynes 26–27 are easily obtained from the imines CF3C(NPG)CO2Me by successive addition of allyl or vinyl magnesium bromide and then of propargyl bromide [30], they have been transformed into bicyclic compounds 28–29 according to the previously described catalytic reaction (Eqs. (18), (19)).
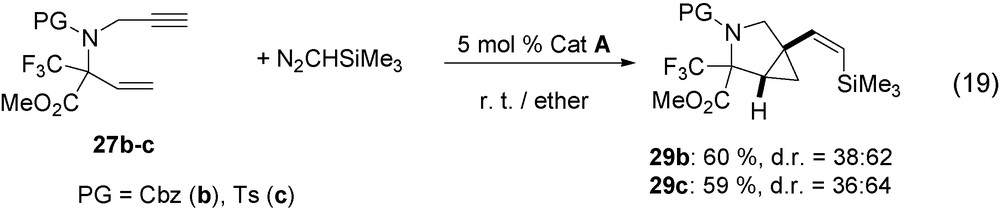
The 1,7-enynes 26a–c on reaction with N2CHSiMe3, in the presence of RuCl(COD)Cp* catalyst precursor A, are completely transformed at room temperature into bicyclo[4.1.0]heptanes 28a–c (Eq. (18)). Both diastereoisomers are obtained but with only the Z configuration of alkenyl group. Analogously, the 1,6-enynes 27b–c with N2CHSiMe3 lead to bicyclo[3.1.0]hexanes 29b–c (Eq. (19)). The same enynes 27b–c also react with N2CHCO2Et but under more drastic conditions at 100 °C and derivatives 30b–c are easily obtained (Eq. (20)). The later always contains an E-alkenyl group.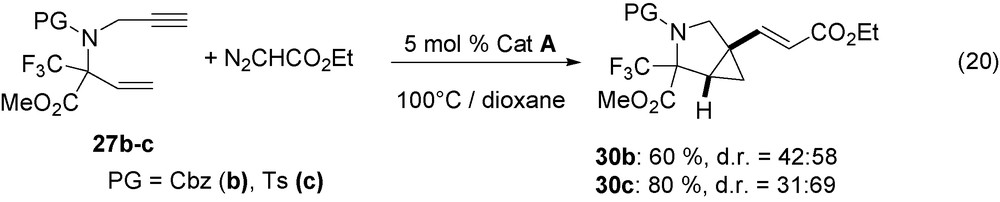
This synthetic method thus offers a straightforward access to fluorinated bicyclic compounds and new bicyclic aminoacid derivatives.
4.2 Direct route to alkenyl alkylidene bicyclo[3.1.0]hexane derivatives
Methylenecyclopropanes offer via Pd(0) and Ni(0) catalysis the direct access to methylene cyclopentanes [31], 5-membered heterocycles [32], alkylidenecyclopentanones [33] and a variety of small cycles [34] such as cyclobutenes [35].
The previous catalytic tandem carbene addition/bicyclisation of allenynes has been applied as a direct route to bicyclic compounds containing the alkylidene cyclopropane moiety. The allenynes 31a–d react at room temperature with N2CHSiMe3 in the presence of catalyst A RuCl(COD)Cp* to give the Z-alkenylalkylidene bicyclic compounds 32a–d in 40–85% yields (Eq. (21)).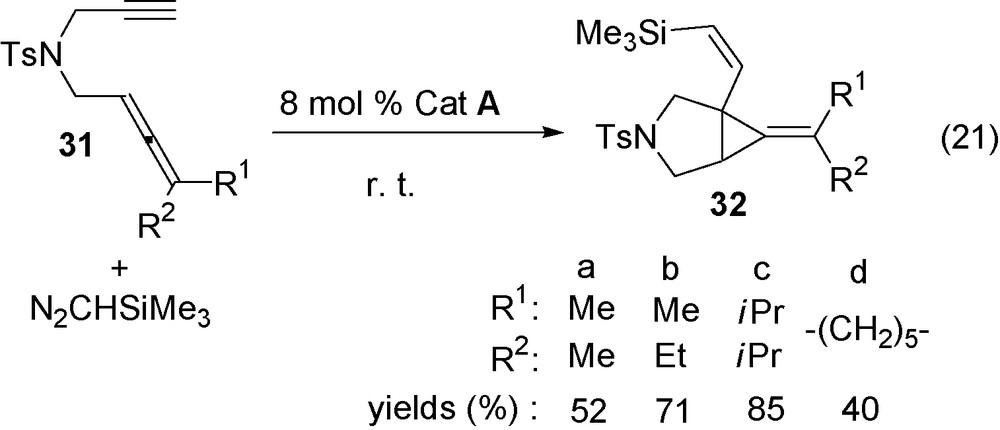
The reaction can be extended to enynes containing a disubstituted triple bond such as 33a–b that are transformed into derivatives 34a–b (Eq. (22)). Analogously, allenyne 35 containing an only-carbon chain was selectively transformed into derivative with a Z-alkenyl chain 36 (Eq. (23)).
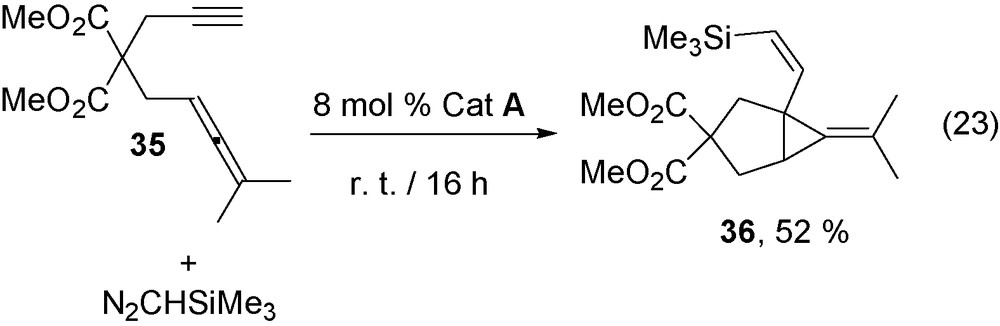
It is noteworthy that the above reaction involves the transformation of the only internal allene double bond. Previous attempts to oxidatively couple allenes with electrophilic alkenes (enones) have led to the coupling of the internal allene double bond [36], whereas for sterically hindered allenylboranes the oxidative coupling with RuCl(COD)Cp* involves the terminal allene double bond [37].
The proposed mechanism of the transformation of allenynes with diazoalkanes is presented in Scheme 7.
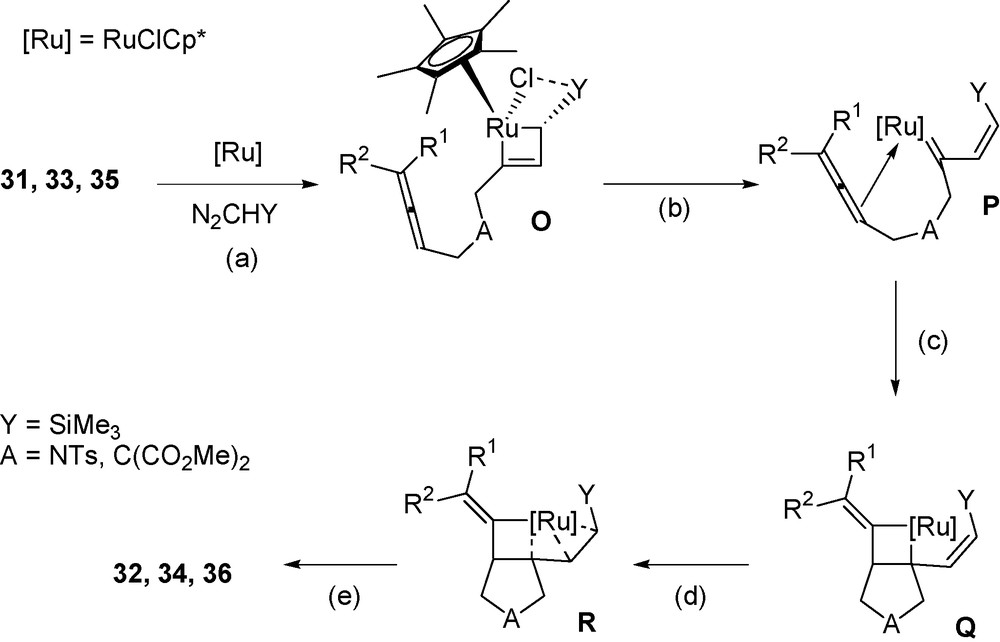
Mechanism for ruthenium catalyzed transformation of allenynes with diazoalkanes.
The mechanism likely involves the following steps:
- • the initial interaction of the RuCHY bond with the triple bond of 31, 33 or 35, to give intermediate O (step a), for which the internal interaction of the chloride with the Me3Si silicon may account for the observed Z-alkenyl configuration in the metathesis product P (step b);
- • the addition of the RuC bond to the internal allene double bond is expected to produce intermediate Q (step c);
- • the coordination of the alkenyl double bond of Q is expected to favour the reductive elimination in step (e), as predicted by DFT calculations [8c], to afford the derivatives 32, 34 or 36 from intermediate R.
5 Catalytic addition of diazoalkane carbene to propargylic compounds: synthesis of functional dienes
Propargylic carboxylates by suitable metal activation are known to generate alkenylcarbene-metal intermediate via the Rautenstrauch rearrangement [38]. The Rautenstrauch rearrangement involves the activation of the triple bond which promotes the carboxylate shift from propargylic carbon to alkyne carbon 2 (Eq. (24)). It was initially performed with palladium catalyst [38] and more recently by Ru(II) [39], Pt(II) [40,41] or Au [41,42] catalysts for the production of cyclopropanes on reaction with alkenes or cyclopentanones, trienes and indenes.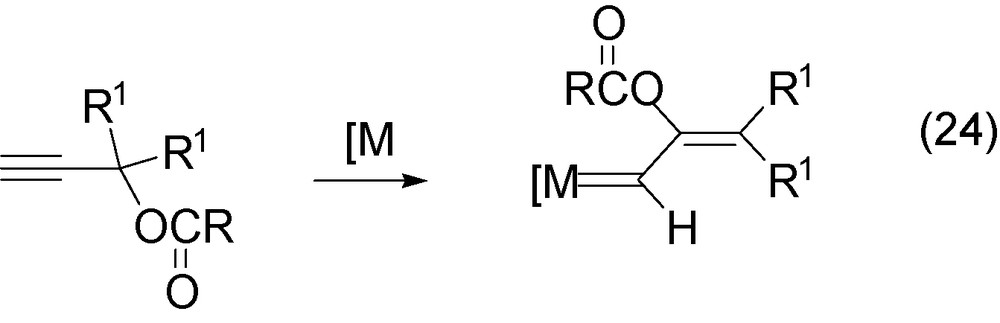
As alkynes have been shown above to initially “insert” into the RuC bond of the RuCHR(Cl)Cp* unit via the metathesis process, we have thus studied the reaction of in situ generated RuCl(CHR)Cp* active species with propargylic carboxylates. The reaction easily takes place and quantitatively affords the functional dienes (Eq. (25)).
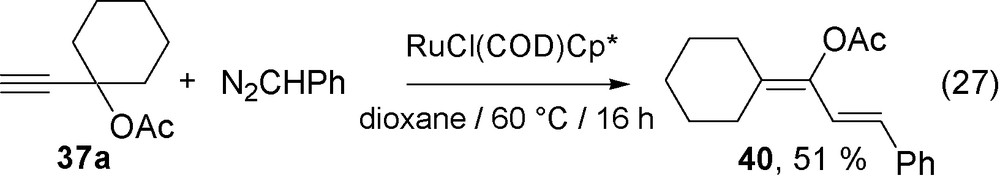
Thus, the reaction of propargylic carboxylates 37a–c react with N2CHSiMe3 at 60 °C in dioxane in the presence of RuCl(COD)Cp* to afford the dienes 38a–c which formally corresponds to the cross-coupling of two carbenes, one arising from the diazoalkane and that resulting from a Rautenstrauch rearrangement as the carboxylate has migrated (Eq. (25)).
From propargylic acetate 37d which has two different substituents at propargylic carbon, the expected two stereoisomers 38d and 38d’ are produced (Eq. (25)).
The corresponding desilylated dienes are produced by the use of in situ generated diazomethane on reaction of N2CHSiMe3 in methanol, as illustrated by the reaction of 37a which directly leads to diene 39 (Eq. (26)). A similar reaction of alkyne 37a with N2CHPh leads to the phenyl substituted diene 40, but requires longer reaction times (Eq. (27)). It is noteworthy that the observed stereochemistry of the silylated dienes 38 is Z whereas that of the phenylated diene 40 is E.
A possible mechanism for this reaction is proposed on Scheme 8.
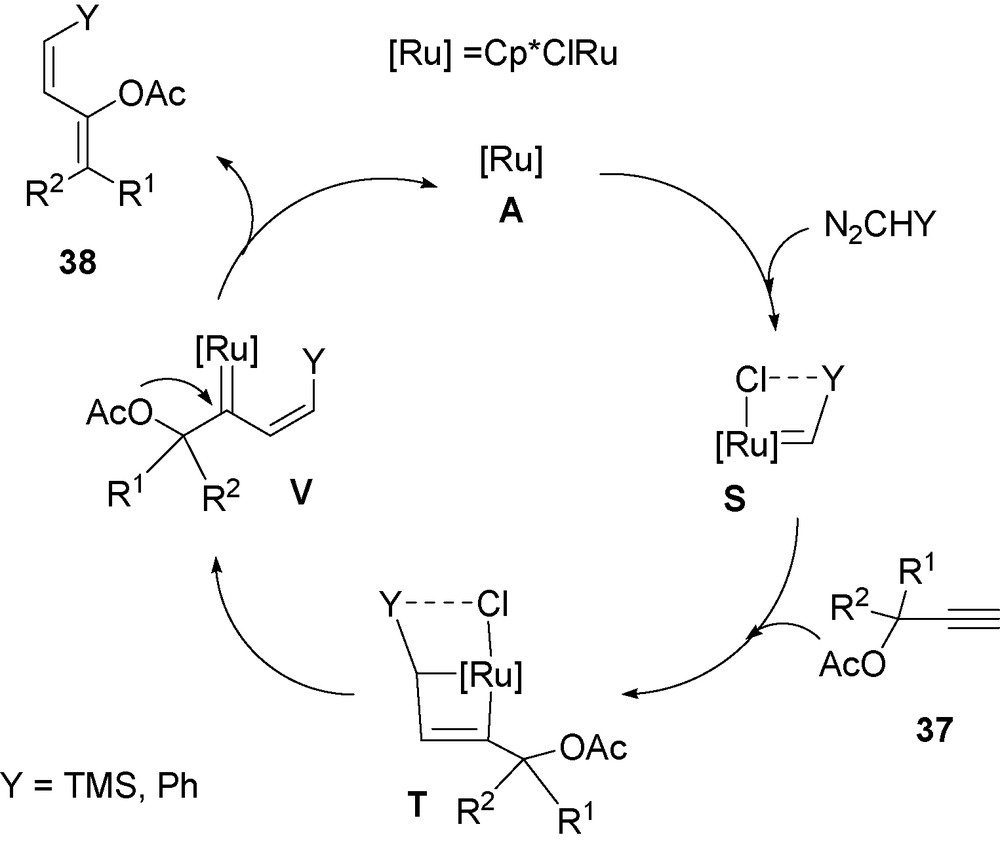
Possible catalytic cycle for the synthesis of dienes from propargylic esters.
This mechanism is based on the previous observations that the RuCHSiMe3 moiety interacts easily with the terminal triple bond. Thus the catalyst A via the formation of RuCl(CHSiMe3)Cp* S is expected to give the 2 + 2 adduct T, rearranging into the metathesis product, the vinyl carbene V. Then the carboxylate is the subject of 1,2 migration from propargylic carbon to the carbene carbon to generate the diene 38. Here again the intramolecular interaction of the chloride and the silicon in S may explain the observed stereoselectivity for the silylated product. The above reaction formally corresponds to the new cross-coupling reaction of two carbene moieties with CC bond formation.
6 Conclusion
This review on the interaction of the catalyst precursor RuCl(COD)Cp* with diazoalkane and several alkynes shows the unique role played by the RuClCp* moiety, with respect to other metal catalysts generating metal carbene intermediates with diazoalkanes.
Although the 16 electrons Ru(Cl)(CHR)Cp* carbene species has never been observed, it explains all the transformations discovered on reactions of diazoalkanes with alkynes. The functionality in the alkyne derivative actually significantly orientates the nature of the formed products:
- • simple alkynes generate dienes via double carbene addition to the triple bond;
- • enynes with terminal triple bond lead to alkenyl bicyclo[x.1.0]alkanes derivatives;
- • 1,6-enynes with disubstituted propargylic carbon afford in priority to alkenyl alkylidene cyclopentanes;
- • 1,6-allenynes offer the direct access to alkenyl alkylidene bicyclo[3.1.0]hexanes;
- • propargylic carboxylates lead to conjugated dienes with 1,2-shift of the carboxylate, with carbene addition to the alkyne terminal carbon atom.
The above chemoselective catalytic reactions offer new examples of the straightforward building of complex architectures from diazoalkanes with alkynes and show the unique activation brought by a RuX(C5R5) moiety.
Acknowledgement
The authors are grateful to the CNRS and the ministère de la Recherche for support, the latter for a PhD grant to CV-LB, the European Union through network IDECAT, the institut universitaire de France for membership (P. H. D.).