

1 Introduction
The basic principles of organometallic chemistry are the basis of the discovery of catalysts and catalytic processes involving CC bond formation or rearrangement, such as olefin and diene polymerizations, olefin and alkyne metathesis, as well as carbonylation reactions.
In this review, we will focus essentially on four typical reactions, which have been the subject of investigations in our lab, aimed at both the improvement of known catalytic processes as well as the establishment of new catalytic reactions.
Namely, this article will deal first with the use of the hemilability concept in catalysis and coordination chemistry, for which reviews have appeared in the literature [1,2]. Two reactions will be described: the linear diene dimerization and codimerization on nickel based catalysts modified by bifunctional ligands and the discovery of an alcoxyether-tungsten based dinuclear complex for its use as precursor for terminal alkyne metathesis.
The second part will focus on transmetalation reactions applied to catalysis. We will first emphasize the role of ligands on lanthanides to produce catalysts used in polymerization, and describe a coordinative chain growth reaction that allows the synthesis of well defined higher Grignard reagents from magnesium dialkyls used in excess via a reversible, transmetalation reaction between the chain growing on lanthanide and magnesium. Applied first to ethylene and α-olefins in our lab, this process will be shown to be generalized to styrene and dienes. The last part will be devoted to rhodium catalyzed carbonylation reaction, by which it will be demonstrated that under suitable conditions, Suzuki-Miyaura type reaction using boronic acids and enones or terminal alkyne may be conducted under CO. As a result, CO insertion into the rhodium-aryl bond formed via transmetalation is occurring, allowing carbon monoxide to be inserted in the following step of the catalytic cycle and produce new carbonylated compounds.
2 Hemilability and catalysis: CarbonCarbon coupling using aminophosphinite–nickel catalysts
2.1 Linear dimerization of conjugated dienes
Linear dimerization of butadiene has been developed in the mid-1960s on nickel carbonyl–phosphine-based catalysts in the presence of alcohols [3] and revisited by Heimbach in the early 1970s, using nickel-based catalysts modified by phosphite ligands in the presence of morpholine [4]. In the same way, Pittman and Smith showed that upon reducing NiBr2(PPh3)2 with NaBH4, linear dimmers are also produced in the presence of ethanol [5]. As compared with the use the same catalytic systems in non protonic, apolar solvents such as toluene, where the reaction course is well known to produce cyclic rather than linear dimers [5] (Fig. 1), we have been interested in the synthesis of a suitable bifunctionnal, phosphorus ligand which would provide a phosphonite moiety to coordinate nickel, and bearing a pendant NH moiety which would act as the proton donor, playing the same role as morpholine and ethanol in the selective production of linear dimers.
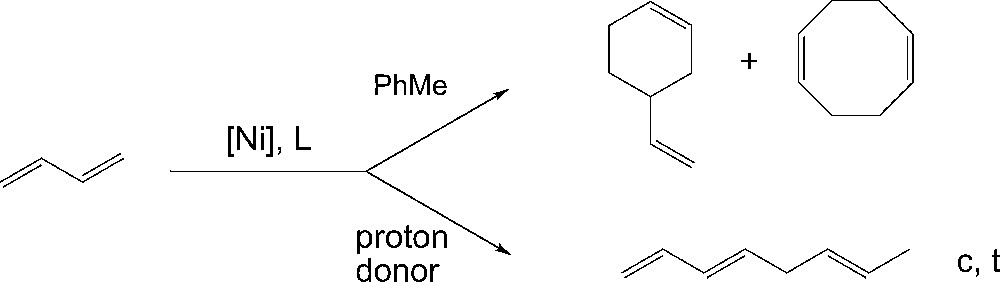
Cyclic vs linear dimerization of butadiene.
To achieve this goal, we started from a chemistry we were developing related to the synthesis of new chiral ligands for asymmetric catalysis, namely that of the so-called AminoPhosphinePhosphinite (AMPP) ligands from aminoalcohols [5], during which synthesis a monophosphinylation could selectively lead in one step to an aminophosphinite ligand bearing a NH moiety on the β carbon atom vs the phosphinite. Such a reaction is depicted in Scheme 1, using ephedrine as the starting aminoalcohol, leading to the new EPHOSNH ligand 1.
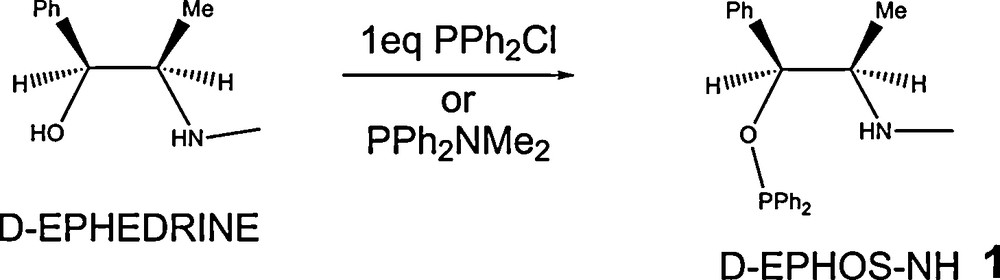
Monophosphinylation of ephedrine: synthesis of the EPHOSNH aminophosphinite ligand.
As shown in Table 1, the reactivity of this system in toluene, where 1 equivalent of the hemilabile EPHOSNH is used as bifunctional ligand for nickel based catalyst obtained by substitution of cyclooctadiene in Ni(COD)2 is found to be two orders of magnitude higher than that of previous catalysts. Turnover rates as high as 5000 h−1 could be attained with butadiene as substrate [6].
Linear dimerization of butadiene into 1,3,6 –octatrienes: comparative results.
| Catalyst | Butadiene/Ni | Conv. (mol%) | Rtion time (T°C) |
| Ni(COD)2/EPHOS-NH (1/1) | 600 | 90 | 15 min, 60 °C |
| Cl2Ni(PPh3)2/NaBH4/EtOH | 600 | 80 | 24 h, 80 °C |
| Ni0/P(OEt)3/Morpholine | 800 | 50 | 24 h, 60 °C |
The role of the secondary amine in that catalytic reaction was determined by using deuterated EPHOSND, leading to propose the mechanism depicted in Scheme 2, where it is obvious that the close proximity of the NH function is the key feature allowing the process to be thus activated, vs the same reaction conducted in a protic solvent or proton donor [7].
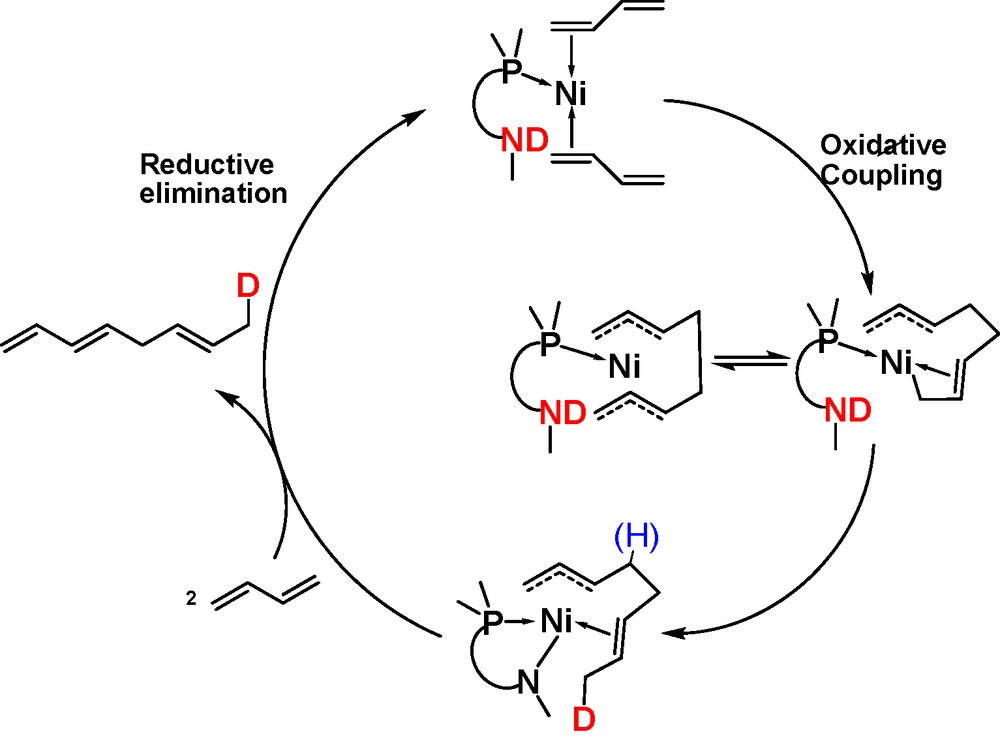
Bifunctional behavior of the EPHOSNH ligand during butadiene linear dimerization.
The same catalytic system was then applied for the linear dimerization of several substituted dienes, as well as their codimerization using mixtures of these.
Interestingly, 1,3-pentadiene was exclusively dimerized via a tail-to-tail linkage into a mixture of E,Z and E,E-2,7-dimethyl-1,3,6-octatrienes, which further isomerize into their conjugated 2,4,6 isomers. Furthermore, thanks to the chirality of the ligand, an asymmetric induction was found, 90% on the E,Z and 35% on the E,E isomers [7].
Other typical results are described in Scheme 3 [8,9].
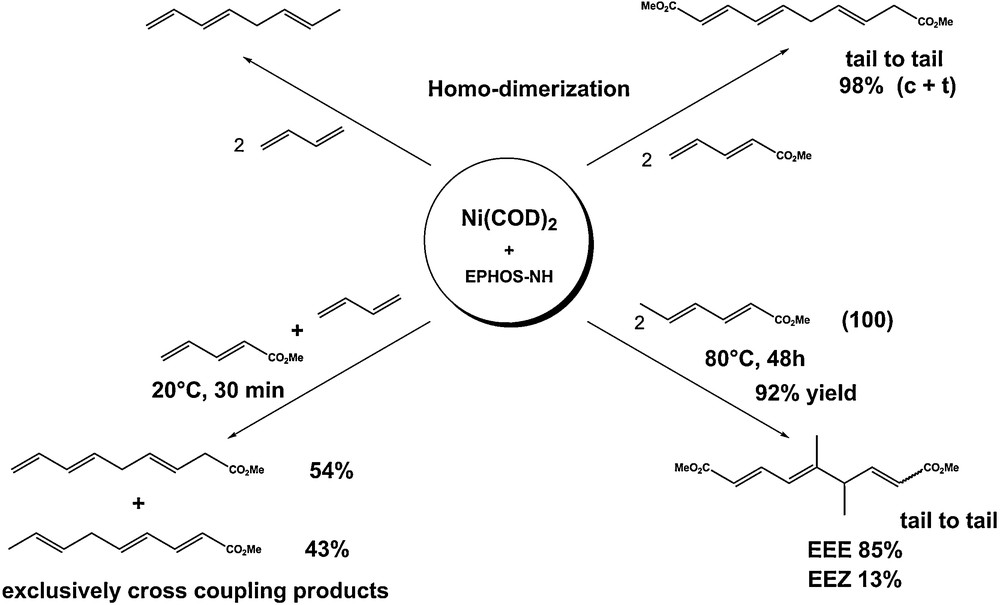
Applications of the Ni(0)/EPHOSNH catalyst.
The homodimerization of methyl pentadienoate was found to take place selectively in a tail-to-tail linkage, whereas its copolymerization with butadiene afforded the cross dimerization product selectively.
Other results have shown that a cross-reaction between butadiene and methyl sorbate only gave the cross coupling products coming from a linear dimerization [9]. Methyl sorbate alone was dimerized linearly, once more via a tail-to-tail linkage.
2.2 Olefin-conjugated diene co-oligomerization using nickel-aminophosphinite catalyst
Further successful experiments were conducted to generalize this CC coupling to co-oligomerize butadiene with acrylates, aimed at the possibility of incorporating the double bond of the acrylate via an insertion into the π allylic intermediate before reductive elimination of the octatriene. Such a process indeed produces C10 trienic esters, as expected (Scheme 4).
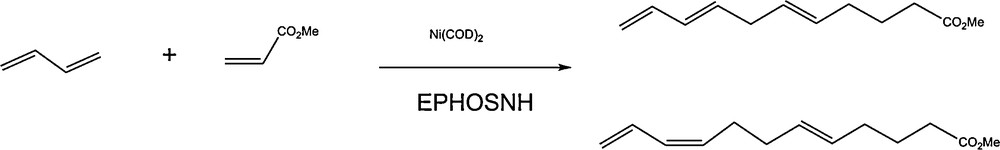
Butadiene-acrylate co-oligomerization using the Ni(0)/EPHOSNH catalyst.
Labeling experiments using EPHOSND followed by analysis (IR, 13C-NMR, MS) of the products at the early stage of the reaction proved that deuterium was transferred at the C2 position of the ester [10]. These results can therefore be explained by the same mechanism as depicted for butadiene homodimerization, where the acrylate insertion would occur before the proton transfer leading to the co-oligomers.
2.3 Conclusion
In this first approach related to the use of the hemilability concept in nickel catalysis, the bifunctional character of the ligand is here well established in a sense that the phosphorus atom allows one first to link the ligand with the metal – conferring the electronic properties of a phosphinite –, and second to provide within the coordination sphere the NH proton donor moiety which one allows to enhance the proton transfer, the rate determining step for the formation of the linear dimers, by two orders of magnitude as compared to the previous catalytic systems. This new system has allowed one not only to dimerize sterically hindered dienes such as methyl sorbate, but also to induce asymmetric induction during CC bond formation thanks to the chirality of the EPHOSNH ligand.
3 Hemilability and catalysis: the challenge of terminal alkyne metathesis
Metathesis of disubstituted alkynes has been established in the late 1960s [11] using heterogeneous catalysts such as WO3/SiO2 (Scheme 5).
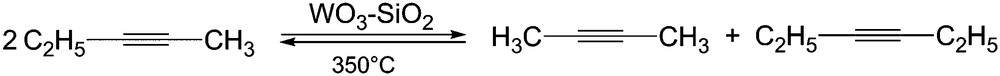
2-pentyne metathesis on silica supported catalysts.
Its homogeneous counterpart was discovered in 1974, using in situ Mo(CO)6/ArOH catalysts [12] whereas Schrock described the first well-defined (tBuO)3W≡CtBu 2 tungsten complex for this reaction [13]. However, little work has been done on the metathesis of terminal acetylenes [14]. At least two major reasons may explain this lack of interest on a reaction, which obviously should also be of interest for organic chemists, due the fact that upon easy removal of acetylene, the reaction may be conducted until completion. The first one comes from the fact that these substrates have been shown to be prone to cyclotrimerization into aromatics using WO3/SiO2 catalysts [15], although a CoOMoO3/SiO2 catalyst has been claimed to be active in metathesis of 1-pentyne [16]. The second and certainly the main one is that using phenylacetylene, such a reaction was found be impossible using Schrock's catalyst, the reason being evidenced by the characterization of a deprotiometallacyclobutadiene complex, arising from a deprotonation of the metallacyclobutadiene intermediates (Scheme 6) [17].
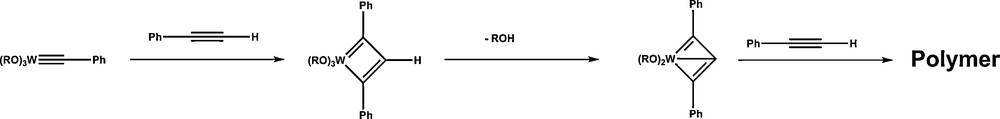
Deprotonation process of tungstacyclobutadienes intermediates.
This irreversible process changes the reaction course, as these species evolve to metallacarbenes, responsible for the polymerization of the alkyne.
In the beginning of the 1990s however, attempts at homogeneous metathesis of terminal alkynes have been successful on (tBuO)3W≡CtBu [18]. The use of alkyl monosubstituted alkynes instead of aromatics led to several turnovers in metathesis at room temperature, which were increased upon using diethyl ether as solvent (Fig. 2).
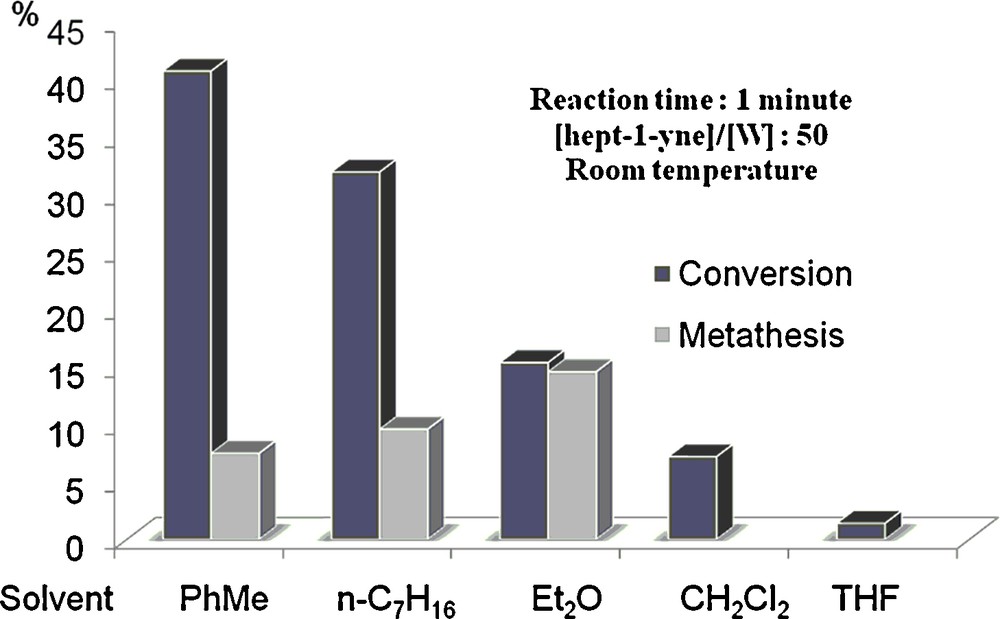
Catalytic metathesis of 1-heptyne: solvent effect.
These successful results prompted us to have a deeper insight into this chemistry [19].
From the observation that a coordinating solvent such as diethyl ether was beneficial, and that bidentate ether ligand such as dimethoxyethane had already been shown to be prone to stabilize tungstacarbyne species [20], the approach was then to look for the use of Schrock's complex together with equimolar amounts of an extra ligand which would be suitable to coordinate the tungsten species to avoid the deprotonation of the metallacyclobutadiene intermediate. Amines have been shown to be good candidates to play such a role, and particularly quinuclidine, by which the selectivity into metathesis products from terminal alkynes has been enhanced to 80% at 80 °C (Scheme 7) [21].
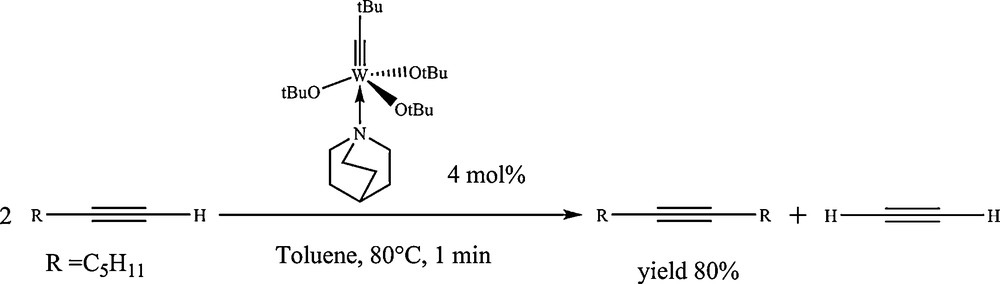
Terminal alkyne metathesis on the (tBuO)3W≡CtBu-quinuclidine complex.
Extension of this system to the cometathesis of terminal and disubstituted alkynes affords the expected cross-reaction products, and the cyclisation of a terminal α-ω, ether containing diyne was also performed.
Our goal was then to try to design a catalyst, which would be able to attain similar selectivities using the hemilability concept. We turned then our attention to the synthesis of a tungsten species possessing the required characteristics for the metathesis reaction: substitution with three sterically hindered alcoxy ligand but in addition bearing a pendant, hemilabile ether function (Scheme 8).
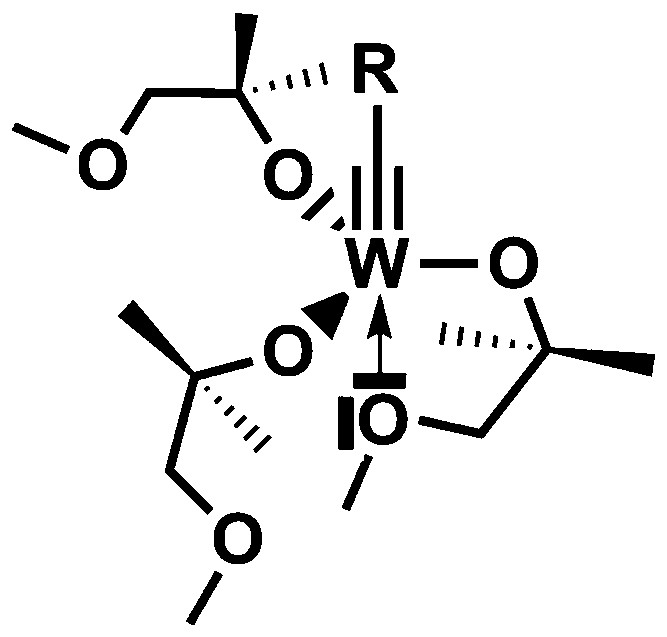
Basic hypothesis for the design of a selective catalyst for terminal alkyne metathesis.
As the cross metathesis between (RO)3W≡W(OR)3 and disubstituted alkynes RC≡CR has been found to occur to produce (RO)3W≡CR carbyne catalytic species, the synthesis of dinuclear tungsten precursors has been performed.
Reacting W2(NMe2)6 in toluene with an excess of the sterically hindered 1-methoxy-2-methylpropan-2-ol (MMPOH) leads to the formation of the bimetallic complex W2(MMPO)6 3 quantitatively (Scheme 9).
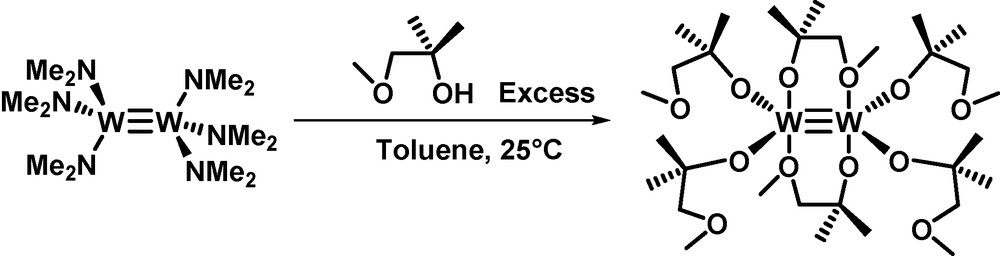
Synthesis of a dinuclear W≡W complex from MMPOH [22].
Complex 3 catalyses internal alkyne metathesis, since non-4-yne is readily transformed at 25 °C into metathesis compounds at equilibrium. 1-phenyl-1-propyne requires a higher temperature to be metathesized (Table 2).
Metathesis of internal and terminal alkynes on the W2(NMPO)6 catalytic precursora.
| Entry | Catalyst | Alkyne | Conversion [%] | Metathesis [%] |
| 1b | 3 | non-4-yne | 50 | 50 |
| 2 | 3 | 1-phenyl-1-propyne | 70 | 70 |
| 3b | 3 | hept-1-yne | 25 | 25 |
| 4 | 3 | hept-1-yne | 86 | 77 |
| 5 | 3+ quinuclidine | hept-1-yne | 50 | 47 |
| 6 | 3 | phenylacetylene | 22 | 10 |
a [Alkyne]/[W] = 28.5; Solvent: Toluene; Temperature = 80 °C; reaction time = 1 h.
b Same conditions, at 25 °C.
This catalyst was then further used for hept-1-yne metathesis. At 25 °C, no polymerization was observed (100% selectivity into metathesis, run 3), but the conversion was limited to 25%. Increasing the temperature to 80° improved the activity, and conducting the reaction under a static vacuum led to a further increase in the metathesis yield, up to 77%. The combination of 3 and one equivalent of quinuclidine was even more selective (94% selectivity, run 5), but resulted in a drop in conversion. Phenylacetylene, which previously has never been metathesized, gave a catalytic metathetic reaction with a low turnover number.
These results show that upon using hemilabile alcoxy ligands, the presence of the extra coordinating moiety in the coordination sphere of these metathesis catalysts undoubtedly plays a major role in the selectivity enhancement. As expected, due to the coordinating properties of the ether function closely localized within the coordination sphere, some decrease in activity was observed. Nevertheless, the activities and selectivities obtained with this new catalyst allow this reaction to be applied to alkyl-substituted terminal alkynes for applications in organic synthesis under mild conditions. Although some interesting results have been obtained using phenylacetylene progress needs to be made in that context to improve the yield of metathesis compound for this substrate. With no doubt, using the hemilability concept with other ligands would help in that context.
4 Transmetallation during polymerization: Reversible Coordinative Chain Transfer Polymerization by Rare Earth Complexes/Dialkylmagnesium systems
Full efficiency reversible chain transfer polymerization was reported for the first time in our group using rare earth-based precatalysts combined to dialkylmagnesium compounds [23–30]. Such processes are known to enable molecular weight control and catalyst atom economy. If the transmetalation occurs reversibly, rapidly, with a high efficiency and in the absence of other termination pathways, the phenomenon is known as a coordinative catalyzed chain growth transfer (CCGT). All polymer chains are bound to the main group metal in the resulting reactive medium in the absence of residual chain transfer agent. Further reactions can be conducted directly taking advantage of the main group metal chemistry, without any workup, and will involve the totality of the macromolecular chains present in the reactive medium. The design of original macromolecular microstructures has also been reported in the frame of coordinative chain transfer polymerization via chain shuttling polymerization notably [31].
The different catalysts used are presented in Fig. 3. The elementary processes involved in the reversible coordinative chain transfer polymerization are presented in Scheme 10. The dialkylmagnesium compound plays a double role, alkylating agent and chain transfer agent. This chemistry was pioneered using ethylene as the monomer and a samarocene precatalyst (2) combined to a dialkylmagnesium (Table 3). A samarium catalyzed polyethylene chain growth on magnesium was obtained, leading to long chain dialkylmagnesium. These macromolecular objects could further be used for the functionalization of the resulting polymer affording carboxylic acid end-capped polyethylene, poly(ethylene-block-ɛ-caprolactone) and poly(ethylene-block-methylmethacrylate) (Scheme 11).
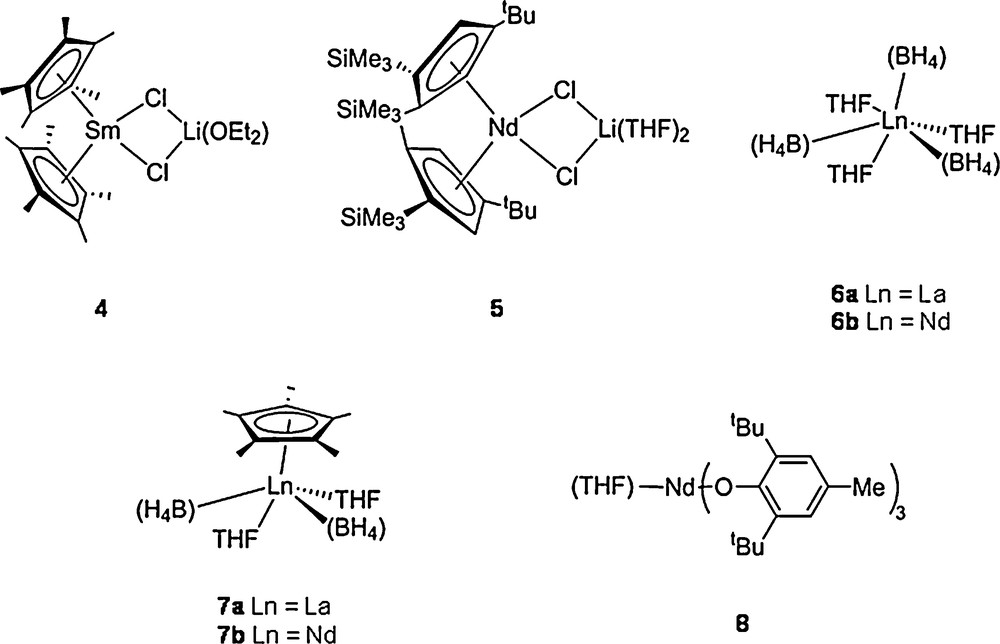
Precatalysts used in our group for coordinative chain transfer polymerisation.
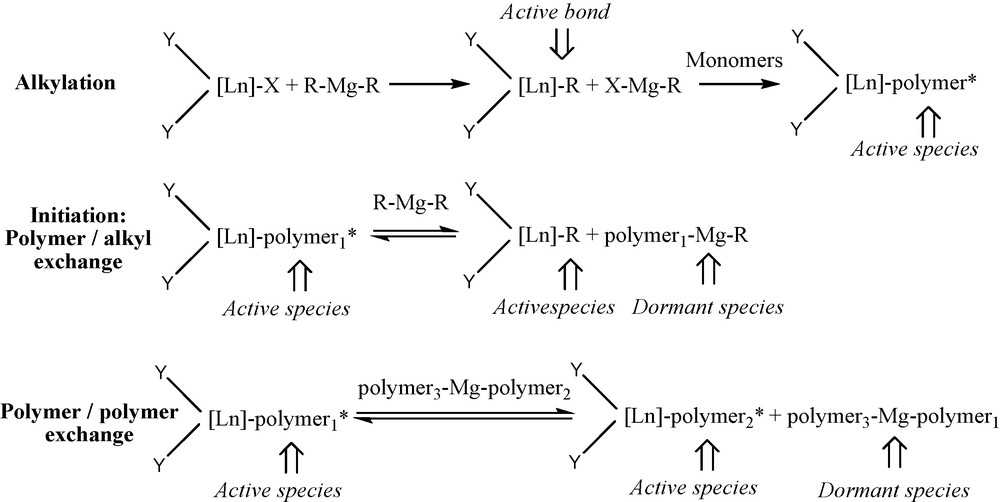
Principle of chain growth transfer mechanism between a lanthanide catalyst and dialkymagnesiums.
Reversible coordinative chain growth transfer polymerization of various monomers by rare earth complexes/n-butylethylmagnesium systems.
| Entry | Monomera | Cata | Mg/Ln | [M]/[Ln] pressure | Solv.b | Time | Temp. (°C) | Yield (%) | thc (g/mol) | d (g/mol) | PDIe | Regio- and stereoselectivity | Comp. (S/I) |
| 1f | E | 4 | 1000 | 1 bar | Isopar | 24 h | 80 | 93g | 460 | 460 | 1.1 | – | – |
| 2g | E | 4 | 20 | 1 bar | Isopar | 2.5 min | 80 | 2.2 g | 1020 | 1070 | 1.3 | – | – |
| 3 | O | 5 | 10 | 640 | – | 24 h | 80 | 74 | 2600 | 1100 | 1.4 | 85% 1,2 | – |
| 4 | O | 5 | 20 | 640 | – | 24 h | 80 | 76 | 1300 | 950 | 1.3 | 85% 1,2 | – |
| 5 | S | 6a | 10 | 1000 | Tol | 24 h | 50 | 52 | 2700 | 1900 | 1.6 | 2,1/atactic | – |
| 6 | S | 6b | 10 | 2000 | Tol | 90 h | 50 | 82 | 8500 | 8300 | 1.2 | 2,1/85% syndiotactic | – |
| 7 | B | 8 | 5 | 100 | Hex | 17 h | 20 | 98 | 540 | 550 | 1.2 | 95% 1,4-trans-5% 1,2 | – |
| 8 | I | 7a | 3 | 2000 | Tol | 20 h | 50 | 78 | 19200 | 18500 | 1.5 | 91% 1,4-trans-9% 3,4 | – |
| 9 | I | 7a | 10 | 2000 | Tol | 40 h | 50 | 86 | 5800 | 6200 | 1.6 | 61% 1,4-trans-39% 3,4 | – |
| 10 | S/I 50/50 | 7a | 3 | 2000 | Tol | 8 h | 50 | 56 | – | 25500 | 1.7 | 95% 1,4-trans-5% 3,4 | 16/84 |
| 11 | S/I 50/50 | 7a | 10 | 2000 | Tol | 40 h | 50 | 58 | – | 8100 | 1.8 | 84% 1,4-trans-16% 3,4 | 32/68 |
a M: monomer, E: ethylene, O: 1-octene, B: butadiene, S: styrene, I: isoprene.
b Tol: toluene, Hex: hexane, Isopar™- L: mixture of heavy liquid parafins.
c Expected molecular weight assuming two growing chains per magnesium atom: .
d Number-average molecular weight measured by SEC.
e Polydispersity index measured by SEC .
f Catalyst 0.1 mmol in 500 ml Isopar L.
g Catalyst 0.05 mmol in 500 ml Isopar L.
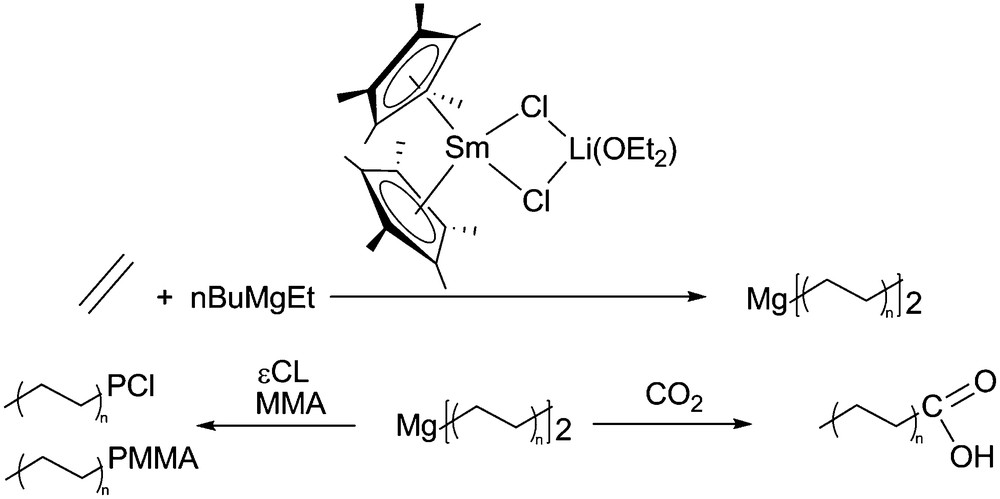
Samarocene catalyzed polyethylene chain growth on magnesium and polyethylene functionalization.
A more hindered neodymocene (5) was used for the chain transfer polymerization of a higher α-olefin (entries 3 and 4 in Table 3). The experimental number average molecular weight of the oligooctenes are in the range of the theoretical value calculated assuming two growing chains per magnesium atom, but are lower. This indicates that effective transmetalation occurred, in the presence of significant βH elimination and/or chain transfer to the monomer occurs. NMR analysis showed ca. 40 to 50% of unsaturated oligooctene chains, comprising around 85% of vinylidene end groups characteristic of a 1,2-insertion mode and around 15% of internal insaturations characteristic of a 2,1-insertion mode.
The reversible coordinative chain transfer of styrene was reported using 6a, 6b, 7a and 7b [24–26] (entries 5 and in Table 3). From the values of the number-average molecular weight, efficient transmetalation occurs for all systems. Unsaturated chain ends were evidenced from MALDI-ToF analysis of the polystyrene obtained using 6a and 6b, highlighting the simultaneous occurrence of βH elimination. Lanthanum-based catalysts were found to be less prone to βH elimination than neodymium based catalysts On the contrary, unsaturated chain ends were scarcely observed using 7a and 7b, highlighting a lanthanide catalyzed polystyrene chain growth on magnesium, and confirming the values of the number-average molecular weight. It is noteworthy that this CCGT is stereoselective, as the resulting polystyrene presents a syndiotacticity ratio of ca. 85%.
Conjugated dienes could also be chain polymerized efficiently using 7a, 7b and 8. A neodymium catalyzed oligobutadiene chain growth on magnesium was reported started from the phenoxide 8, and a lanthanum catalyzed polyisoprene chain growth on magnesium was reported, starting from the half-lanthanocene 7a. The selectivity of the reaction was found to lead mainly to 1,4-trans polydiene. Increasing the amount of chain transfer agent leads to a decrease of the 1,4-trans selectivity at the expense of 3,4 polyisoprene.
As borohydrido half-sandwich complexes are known on the one hand to copolymerize efficiently isoprene and styrene [32] and, on the other hand, to afford an exceptional dual transmetalation in the course of the polymerization of both isoprene and styrene, the reversible coordinative chain transfer was performed in the course of the statistical copolymerization of isoprene and styrene (entries 10 and 11 in Table 3). From the values of the experimental number average molecular weight, it can be deduced that transmetalation occurs in the course of the reaction. In a similar way to the homopolymerization of isoprene in the presence of an excess dialkylmagnesium, the selectivity of the polymerization decreases with increasing the quantity of chain transfer agent, in a lesser extent however. Interestingly, the amount of styrene inserted in the copolymer is increased by a factor 2 when the amount of n-butylethylmagnesium increases from 3 to 10. The composition of the copolymer can thus be easily changed by modifying the ratio of the chain transfer agent in the reactive mixture. The comonomer content in a copolymer is usually tailored by reactor feed, catalyst modification or process considerations. The cotransmetalation can thus be viewed as an original route for the control of the composition of a statistical copolymer.
The above examples show how using the concept of the well-controlled reversible transfer of a propagating chain to the alkylating reagent may result into the synthesis of magnesium dipoly(alkenyl, dienyl, styryl, or mixtures of those) Grignard higher molecular weight compounds, and into the development of new concepts for the design of polymeric architectures. The lanthanide complexes must be considered as catalysts for these transformations, thanks to a fine tuning of the ligand environment, leading first to a propagation reaction on the lanthanide catalyst and second to a good match between the Ln- and Mg-carbon bonds to provide a rapid exchange between those, at a higher rate than the propagation.
5 Transmetallation and CO chemistry: Carbonylative addition of arylboronic acids to unsaturated compounds
The transmetallation of an organic moiety from boron to a transition metal has recently found a wide development (Scheme 12) [33]. That elemental step is generally used with palladium in cross-coupling reactions such as the well-known Suzuki coupling reaction. Rhodium has also proved to be suitable and the transmetallation involving organoboronic acid and rhodium catalysts is the basis of the development of several new efficient transformations. The rhodium catalysed arylation of α,β-unsaturated ketones was one of the early examples based on this methodology, rapidly followed by several other addition type transformations involving acrylic esters or amides as activated olefins and allenes or alkynes as alternative unsaturated compounds [34].
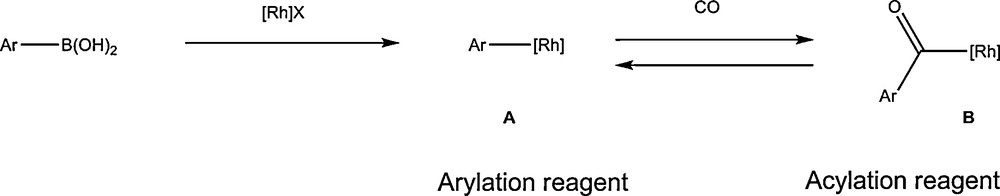
The transmetalation reaction involving an organoboronic acid and rhodium.
Rhodium catalysts are also well known to be suitable for carbonylation reactions and are industrially used in several large-scale industrial processes [35]. Both the hydroformylation of olefins and carbonylation of methanol involve the insertion of carbon monoxide in a metalcarbon bond. We thus expected that under CO pressure, the aryl-rhodium intermediate A obtained from the transmetalation step would be readily carbonylated to yield the potential aroyl acylating reagent B. Although the kinetics of this CO insertion reaction may not have been suitable as compared with that of a further reaction of the aryl-rhodium species, this idea proved to be fruitful as in our hands, the reaction between phenylboronic acid 9 and methylvinylketone 10 under CO pressure catalyzed using a simple rhodium precursor effectively affords the expected product of hydroacylation, the 1,4-diketone 11 (Scheme 13) [36]. However, the presence of carbon monoxide does not prevent the non-carbonylative process as the arylation product 12 is also obtained. In addition to this side-product, diarylketone 13 was obtained from the carbonylative homocoupling of two arylboronic acids.
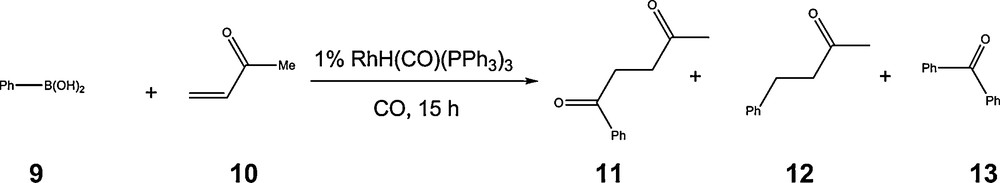
Rhodium catalysed aroylation of methylvinylketone with phenylboronic acid and carbon monoxide.
The optimum reaction condition was found to be 80 °C. Below 60 °C, the reaction rate is strongly reduced and at 100 °C or higher, the formation of the diarylketone 13 becomes strongly limitative as two equivalents of phenylboronic acid are used for the formation of one equivalent of 13 (Table 4).
Carbonylative addition of phenylboronic acid to methylvinylketonea.
| Entry | T (°C) | PCO (atm) | Solvent | 11 (%)b | 12 (%)b | 13 (%)b |
| 1 | 60 | 20 | MeOH | 45 | 3 | 2 |
| 2 | 80 | 20 | MeOH | 78 | 9 | 7 |
| 3 | 100 | 20 | MeOH | 44 | 7 | 18 |
| 4 | 80 | 1 | MeOH | 52 | 21 | 7 |
| 5 | 80 | 20 | THF | 42 | < 2 | 7 |
| 6 | 80 | 20 | THF/H2O | 46 | 3 | 9 |
a The reaction was carried out using phenylboronic acid (1.5 mmol), methylvinylketone (3.2 mmol), 1% HRh(CO)(PPh3)3 and 10 mL solvent for 18 h.
b Yields determined by GC based on phenylboronic acid.
At atmospheric CO pressure, the non-carbonylated product 12 is formed in a significant yield. Higher CO pressure allows a higher selectivity into the carbonylated derivative 11. The CO insertion is generally an equilibrated step and an increase of the pressure shifts the equilibrium to the formation of the aroyl-rhodium B to the detriment of the aryl-rhodium intermediate A (Scheme 12).
The reaction is, however, restricted to unsubstituted α,β-unsaturated kenones such as alkyl- or aryl-vinylketones. The reaction failed with cyclohexenone or chalcone, even at higher temperature. Other unsaturated compounds were also evaluated. The activation of the olefin with a withdrawing group is essential and 1-hexene does not react at all. Using very similar reaction conditions, the carbonylative addition of arylboronic acids to butyl acrylate or acrolein affords the corresponding ketoester or ketoaldehydes with modest yields.
The reaction could be enlarged to alkyne containing derivatives [37]. The first successful experiments were made with 1-hexyne 14 as model reagent. The reaction of carbonylative addition of phenylboronic acid to 1-hexyne affords the corresponding α,β-unsaturated ketone 15 with, in all cases, only traces of diphenylketone, and less than 10% of the cyclopentenone 16 obtained as a mixture of isomers (Scheme 14). On the contrary to the precedent observations with methylvinylketone, in the case of alkynes, the non-carbonylated product has never been observed. The reaction is characterised by a competition with the polymerisation of the terminal alkyne which is known to be promoted by rhodium catalysts. This side reaction implies the use of an excess of terminal alkyne in the experiments and 2.3 equivalents were commonly used. In addition, the appropriate choice of the reaction conditions is of importance in order to tune the rate of polymerisation versus the rate of aroylation reaction (Table 5). Compared to the reaction with methylvinylketone, several reaction conditions gave similar results such as the temperature of 80 °C or the choice of MeOH as solvent. The pressure parameter is in the case of terminal alkynes more critical and higher pressures are greatly detrimental for the yield in α,β-unsaturated ketone. An optimum pressure of 5 bars was found to give the best results (Scheme 15).
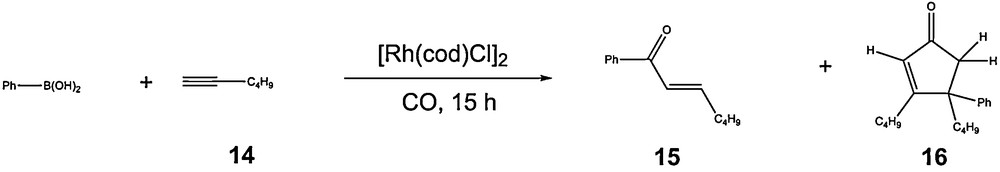
Rhodium catalysed aroylation of 1-hexyne with phenylboronic acid and carbon monoxide.
Carbonylative addition of phenylboronic acid to 1-hexynea.
| Entry | T (°C) | PCO (atm) | Solvent | Conversion (%)b | 15 (%)c |
| 1 | 60 | 5 | MeOH | 46 | 16 |
| 2 | 80 | 5 | MeOH | > 90 | 57 |
| 3 | 100 | 5 | MeOH | > 90 | 46 |
| 4 | 80 | 10 | MeOH | > 90 | 52 |
| 5 | 80 | 20 | MeOH | > 90 | 32 |
| 6 | 80 | 5 | Toluene | > 90 | Traces |
a The reaction was carried out using phenylboronic acid (1.5 mmol), 1-hexyne (2.3 mmol), 0.5% [Rh(cod)Cl]2 and 10 mL solvent for 18 h.
b Conversion of 1-hexyne determined by GC.
c Yields determined by GC based on phenylboronic acid.
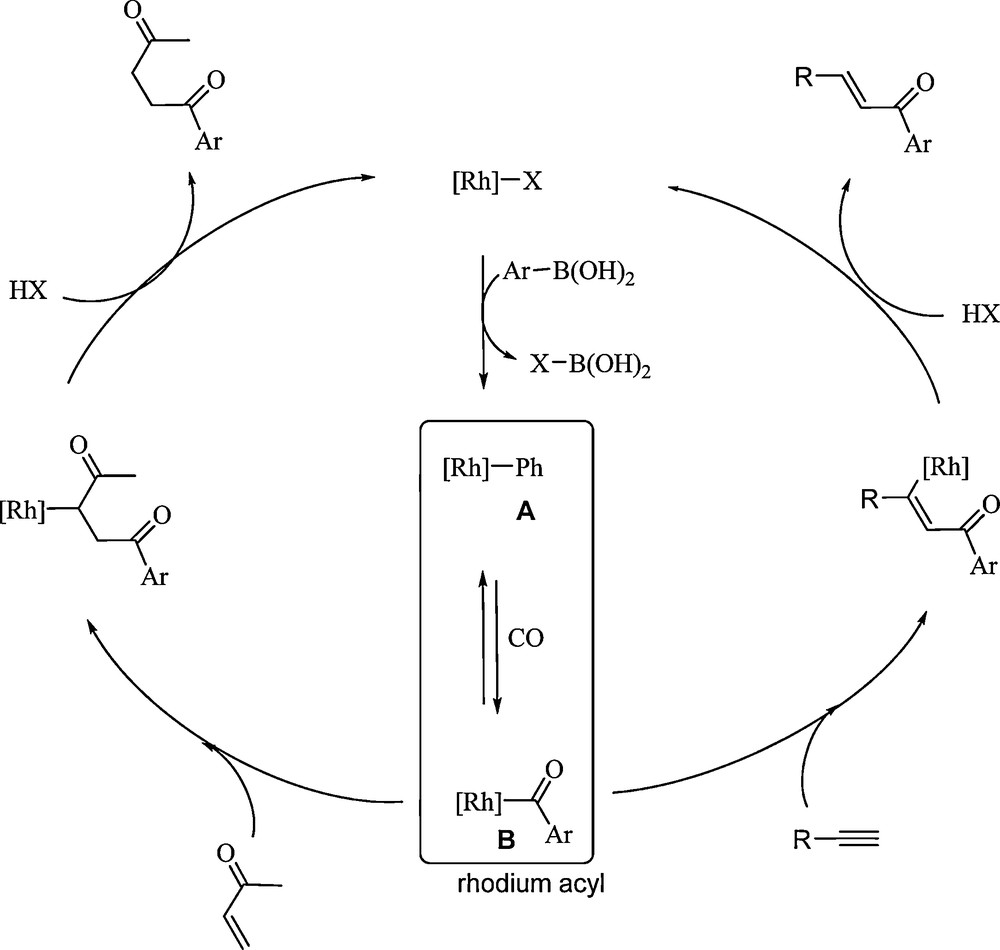
Proposed catalytic cycles for the rhodium catalyzed hydroacylation reaction of methylvinylketone and a terminal alkyne.
Both reactions showed strong similarities, specificity to methanol, reaction temperature. In addition, the yields in product of hydroacylation are varying in the same extent depending on the nature of the arylboronic acid used (Table 6).
Yields of product for the carbonylative addition of arylboronic acids to methylvinylketone and 1-hexyne.
| Entry | ArB(OH)2 | Hydroacylation of MVK (%)a | Hydroacylation of 1-hexyneb (%) |
| 1 | 78 | 57 | |
| 2 | 81 | 69 | |
| 3 | 80 | 70 | |
| 4 | 62 | 45 | |
| 5 | 38 | 27 |
a Isolated yield, the reactions were carried out using arylboronic acid (1.5 mmol) and 1% RhH(CO)(PPh3)3 in 10 mL MeOH with 20 bars CO and at 80 °C for 18 h.
b Isolated yields, the reactions were performed with 1.5 mmol arylboronic acid, 2.3 mmol methylvinylketone or 1-alkyne and 0.5% of [Rh(COD)Cl]2 in 10 mL of MeOH under 5 b CO at 80 °C for 18 h.
Electrodonating substituents on the paraposition of the phenyl group lead to the highest yields in ketones and electrowithdrawing groups such as chlorides to the lowest. CO insertion in a metalcarbon bond is known to be easier if the organic moiety is an electron-rich heterocycle. With methylvinylketone, this is clearly illustrated by the fact that lower yields of carbonylated product are accompanied by higher amounts of non-carbonylated derivatives. The selectivity of the reaction can thus be improved by using higher CO pressures.
Those strong similarities between the two reactions suggest that the corresponding catalytic cycles involve common intermediates. We thus proposed two related catalytic cycles that both involve intermediates A and B obtained from the transmetalation and the carbon monoxide insertion steps.
The unsaturated compound (1-alkyne or methylvinylketone) can then insert in the rhodiumcarbon bond to generate an intermediate which, after protonation, liberates the corresponding products (α,β-unsaturated ketone or 1,4-diketone). This final step leads to the formation of a rhodium species able to participate in a new transmetalation step. CO insertion, as well as that of the unsaturated compound, can be expected at several stages of these catalytic cycles. That reactivity would lead to the formation of oligomers or polymers containing or not carbon monoxide. In the case of methylvinylketone, such supplementary insertion is greatly limited by the stability of the enolate and no product resulting from this process has been observed. Such limitation on the number of insertion is not as evident with terminal alkynes. This is clearly shown by the formation of the cyclopentenone 16, albeit in usually very low yield, which can be explained by the consecutive insertion of two equivalents of alkyne and one equivalent of carbon monoxide starting from intermediate A. This double insertion process can be considered as the first step to the formation of higher oligomers, which likely explains the usually lower yields obtained with terminal alkynes compared to those obtained with methylvinylketone.
These two examples show that transmetalation reactions involving rhodium complexes and organoborons open opportunities for the development of new catalytic reactions. Carbonylative transformations are particularly advantageous as they allow the one step synthesis of functionalized products by combining three or more simple reactants. In the present work, we have shown the fast synthesis of α,β-unsaturated ketones or 1,4-diketones starting from carbon monoxide, arylboronic acids and methylvinylketone or a terminal alkyne. However, the complexity of such transformation necessitates a fine choice of the reaction conditions and catalysts used in order to achieve the best yields.
6 Conclusion
These examples have demonstrated that several homogeneous catalytic reactions can be enhanced either in terms of activity and selectivity, or interestingly, new catalytic reactions or processes can be discovered, via either the molecular design of new catalysts or by simply using basic principles of organometallic chemistry.
Further work is in progress in that sense in our laboratory, as to our opinion these are concepts by which the chemist involved in homogeneous catalysis will pave the way for new applications in sustainable organic chemistry in the future, directed towards the reduction of the E factor, ideally towards a 100% atomic yield whenever possible [38].
Acknowledgments
We are indebted to our colleagues and coworkers whose names appear in the references. The Institut Universitaire de France, the Ministry of Research of Technology and the CNRS are greatly acknowledged for their financial support, as well as the European Science Foundation (COST-D24 and D-40 programs).