

1 Introduction
Allyl-lanthanide complexes are compounds of fundamental interest, notably due to their intermediacy in a number of important catalytic processes. This class of compounds has been studied since the 1960s but “simple” compounds have remained elusive for a long time. For instance, homoleptic tris(η3-allyl)-lanthanide complexes, which have remained as other homoleptic tris(alkyl)-lanthanides over the years one major challenge in organolanthanide chemistry, were unknown until the late 1990s. In fact, the chemistry of allyl-lanthanide complexes has made significant progress in recent years in terms of preparative results as well as of applications in organic synthesis and catalysis. The aim of this short review article is to outline general trends around discrete, well-defined allyl-lanthanide complexes1 in terms of compounds diversity, synthetic procedures and main structural and reactivity features.2 Brief comments on the catalytic performances of some key compounds in polymerization processes are also provided.
2 Allyl-lanthanide complexes: simple homoleptic and pseudo-homoleptic neutral, “ate” and cationic complexes
2.1 Synthesis, reactivity and structural features of unsubstituted allyl complexes
The synthesis of homoleptic and pseudo-homoleptic neutral tris(allyl)-lanthanide, anionic “ate” tetrakis(allyl)-lanthanide and cationic allyl-lanthanide complexes was essentially stimulated from the very beginning by the valuable performance of some mixed rare earth-based systems in the stereoselective polymerization of conjugated dienes and the assumed intermediacy of allyl-lanthanide species in those processes (vide infra, section 2.1). Early works focused accordingly on the isolation of putative active species or on the synthesis of model complexes.
The first reports on homoleptic “ate” tetra(allyl)-lanthanide complexes appeared in the 1980s. Mazzei and Lugli prepared [Li(1,4-dioxane)n][Ln(allyl)4] (Ln = Ce, Pr, Nd, Sm, Gd, Dy; n = 2) complexes from the reaction of LnCl3 and 4 equiv. of allyllithium (Scheme 1) [2,3]. Those compounds were characterized spectroscopically, in particular by variable temperature NMR studies that evidenced the dynamic behavior of these species in solution. At the same time, but using five equiv. of allylithium, Wu et al. reported the synthesis of [Li2(μ-C3H5)(1,4-dioxane)n][Ln(η3-C3H5)4] complexes (Ln = Ce, Nd, Sm, Gd, Dy, Ho, Er; n = 1, 3), of which the cerium congener constituted the first structurally characterized example of this type of anionic allyl-lanthanide complexes [4]. Using an analogous synthetic procedure involving the in situ preparation of the allyllithium reagent from tetraallyltin and n-butyllithium, Taube and Schumann prepared [Li(1,4-dioxane)1.5][La(η3-C3H5)4] (Scheme 1), which was also characterized in the solid state by single-crystal X-ray diffraction studies [5]. The structure of this compound features a pair of discrete ions, with the La center in the anion coordinated in a strongly distorted tetrahedral environment (range of C–La–C bond angles involving the central C atom of each allyl group: 78.7–133.4°) by four η3-bonded allyl groups (range of La–C bond distances involving all C atoms of the allyl groups: 2.722(9)–2.84(1) Å).
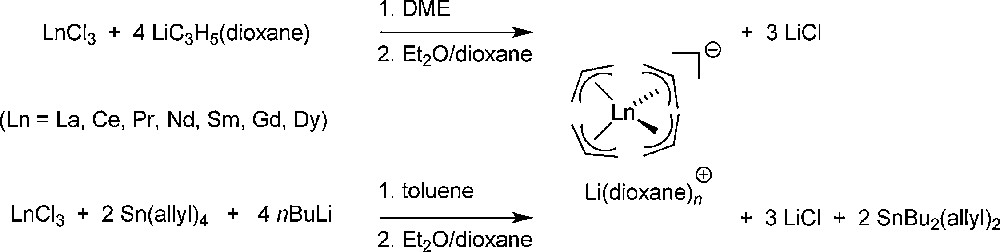
Preparation of [Li(1,4-dioxane)n][Ln(allyl)4] “ate” complexes [2,3,5].
A significant breakthrough in this chemistry came from the group of Taube who disclosed an effective entry toward homoleptic neutral tris(allyl)-lanthanide complexes. A first route involves allyllithium abstraction of [Li(1,4-dioxane)n][Ln(η3-C3H5)4] (Ln = La or Nd) with BEt3 as the Lewis acidic acceptor, in dioxane, affording [Ln(η3-C3H5)3.x dioxane]n (Ln = La, x = 1.5; Ln = Nd, x = 1) in 60–70% yield (Scheme 2) [6]. As revealed by NMR and X-ray crystal structure analyses, in both complexes, the three allyl anions are η3-coordinated. In the solid state, the lanthanum compound features a dimeric structure, [{La-(η3-C3H5)3(η1-1,4-dioxane)}2(μ-1,4-dioxane)], due to the bridging coordination mode of one dioxane molecule, while the same phenomenon induced a polymeric structure for the neodymium complex, [Nd(η3-C3H5)3(μ-1,4-dioxane)]∞.
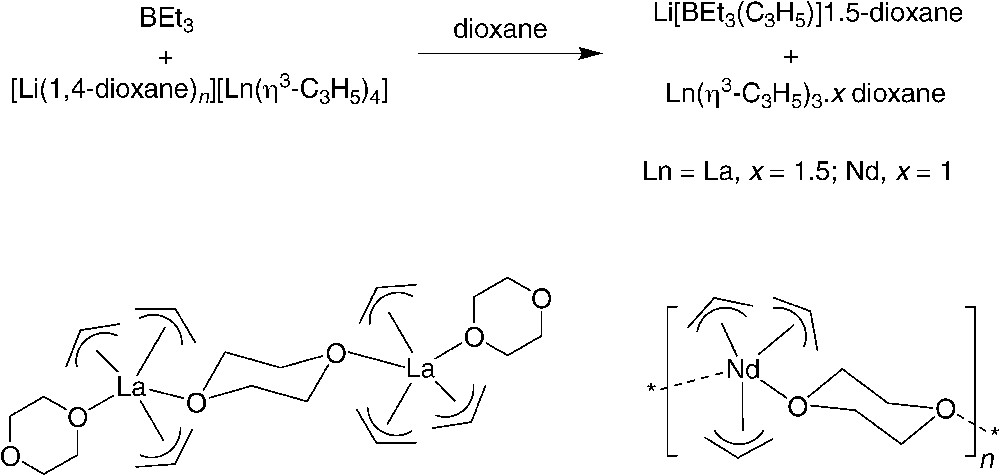
Synthesis and structure of homoleptic neutral [Ln(η3-C3H5)3.x dioxane]n complexes [6].
The dioxane molecules can easily be removed from these complexes at 50 °C without further decomposition to give the corresponding unsolvated tris(allyl) complexes Ln(η3-C3H5)3 (Ln = La or Nd) (Scheme 3) [6]. In the presence of THF, DME or dioxane, they form the corresponding solvates again. This series of neutral tris(allyl)s, as well as the anionic allyl-lanthanum complexes, has been studied by 139La NMR spectroscopy. Typical ranges of the chemical shifts were found for the different types of complex independent of the number and the nature kind of organic groups directly bonded to lanthanum [7].

Synthesis of unsolvated homoleptic Ln(η3-C3H5)3 complexes [6,8].
Another straightforward preparation of the dioxane adduct of tris(allyl)neodymium involves the reaction of NdI3(THF)3.5 with three equiv. of C3H5MgBr (or allylmagnesium iodide) in THF, followed by recrystallization from dioxane. This procedure affords the halide-free product in more than 70% yield [8]. Vacuum drying further affords the unsolvated Nd(η3-C3H5)3 (Scheme 3).
The dimeric molecular structure of (μ-dioxane)[La-(η3-C3H5)3(dioxane)]2 can be broken up into monomeric units by addition of hexamethylphosphoramide (HMPA) or chelating ligands such as 1,2-dimethoxyethane (DME) or tetramethylethyenediamine (TMEDA). The mononuclear nature of La(η3-C3H5)3(TMEDA) was confirmed by a single-crystal X-ray structure analysis [9].
The solvated tris(allyl)lanthanide complexes serve as a source toward neutral bis- and mono(allyl)-lanthanide halides, either by partial protolysis with trimethylammonium halides as the Brønsted acid or by comproportionation with the lanthanide trichloride in the corresponding 2:1 and 1:2 molar ratio, respectively (Scheme 4) [10,11]. The structures of the bis(allyl)-lanthanide chloride complexes [{Ln(η3-C3H5)2Cl(THF)2}2] (Ln = La, Nd) were determined to be dimeric in the solid state, via μ-bridging chloride ligands.
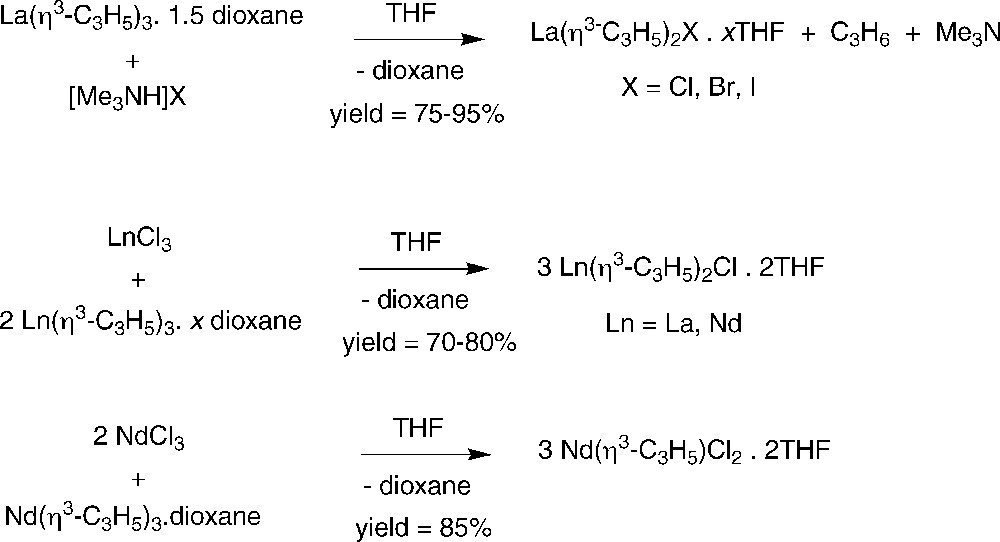
Synthesis of neutral bis- and mono(allyl)-lanthanide halides [10,11].
The group of Taube further demonstrated that these solvate tris(allyl)lanthanide complexes are also valuable precursors for the synthesis of cationic bis(allyl)-lanthanide derivatives [Ln(η3-C3H5)2L4]X (X = B(C6H5)4, B(C6F5)4). The preparation involves partial protolysis with trimethylammonium salts of the tetrakis(phenyl)- or tetrakis(pentafluorophenyl)borate anion as “non”-coordinating counteranion, in proper donor solvents (L) (Scheme 5) [8,12]. A set of such compounds was fully characterized and single-crystal structures were obtained for the lanthanum complexes. The latter compounds exist in the solid state as well-separated ion pairs, with the lanthanum center in the cation coordinated by two η3-allyl groups and four η1-THF ligands.
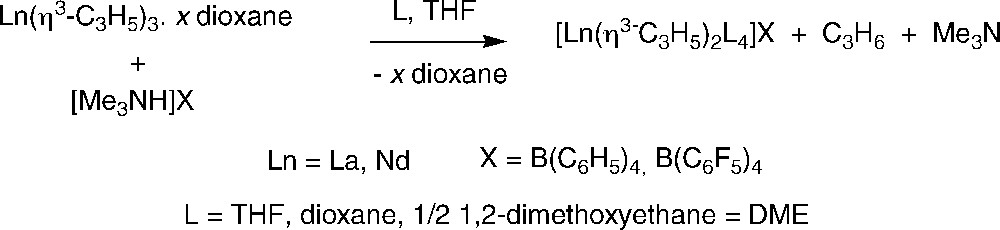
Synthesis of cationic bis(allyl)-lanthanide derivatives [Ln(η3-C3H5)2L4]X [12].
Very recently, Okuda et al. reported the synthesis and comprehensive structural characterization of a series of mono- and dicationic allyl complexes [13]. Monocationic bis(allyl) complexes [Ln(η3-C3H5)2(THF)3][B(C6X5)4] (Ln = Y, La, Nd; X = H, F) and dicationic mono(allyl) complexes of yttrium and the early lanthanides [Ln(η3-C3H5)(thf)6][BPh4]2 (Ln = La, Nd) were prepared by protonolysis of the trisallyl complexes [Ln(η3-C3H5)3(dioxane)] (Ln = Y, La, Ce, Pr, Nd, Sm), which were isolated and structurally characterized as a 1,4-dioxane bridged dimer (Ln = Ce) or THF adducts [Ln(η3-C3H5)3(THF)2] (Ln = Ce, Pr) (Scheme 6). Allyl abstraction from the neutral tris(allyl) complex by a Lewis acid, ER3 (Al(CH2SiMe3)3, BPh3) gave the ion pair [Ln(η3-C3H5)2(THF)3][ER3(η1-CH2CH = CH2)] (Ln = Y, La). Also, it was found that benzophenone inserts into the La–C(allyl) bond of [La(η3-C3H5)2(THF)3][BPh4] to form the alkoxy complex [La{OCPh2(CH2CH = CH2)}2(THF)3][BPh4].
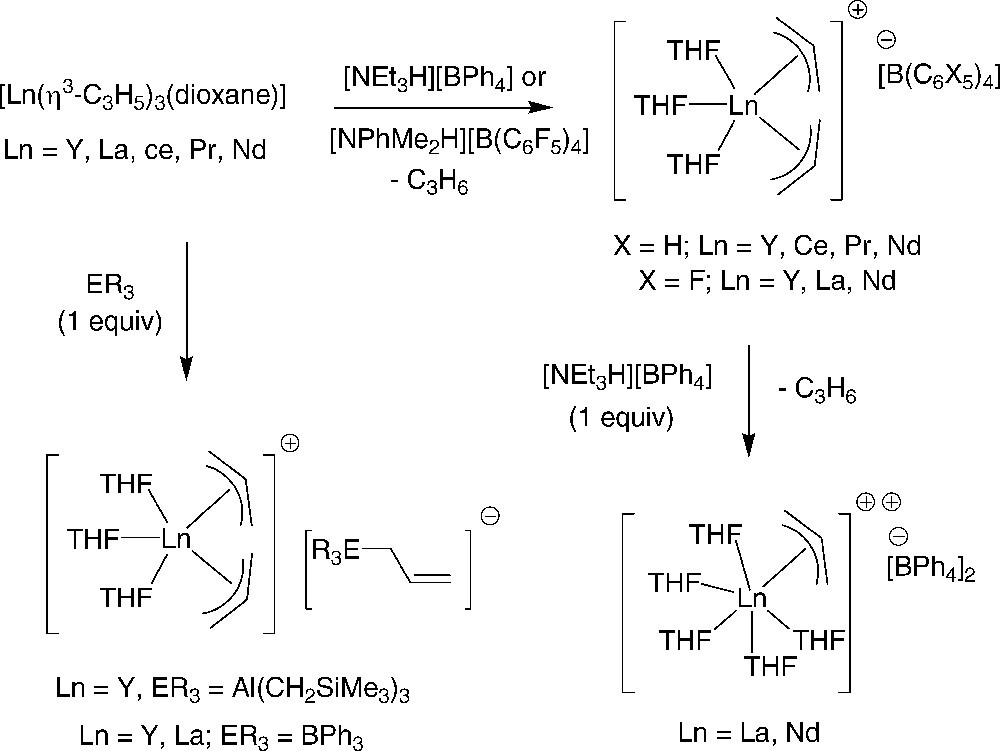
Synthesis of monocationic bis(allyl)- and dicationic mono(allyl)-lanthanide derivatives [13].
Another interesting contribution, considering the possible relevance of the prepared complexes to Ziegler-Natta polymerization catalysis, came from Wu et al. [14]. They prepared a series of mixed halide-allyl-lanthanide-magnesium complexes, (η3-C3H5)2LnX5Mg2(Et2O) and (η3-C3H5)2LnX5Mg2(TMEDA) (Ln = La, Ce, Pr, Nd, Sm; X = Cl, Br), by the reaction of anhydrous LnC13 with allyl Grignard reagents in diethylether, or in THF in the presence of TMEDA (Scheme 7). The preparation of such complexes was further extended to Y, La and Nd derivatives with THF ligands [15]. Because in some cases, lanthanide trischlorides and allyl-magnesium bromide were used, compounds having both Cl and Br were obtained, making X-ray diffraction studies somewhat complicated [14]. The determination of the solid-state structure of three of such compounds established that they all have triangular metallic (i.e., two Mg and one Ce or Nd) skeletons bonded together by three bridging and two capping halide atoms. The two allyl groups are η3-bonded to the lanthanide ions. The quite high decomposition temperatures (higher than 160 °C) of these complexes indicated the stabilities of such heteropolymetallic structures.
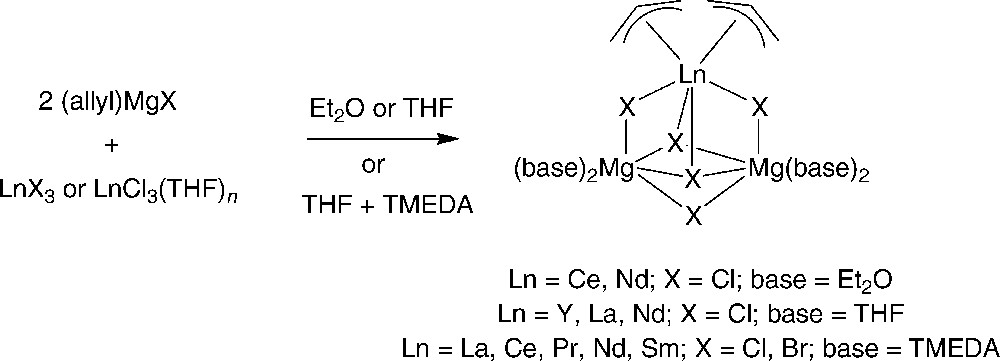
Synthesis of mixed halide-allyl-lanthanide-magnesium complexes (η3-C3H5)2LnX5Mg2(base) [14,15].
Bochmann et al. showed that the reaction of LnI3(THF)n (Ln = La, n = 4; Ln = Y, n = 3.5) with three equiv. of allylMgI in THF/1,4-dioxane proceeds in moderate yields to give the unexpected mixed-metal complexes [Ln(η3-C3H5)3(μ-dioxane).Mg(η1-C3H5)2(μ-dioxane)1.5]∞. The solid-state structures of these compounds contain distorted trigonal-bipyramidal lanthanide centers with three η3-allyl ligands and distorted trigonal-bipyramidal magnesium centers with two η1-bonded allyl groups. Both metal centers are connected by bridging 1,4-dioxane molecules building a planar polymeric network (Scheme 8) [16].
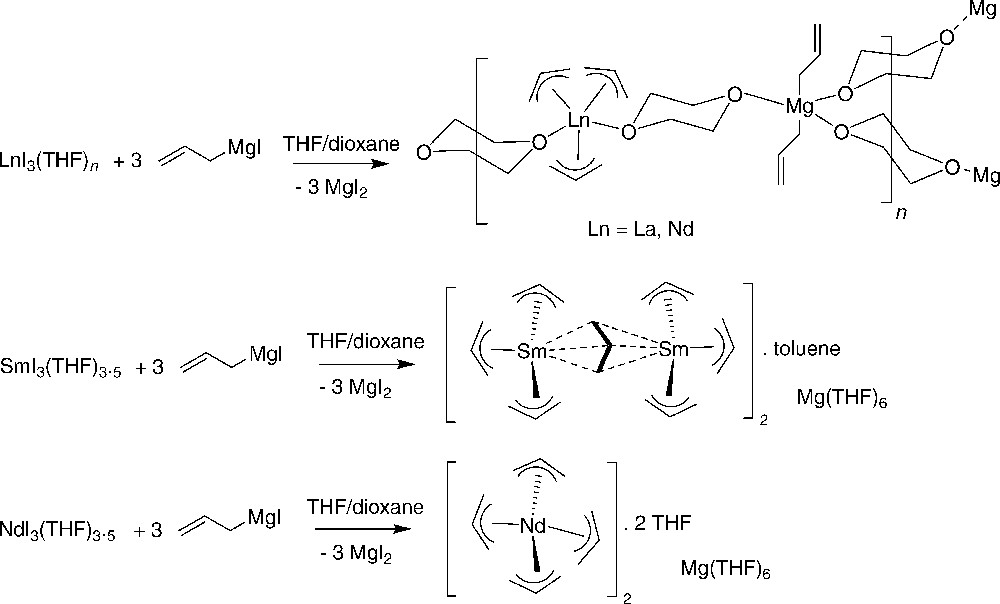
Preparation of mixed-metal complexes [Ln(η3-C3H5)3(μ-dioxane).Mg(η1-C3H5)2(μ-dioxane)1.5]∞ complexes [16].
In contrast, samarium(III) iodide gives a salt containing a new allyl-bridged anion, [Mg(THF)6][Sm2(η3-C3H5)6(μ-η3: η2-C3H5)]2.toluene, while neodymium(III) iodide affords [Mg(THF)6][Nd(η3-C3H5)4]2.2THF under the same reaction conditions (Scheme 8) [14]. The solid-state structure of the samarium compound consists of well-defined discrete ions, with an unprecedented type of binuclear anion containing two samarium centers bridged by a η3: η3-allyl ligand.
2.2 Catalytic properties of trivalent homoleptic and pseudo homoleptic allyl-lanthanide complexes in stereospecific diene polymerization
A significant part of synthetic rubber is currently produced by homogeneous catalytic liquid phase stereospecific polymerization of 1,3-dienes (typically butadiene and isoprene) to 1,4-cis-polydienes using Ziegler–Natta mixed catalysts based on neodymium precursors, Lewis acidic co-catalysts such as AlEt2Cl and possible other promoters (Scheme 9) [17]. The nature of the active rare-earth-metal species generated from these (binary, ternary and quaternary) systems has been controversial since the early discoveries of their superior performance in these polymerization processes. As mentioned in the introduction, the synthesis and isolation of the putative active species have been a driving force for the preparation of well-defined allyl-lanthanide compounds. The aim of this short section is to give a brief overview of the catalytic performance that the aforementioned different classes of simple allyl-lanthanide compounds feature in stereospecific polymerization of dienes. For more comprehensive studies and detailed comments on mechanistic issues, the readers shall refer to some leading articles [12,17–19].
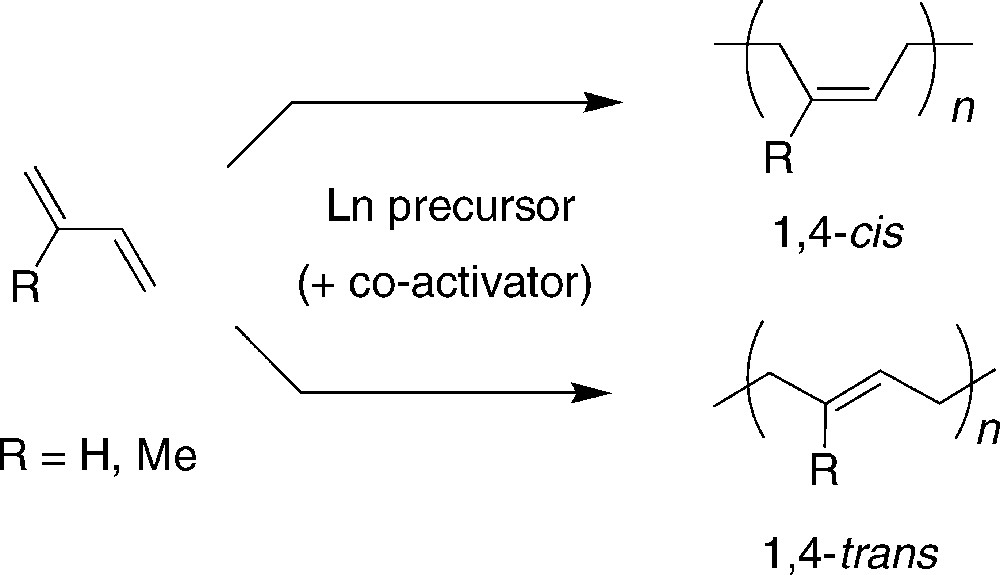
Stereospecific polymerization of 1,3-dienes promoted by Ziegler-Natta mixed catalysts.
Anionic and neutral allyl-lanthanide complexes with or without different donor ligands are active homogeneous catalysts for the polymerization of butadiene (BD) (and isoprene) in solution. Usually, these complexes catalyze the trans-polymerization of butadiene, while the addition of Lewis-acidic aluminum organic compounds results in a change of the selectivity to cis-polybutadiene. Neodymium-based compounds most often lead to the highest stereoselectivities, as compared to other rare-earths, regardless of the ionic radius (lanthanum, yttrium….). For instance, La(η3-allyl)3.1.5(dioxane), Nd(η3-allyl)3.(dioxane) and Nd(η3-allyl)3 polymerize butadiene in toluene at 50 °C with turnovers frequencies (TOFs, in mol(BD).mol(Ln)−1.h−1) of 500, 600, and 1,300, and trans selectivities of 83, 94 and 89%, respectively [12]. Obviously, donor ligands act as competitors/inhibitors in the catalytic reaction and also diminish the cis selectivity.
Once the tris(allyl)lanthanides are combined with a Lewis acid like AlEt2Cl or MAO (methylalumoxane) (2–30 equiv. vs Ln), a significant increase in activity and a dramatic reversal in stereoselectivity, to the benefit of the desired 1,4-cis polymer, are observed. Thus, La(η3-allyl)3.1.5(dioxane) and Nd(η3-allyl)3.(dioxane) combined with 30 equiv. of MAO give TOFs of 4700 and 14,000 h−1 and cis selectivities of 65 and 79%, respectively. Using the bis and mono(allyl)neodymium chloride precursors Nd(η3-allyl)2Cl.1.5(THF) and Nd(η3-allyl)Cl2.2(THF), still combined with 30 equiv. of MAO, afforded even better performances; i.e., TOFs of 575,000 and 570,000 h−1 and cis selectivities of 98 and 98%, respectively [12]. Very high performances were also reached with bimetallic precursors of the type (η3-allyl)2NdCl5Mg2(THF) combined with MAO [15]. These results strongly suggested that the active species in the stereoselective polymerization of dienes is a dicationic mono(allyl)-lanthanide fragment, probably stabilized by coordinative interactions with donor atoms of the activator (e.g., heterobridging chloroaluminate ligands) [20].
The cationic bis(allyl)-lanthanide complexes developed by Taube et al. are single-component active catalysts, i.e., they do not require any coactivator that can perform a high cis selectivity. Thus, [Nd(η3-allyl)2(dioxane)4][B(C6F5)4] feature, under the same conditions as above, TOF up to 11,200 h−1 and a cis selectivity higher than 90% [12]. On the other hand, no control of the stereoselectivity was observed with the THF complexes [Ln(η3-allyl)2(THF)4][B(C6F5)4] (Ln = La, Nd) (cis selectivity = 27–36% and 42–45%, respectively). In addition, comparison of theoretical and experimental molecular weights established that all the lanthanide centers in those compounds are active, and that two polymer chains are formed per lanthanum while a single chain is formed at the neodymium. The authors interpreted those results as a corroboration that the cis selectivity in the allyl-lanthanide-catalyzed diene polymerization is structurally determined by the formation and reactivity of a dicationic mono(allyl)-lanthanide species [12]. However, the reaction pathway leading to the latter dicationic mono(allyl) species from the cationic di(allyl) species (in the absence of other activator) was not addressed.
Controlled 1,3-butadiene polymerization was observed using the cationic allyl complexes prepared recently by Okuda et al. [13] (Scheme 6), giving polybutadiene with ca. 90% 1,4-cis stereoselectivity. The results further corroborated the assumption of a dicationic rare-earth-metal center in [Ln(η3-C3H5)(s-cis-C4H6)2]2+ as the active site of insertion. Somewhat unexpectedly, the yttrium catalysts were found to be more active than their homologues based on the larger lanthanum and neodymium centers. Narrower molecular weight distributions were achieved by the less active lanthanum and neodymium catalysts.
2.3 Homoleptic and pseudo-homoleptic neutral lanthanide complexes bearing silyl-substituted allyl ligands
Studies on the coordination of silyl-substituted allyl groups, typically 1,3-bis(trimethylsilyl) allyl, onto lanthanide metal centers arose from two main expectations: First, it is known that the application of such ligand imparts additional thermal stability relative to the parent transition metals complexes. Furthermore, the confirmed ability of 1,3-bis(trimethylsilyl) allyl to adopt a σ geometry is in line with the potential for σ/π interconversion of the parent allyl moiety to open up coordination sites for catalytic reactions.
The group of Bochmann intensively investigated the preparation of lanthanide complexes supported by allyl ligands having trisalkylsilyl substituents at diverse positions. As for their unsubstituted analogues, those complexes were usually prepared by direct salt metathesis reactions of the adequate allyl anion (as the potassium salt) with trishalide precursors LnX3(THF)n or LnX3 in THF. The nature of the final compound(s) recovered in these reactions varied considerably according to the nature of the lanthanide precursor. Thus, the reaction of trischloride group 3 metal precursors (Ln = Sc, Y, La) with two equiv. of the bulky 1,3-bis(trimethylsilyl)allyl anion, [1,3-C3H3(SiMe3)2]−, yielded the corresponding bis(allyl) compounds (Scheme 9). “Ate” or neutral mononuclear complexes, with a variable number of THF donor ligands, were obtained according to the nature of the metal center [21]. A similar reaction with NdI3(THF)3.5 yielded a mixture of the mono(allyl) and bis(allyl) iodide complexes (Scheme 10) [22]. The latter complex adopts in the solid state a distorted trigonal-bipyramidal structure with the THF ligands in trans position.
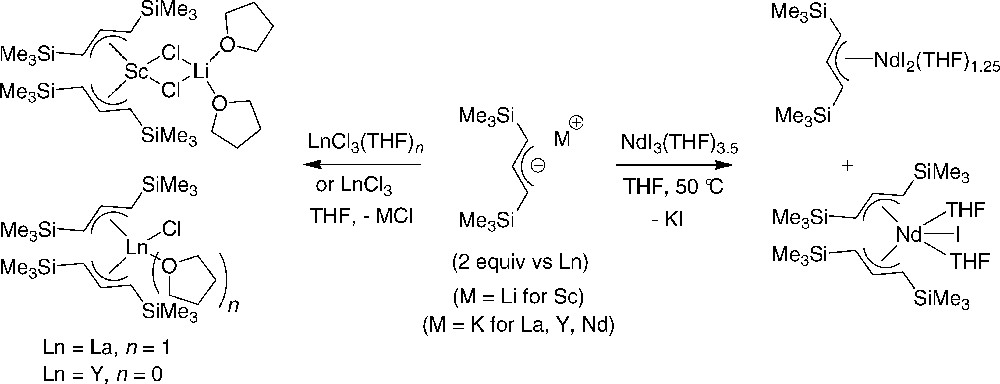
Synthesis of silylated allyl-lanthanide-halide complexes [21,22].
The analogous reaction of [1,3-C3H3(SiMe3)2]− with SmI2(THF)2 proceeded to give a dark green solution from which a compound of composition KSm(C3H3(SiMe3)2)3(THF)2 was isolated. Single-crystal X-ray diffraction showed that this compound is the allyl-bridged dimer [Sm(η3-1,3-C3H3(SiMe3)2)3{μ-K(THF)2}]2 (the first structurally authenticated Sm(II) allyl complex), which possesses a cyclic structure, comprising two samarium and two potassium ions bridged by four η3-allyl ligands (Scheme 11) [23]. Interestingly, the addition of THF-d8 to a toluene solution of this compound had very little effect on the NMR spectra, suggesting that the cyclic structure is maintained in solution. [Sm(η3-1,3-C3H3(SiMe3)2)3{μ-K(THF)2}]2 is an highly effective single-component catalyst for the polymerization of methyl methacrylate (TOFs > 80,000 mol(MMA).mol(Ln)−1 h−1 at 0 °C) and of ɛ-caprolactone (TOFs > 58,000 h−1 at 20 °C) (the bis(allyl) iodide complex [Nd(η3-1,3-C3H3(SiMe3)2)2I(THF)2] is also highly active in this process), producing high molecular weights polymers with relatively narrow molecular weight distributions.
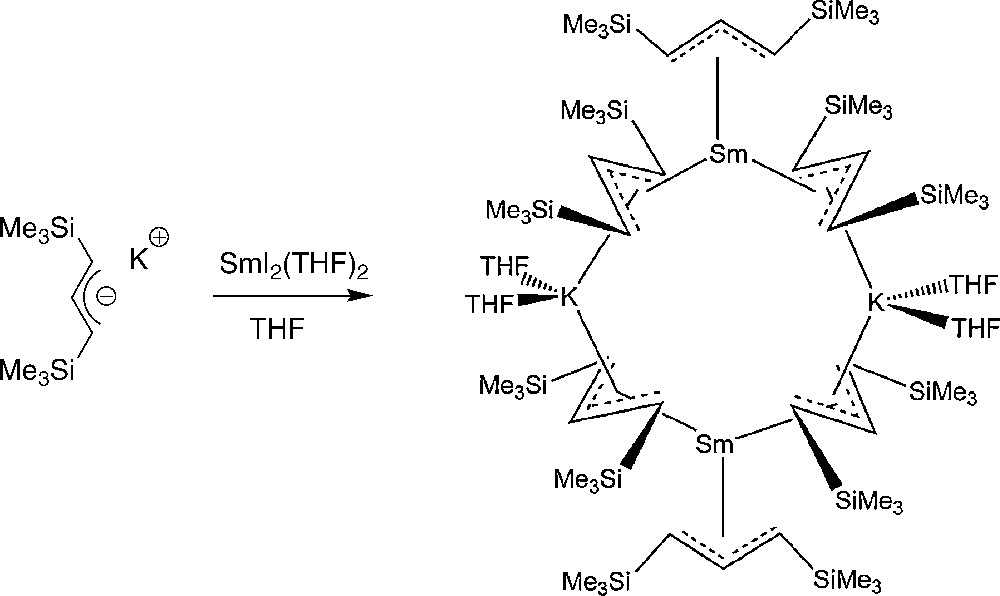
Synthesis of [Sm(η3-1,3-C3H3(SiMe3)2)3{μ-K(THF)2}]2 [23].
Note that earlier, Ihara et al. reported samarium(II) complexes formulated as (1,3-C3H3R2)2Sm(THF)2 (R = Ph, SiMe3), obtained by reaction of K[1,3-C3H3R2] with SmI2(THF)2 [24]. However, these compounds were not fully characterized and did not show the same catalytic reactivity towards methyl methacrylate than the aforementioned Sm-K-allyl complex.
All attempts to obtain halide-free products and generate neutral homoleptic tris(allyl) complexes of the type Ln{1,3-C3H3(SiMe3)2}3 from LnX3 failed (e.g., Ln = La, Y; X = Cl, [23]). For instance, Kuehl et al. isolated allyl-lanthanide salts of the type [K(THF)4][{η3-1,3-C3H3(SiMe3)2}3LnI] (Ln = Ce, Pr, Nd, Gd, Tb, Dy, Er) by reaction of three equiv. of K{1,3-C3H3(SiMe3)2} with LnI3 in THF [25]. Similarly, when lanthanide trisiodides and the lithium allyl Li{1,3-C3H3(SiMe3)2} were combined, the corresponding lanthanate species [Li(THF)4][{η3-1,3-C3H3(SiMe3)2}3LnI] were formed [26]. The former cerium(III), terbium(III) and erbium(III) congeners were characterized by X-ray diffraction studies and found to be isostructural. In these three complexes, all three allyl ligands are bonded to the lanthanide center in a trihapto, slightly asymmetric fashion, with the SiMe3 groups from all three allyl ligands in a syn,syn configuration. These authors also observed that the reaction of four equiv. of K{1,3-C3H3(SiMe3)2} with LnI3 does not lead to [K(THF)X][{1,3-C3H3(SiMe3)2}4Ln] (which could have been precursors toward homoleptic neutral tris(allyl) complex by allyl lithium abstraction following Taube's procedure [6], vide supra), but only [K(THF)4][{1,3-C3H3(SiMe3)2}3LnI] and excess K{1,3-C3H3(SiMe3)2} were obtained.
Successful preparation of neutral homoleptic complexes bearing silylated-allyl ligands was achieved using triflate salts. Hence, a series of complexes {1,3-C3H3(SiMe3)2}3Ln(THF)n could be isolated in moderate yields from the reaction of rigorously anhydrous Ln(O3SCF3)3 with three equiv. of the 1,3-bis(trimethylsilyl)allyl anion (Scheme 12) [25,26].
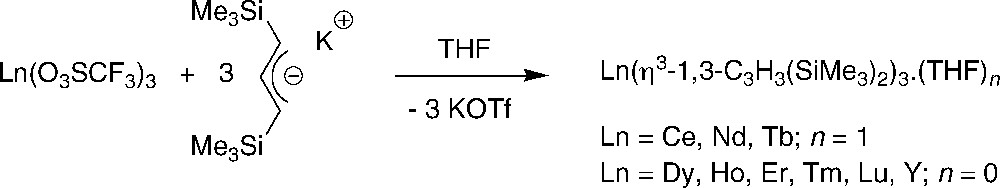
Synthesis of neutral homoleptic complexes {1,3-C3H3(SiMe3)2}3Ln(THF)n [25,26].
Thanks to a significant number of crystal structures obtained in the studies mentioned above, trends in the bonding of lanthanide allyl complexes with the 1,3-bis(trimethylsilylated)allyl ligand could be explored and compared with those of cyclopentadienyl lanthanide complexes [26]. It was concluded that despite the geometrically irregular shape of the substituted allyl anion, metal–ligand distances in monomeric lanthanide allyl complexes appear to be reasonably predictable based on metal radius and oxidation state. This is an expected consequence of a high degree of ionic character in the bonding.
Traces of coordinated water in holmium triflate proved to have a major influence in the fate of reactions with the 1,3-bis(trimethylsilyl)allyl anion (Scheme 13); this eventually leads to the unexpected generation of dimethylsilylene and allylidene holmium complexes [27]. It was found that, even long vacuum-dried, Ho(O3SCF3)3 may retain up to ca. 3% of water that induces the formation of a complex isolated in 74% yield, which is quite different from the expected homoleptic tris(allyl) compound. An X-ray diffraction study revealed a dimer in which one η3-coordinated allyl ligand is bound to each metal, but hydrogen abstraction from a trimethylsilyl group had occurred on a second allyl ligand associated with each metal center, forming dimethylsilylene units that bridge the holmium atoms (Scheme 13). When the reaction that produced the dimeric dimethylsilylene complex was prolonged for more than 20 h before workup, a second yellow-orange dinuclear product was isolated. The X-ray structure analysis revealed that, in addition to two dimethylsilylene bridges, the metal centers are joined with a μ–η1,η3-allylidene ligand (Scheme 13). This study clearly showed that trimethylsilyl groups on allyl ligands cannot always be assumed to be completely unreactive in f-element complexes.
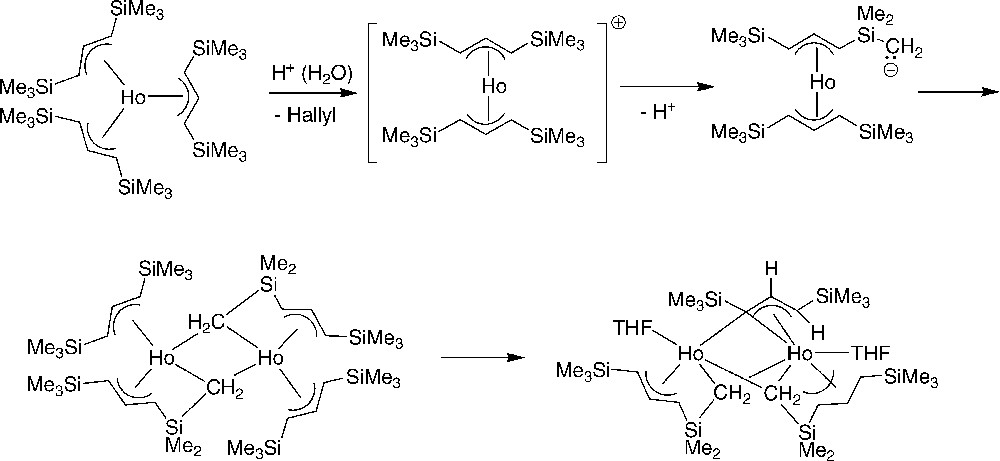
Decomposition pathway of the tris(silylated allyl)holmium complex [27].
Salt metathesis of silylated allyl anions with YbI2 afforded the corresponding homoleptic bis(allyl)-ytterbium complexes as THF solvates. These were subsequently reacted with a series of bulky terpyridines to determine the effect of allyl ligands on the internal charge-transfer process that exists in these terpyridyl-bis(allyl) complexes (Scheme 14) [28]. Single-crystal X-ray characterization of [η3-1,3-C3H3(SiMe3)2]2Yb(tpy) revealed that the allyl moiety possesses shorter Yb–C and Yb–N bond lengths than the Cp* analogue. The magnetic susceptibility data for [η3-1,3-C3H3(SiMe3)2]2Yb(tpy) departs dramatically from the Curie law, with a room-temperature magnetic moment of 2.95 μB.
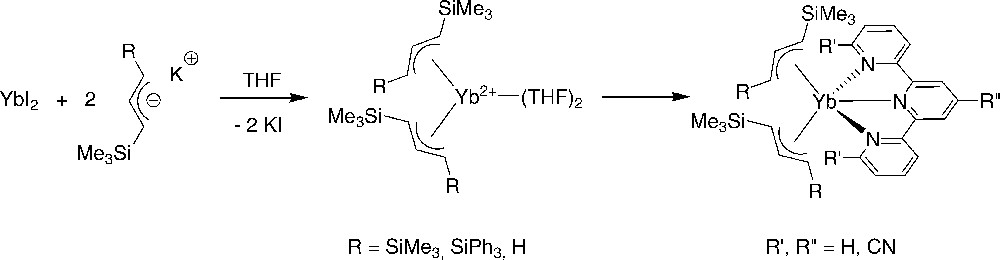
Synthesis of terpyridyl-bis(silylated-allyl)ytterbium(II) complexes [28].
In contrast to mono(allyl) ligands, the ansa-bis(allyl) ligand {Me2Si(CHCHCHSiMe3)2}2− readily gives halide-free compounds. Thus, the reactions of lanthanide trishalides with an equimolar amount of the dipotassium salt K2{3-(C3H3-SiMe3-1)2SiMe2} in THF lead to the formation of the ansa-bis(allyl)-lanthanide “ate” complexes [Ln{(3-(η3-1-C3H3SiMe3)2SiMe2}2-{μ-K(THF)}·(THF)n]∞ (Ln = La, n = 0.5; Sc, Y, n = 1) (Scheme 15) [29]. These compounds are highly effective single-component catalysts for the polymerization of methyl methacrylate (TOFs > 80,000 mol(MMA).mol(Ln)−1 h−1 at 0 °C), producing high molecular weights polymers with relatively narrow molecular weight distributions.
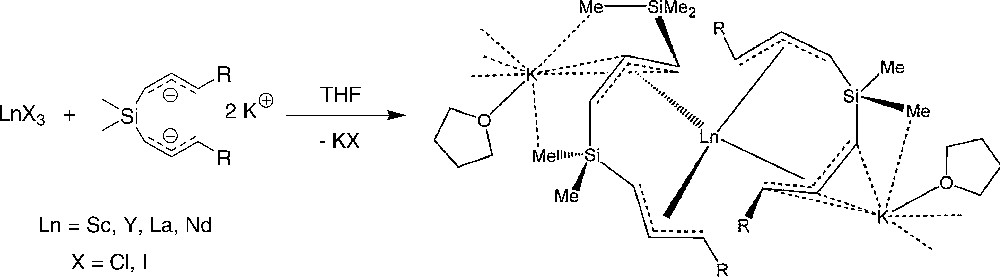
Synthesis of ansa-bis(allyl)-lanthanide “ate” complexes [Ln{3-(η3-1-C3H3SiMe3)2SiMe2}2{μ-K(THF)}·(THF)n]∞ [29].
An X-ray diffraction study on the lanthanum complex revealed that each lanthanum atom is coordinated to two ansa-bis(allyl) ligands, both of which bind to the metal in a η3 fashion. The coordination environment of the lanthanum consists of 12 carbon atoms, with La–C bond lengths (2.769(3)–2.902(3) Å) similar to those observed in the related unsubstituted lanthanum complex [Li(1,4-dioxane)1.5][La(η3-C3H5)4] (2.721–2.842 Å) [5] and slightly larger than those observed in the neutral complex [{La-(η3-C3H5)3(η1-1,4-dioxane)}2(μ-1,4-dioxane)] (2.751(3) to 2.829(5) Å) [6]. The allyl ligands adopt an anti,syn arrangement and feature bond lengths very similar to the aforementioned unsubstituted allyl compounds. A potassium cation, that balances the charge of the tetra(allyl)-lanthanide “ate” moiety, is coordinated to two allyl moieties of two different lanthanum atoms, resulting in the formation of a coordination polymer with infinite chain structure. This potassium ion binds one THF ligand and features as well complex bonding interactions with the allyl ligands and close K…CH3Si contacts.
Although X-ray quality crystals for the analogous Sc and Y complexes were not obtained, all other methods of analysis suggested they have a structure closely similar to that of the lanthanum compound [29]. Also, similar reaction of K2{3-(1-C3H3SiMe3)2SiMe2} with NdI3(THF)3.5 and subsequent recrystallization from toluene afforded the polymeric complex [Nd{η3-C3H3SiMe3)2SiMe2}2-{μ-K(THF)}(THF)(toluene)]∞, which can be transformed upon drying under vacuum into the solvent-free coordination polymer [Nd{3-(η3-1-C3H3SiMe3)2SiMe2}2-{μ-K}]∞.
On the other hand, treatment of SmI2(THF)2 with Li2{3-(1-C3H3SiMe3)2SiMe2}/KOtBu afforded, in small yield, the samarium(III) complex [Li(OEt2)4][Sm{3-(η3-1-C3H3SiMe3)2SiMe2}2]; this one shows up in the crystal structure as discrete ions, with no close interionic contacts (Scheme 16) [29]. Apparently, the lithium salt crystallized more easily and this change in the nature of the alkali cation accounts for the formation of a non-polymeric complex. The arrangement of the allyl ligands around the samarium atom ion in this compound is closely similar to that observed in the polymeric lanthanum complex, with the same anti,syn conformation.
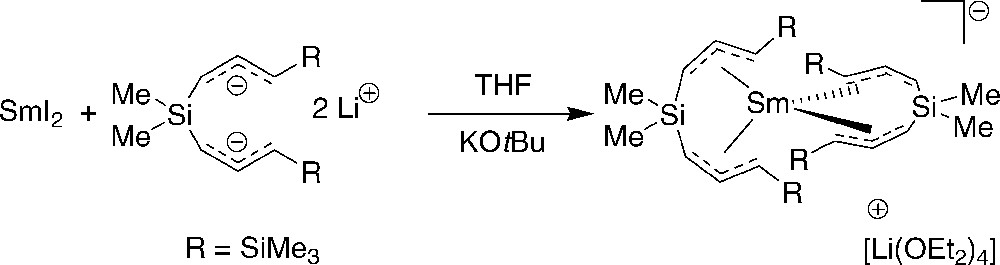
Formation of samarium(III) complex [Li(OEt2)4][Sm{3-(η3-1-C3H3SiMe3)2SiMe2}2] [29].
2.4 Towards allyl-lanthanidocenes
Installation of such ansa-bis(silylated allyl) ligands into lanthanidocenes proved to be difficult. The reaction of (Cp′ = 1,3-C5H3(SiMe3)2; Ln = Y or Sm) with K2{3-(1-C3H3SiMe3)2SiMe2} in THF leads, upon workup (crystallization from diethyl ether), to ligand redistribution, to give [Li(OEt2)(THF)3][Ln{3-(η3-1-C3H3SiMe3)2SiMe2}2] (Scheme 17) [22]. The latter yttrium compound was characterized by X-ray single crystal diffraction, which revealed a structure close to that observed for the abovementioned samarium(III) complex [Li(OEt2)4][Sm{3-(η3-1-C3H3SiMe3)2SiMe2}2], with the same anti,syn arrangement of the ansa-bis(allyl) ligands around the metal [29]. Ligand redistribution was also encountered in the reaction of with K2{3-(1-C3H3SiMe3)2SiPh2}. In this case, a potassium atom was retained as the counteranion, even in the presence of lithium cations, to afford the polymeric complex [Y{3-(η3-1-C3H3SiMe3)2SiPh2}2{μ-K}(THF)0.5(Et2O)1.5]∞ [22].
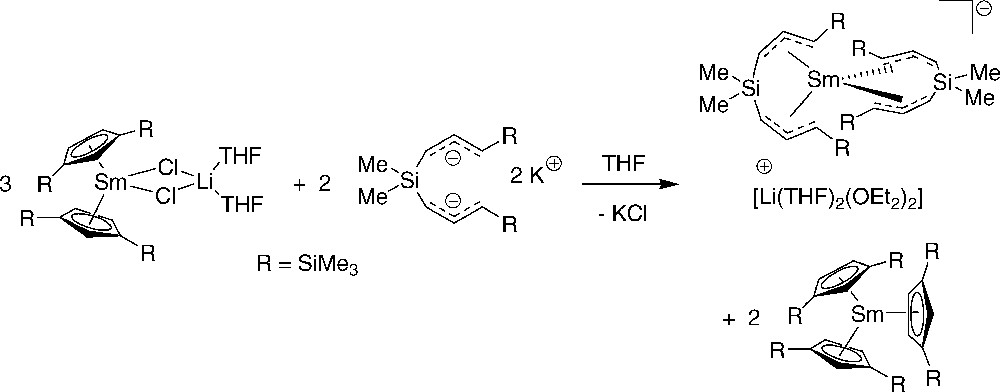
Ligand redistribution processes in reaction of chlorolanthanidocenes with bis(silylated allyl) dianion [22].
Only in the reaction of and K2{3-(1-C3H3SiMe3)2SiPh2}, a red crystalline solid was isolated, which was authenticated by NMR and single-crystal X-ray analysis as the heteroleptic mono-Cp complex [Cp′{{3-(η3-1-C3H3SiMe3)2SiMe2}2(μ-Cl)Li(THF)3}] (Scheme 18) [22]. The formation of this compound is an unusual example of the displacement of a cyclopentadienyl ligand while a chloride ligand has been retained, a phenomenon which was propose to arise from the steric congestion at the lanthanide center. Again, as in other ansa-allyl complexes, the allyl ligand in this heteroleptic compound adopts an anti,syn configuration, with bond lengths very similar to those observed in related samarium(III)-allyl complexes.
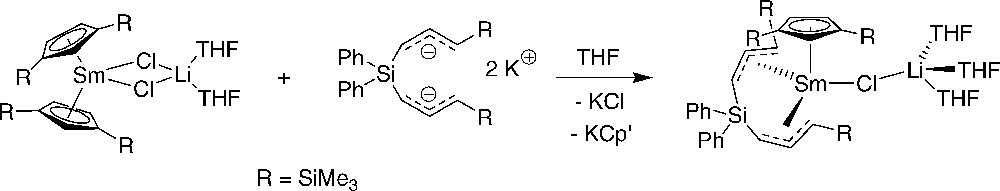
Preparation of a silylated bis(allyl)-samarocene complex [22].
3 Allyl-lanthanidocenes and cyclopentadienyl-supported allyl-lanthanides
3.1 Neutral and ate allyl-lanthanidocenes and half-sandwich complexes
Allyl-lanthanidocenes and related complexes supported by cyclopentadienyl ancillaries were actually the first allyl-lanthanide compounds reported. The field was pioneered by Tsutsui in 1975 who reported that reactions of Cp2LnCl complexes (Ln = Sm, Ho, Er) with allylmagnesium bromide in THF/ether solution at −78 °C, followed by workup with dioxane to precipitate MgCl2, afford the corresponding allyl-lanthanidocene derivatives, Cp2Ln(C3H5) [30] (Scheme 19). Although very sensitive to air and moisture, these compounds were found to be quite thermally stable, decomposing at temperatures above 200 °C. IR studies indicated a η3-coordination mode of the allyl moiety. The IR spectrum of the samarium(III)-allyl complex in THF showed the C–C stretch to be essentially unshifted, indicating that there is little tendency for the allyl group to undergo a σ–π rearrangement in the presence of a strongly coordinating solvent.
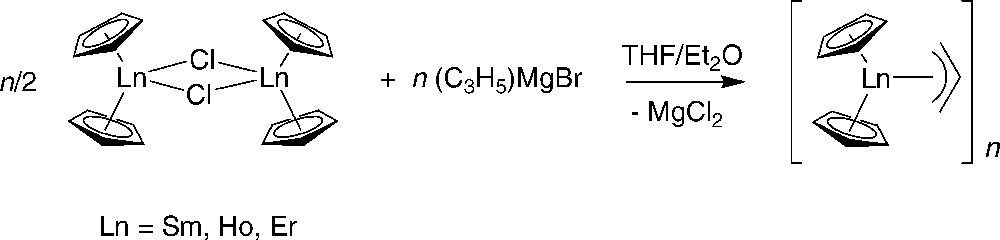
Synthesis of allyl-lanthanidocenes [30].
This methodology proved to be quite general and a variety of allyl-lanthanidocenes was prepared efficiently following similar transmetallation protocols.
For instance, Baudry et al. described the synthesis of neutral allyl complex (Cp′ = tBuC5H4) and ansa-“ate” allyl complexes [Me4C2(C5H4)2]Ln(C3H5)2Li(base) (Ln = Sm, Nd; base = DME, dioxane) from and the magnesium derivatives [Me4C2(C5H4)2]LnCl.MgCl2(THF)4, respectively (Scheme 20) [31,32]. The anionic ansa-samarocene complex exhibited moderate activity for the polymerization of isoprene (TOF = 260 mol(IP) mol(Ln)−1 h−1 at 50 °C), giving 1,4-trans-enriched (95%) polymers. The polymerization of an equimolar mixture of isoprene and 1-hexene with [Me4C2(C5H4)2]Ln(C3H5)2Li(DME) (Ln = Sm, Nd) as single component catalysts led to a co-polymer (1-hexene/isoprene = 1:10) essentially formed of 1,4-trans-polyisoprene blocks separated by only one inserted hexyl group [33]. It was proposed that these polymerization reactions proceed via prior dissociation of allyl-lithium [18], generating a neutral species “[Me4C2(C5H4)2]Ln(η3-C3H5)”3 having a vacant site for isoprene coordination and subsequent polymerization (Scheme 18); allyl-lithium cogenerated in this equilibrium would account for the imperfect 1,4-trans stereoselectivity, as this compound was shown to produce 1,4-cis-polyisoprene under those conditions.
Also, it was observed that [Me4C2(C5H4)2]Nd(C3H5)2Li(DME) is a moderately active catalyst for the polymerization of styrene, affording isotactic-enriched polystyrenes [32]. On the other hand, the neutral allyl derivative was inactive towards isoprene. The lack of reactivity of this compound was related to its associated structure: isoprene probably does not possess a sufficiently strong coordination ability [unlike, for example, the nucleophilic carbene C(NiPr)2(CMe)2] to dissociate the starting polymeric material.
Our group also reported the preparation via salt metathesis protocols of well-defined allyl-ansa-lanthanidocenes that proved especially useful for styrene polymerization. Neutral allyl complexes bearing ansa-bridged fluorenyl/cyclopentadienyl ligands [FluEMe2(3-R-Cp)]Ln(η3-C3H5)(THF) (E = C, R = H, Ln = Y, La, Nd, Sm; R = tBu, Ln = Y, Nd; E = Si, R = H, Ln = Y, Nd) were prepared in good yields (Scheme 21) [35]. The complexes were characterized by elemental analysis, NMR spectroscopy for diamagnetic complexes, and single-crystal X-ray diffraction studies for several members. The allyl fragment is coordinated in an η3-mode, though in a rather non-symmetric manner, as judged by the ca. 0.1–0.2 Å difference in the M–C(allyl) and also the C–C bonds in the allyl ligands distances. This peculiarity could arise from either severe steric congestion induced by the neighboring coordinated THF molecule and/or non-equal participation of the sp2-carbon atoms in bonding with the metal center. Considering a η3-coordination mode for the allyl fragment and fluorenyl ligand, those eight-coordinate allyl complexes are formal 16-electron species.
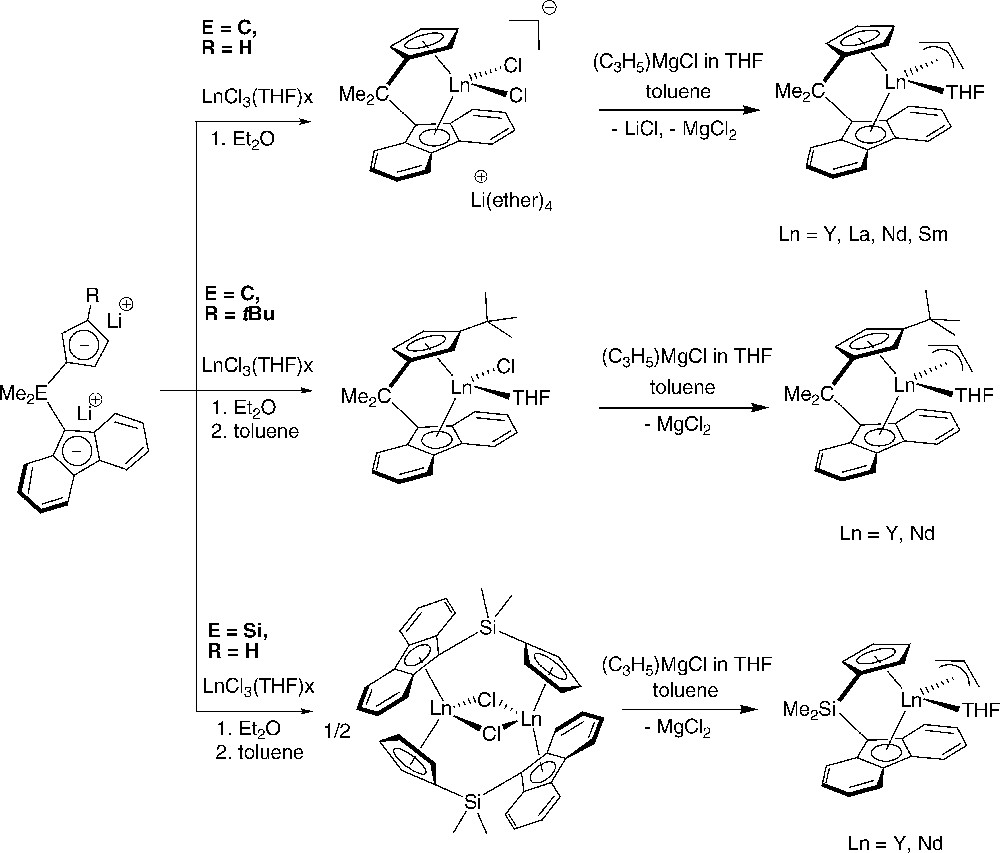
Synthesis of neutral allyl ansa-lanthanidocenes supported by fluorenyl-cyclopentadienyl ligands and used in syndiospecific homo and copolymerization of styrene [35].
Some of these allyl ansa-lanthanidocenes, especially {FluCMe2C5H4)}Nd(η3-C3H5)(THF), are effective single-component (i.e., they do not require any activator) catalysts for the polymerization of styrene, giving pure syndiotactic polystyrenes (rrrr > 99% on the crude unfractionated polymers) with high molecular weights and narrow polydispersities [35].4 Note that the corresponding alkyl and hydrido complexes [{FluCMe2C5H4)}Nd(R)(THF)]n (R = CH2SiMe3, CH(SiMe3)2, n = 1; R = H, n = 2) are inactive toward styrene [37]. The allyl-lanthanidocene catalysts are remarkably stable, capable of polymerizing styrene up to 120 °C, while maintaining high activities and high syndiotacticity. Also, highly effective copolymerization of styrene with ethylene was achieved (activity up to 2530 kg(PS–PE) mol−1 h−1) to give, for styrene-rich true copolymers void of homopolymers with high molecular weights and narrow polydispersities. These P(S-co-E) copolymers have a microstructure predominantly made of long highly syndiotactic PS sequences separated by single or few ethylene units. The mechanism of the initiation step of styrene polymerization promoted by those single-component allyl ansa-lanthanidocene catalysts has been studied from a combined experimental/computational (DFT) perspective [38]. Key results include that (i) insertion of styrene proceeds directly in the η3-coordinated allyl group and (ii) the insertion of styrene proceeds selectively in a secondary (2,1) fashion, both at the initiation and propagation steps.
C2-symmetric allyl ansa-lanthanidocenes supported by a bis(indenyl) ligand were efficiently prepared using a similar salt metathesis approach (Scheme 22) [39]. The one-pot reaction proceeded with selective rac-coordination of the indenyl ligand. A diffraction study performed on the bis(silylated)allyl-yttrium complex showed that the allyl group is η3-coordinated with the SiMe3 groups arranged in a syn,syn fashion; i.e., the same conformation as in {η3-1,3-C3H3(SiMe3)2}2Sc(μ-Cl)2Li(THF) [21]. These species turned out to be efficient single-component catalysts for highly isospecific polymerization of styrene (> 99% mmmmmm on the crude unfractionated polymers, even up to 100 °C), operable under industrially relevant conditions (bulk, 60–120 °C).
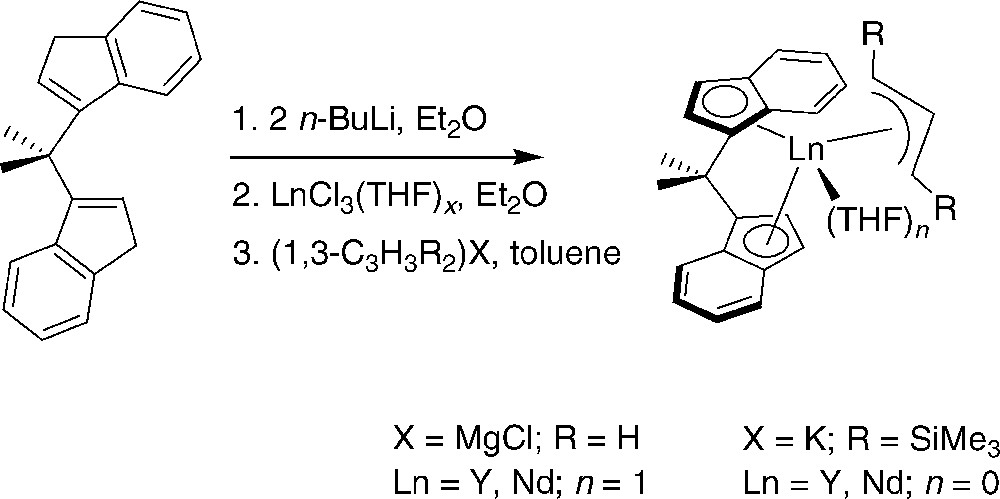
Synthesis of neutral allyl ansa-lanthanidocenes supported by bis(indenyl) ligands and used in isospecific polymerization of styrene [39].
Another synthetic entry to allyl-lanthanidocenes involves partial protolysis of one allyl ligand in homoleptic tris or tetra(allyl)-lanthanide compounds with a proteo cyclopentadiene derivative Cp′H. Thus, [Li(μ-dioxane)1.5][Ln(η3-C3H5)4] (Ln = La, Nd) reacts with a stoichiometric amount of Cp′H (Cp′ = C5H5, C5Me5 = Cp*, indenyl, fluorenyl) to yield selectively the complexes [Li(dioxane)2][Cp′Ln(η3-C3H5)3] (Scheme 23) [40,41]. Similarly, the synthesis of Cp*Nd(η3-C3H5)2(dioxane) has been achieved by protonation of Nd(η3-C3H5)3(dioxane) with pentamethylcyclopentadiene [8]. The ease of these protolytic reactions reflects the predominantly ionic nature of the tris(allyl)-lanthanide precursors.
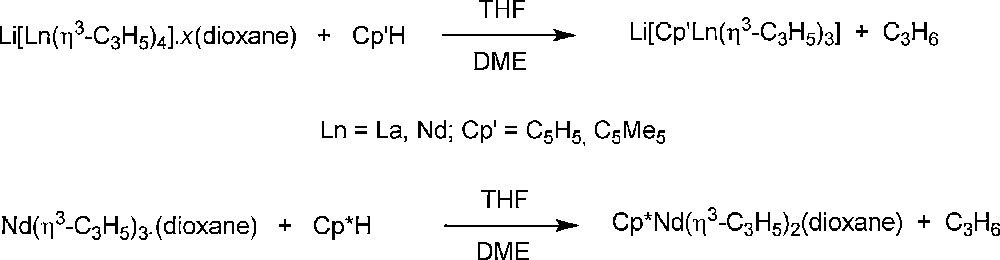
Protolysis of homoleptic lanthanide-allyls with Cp′H pro-ligands [8,40,41].
A most important, pioneering study in the field of early metallocene chemistry was reported by Watson et al. at Dupont [42]. This group conducted spectroscopic studies aimed at the establishment of propene polymerization by and obtained at that time one of the clearest experimental model for coordination catalysis of olefin polymerization. Among the numerous interesting results gathered in this study, it was demonstrated that termination processes occur by C–H activation; i.e., abstraction of an allylic methyl C–H from propene or isobutylene gives allylic complexes, which do not react further with propene (Scheme 24).
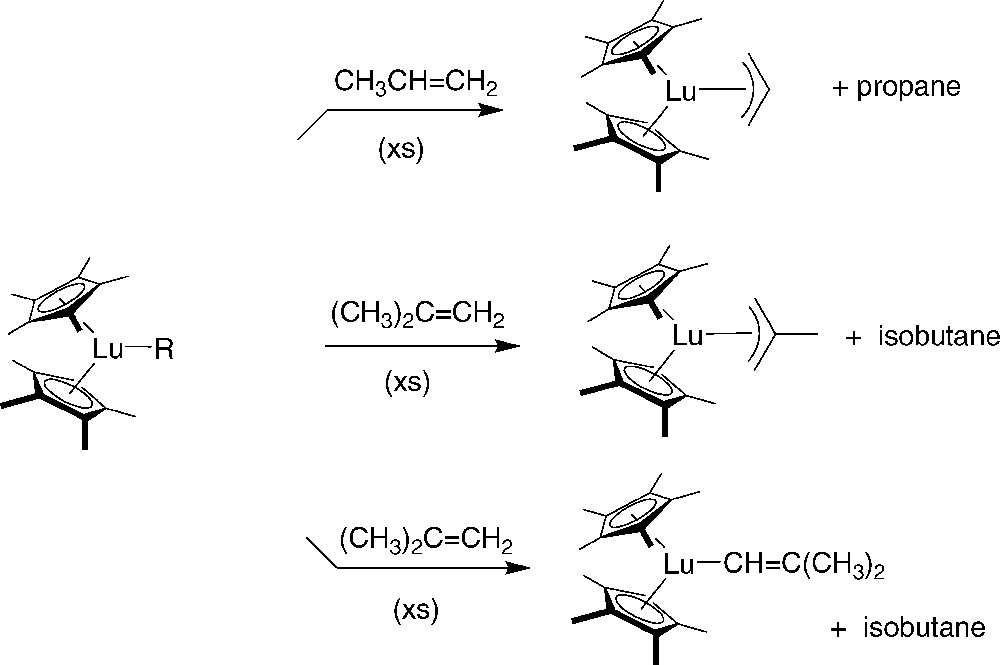
Reaction of alkyl-lanthanidocene with α-olefins [42].
Marks and Schumann [43] and later Evans et al. [44] further expanded on these results, demonstrating that (Ln = La, Nd) and react with propene, butene, and allylbenzene in hexane to form the corresponding allyl complexes in high yields (Scheme 25). In these reactions, the corresponding alkane was formed as a byproduct in equal quantities, as observed in the lutetium case (vide supra). All of these allyl complexes were fully characterized, in particular in the solid state by X-ray diffraction studies for the samarocene compounds which confirmed the trihapto coordination of the allyl moieties. The presence of two Cp* signals and separate signals for the syn and anti protons of the allyl group in the 1H NMR spectrum of indicated that, in the absence of coordinating solvents, this compound exists in solution as a static η3-allyl structure. The addition of a coordinating solvent, such as THF, causes both the C5Me5 signals and the syn and anti signals to coalesce. This coalescence was proposed to occur through a THF adduct in which the allyl moiety can shift to a η1-mode of binding. The chemistry of is indeed consistent with the availability of an η1-allyl binding mode. For example, dissolved in hexane reacts with 1 atm of H2 to produce , it reacts with toluene to form the corresponding benzyl complex with elimination of propene (Scheme 25), and catalyzes the polymerization of ethylene.
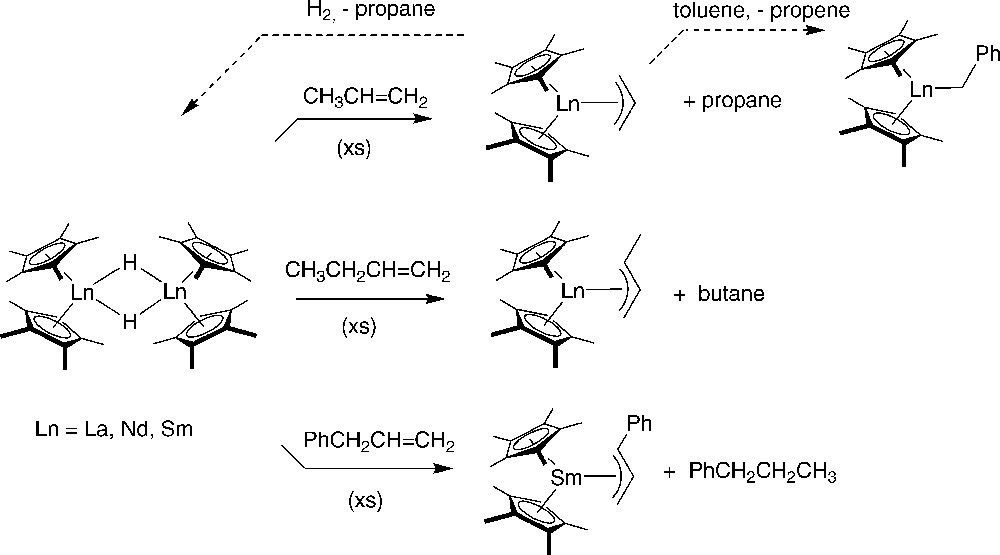
Reaction of with α-olefins, nature of the structurally characterized allyl-samarocene products and reactivity of [43,44].
Later on, Casey et al. conducted a similar detailed study on the reactivity of yttrium alkyl complexes of the type with α-olefins [45]. The selectivity of the reaction of with C–H bonds is allylic CH3 > vinyl C–H > allylic CH2, leading eventually to either allyl or vinyl yttrium complexes. Less substituted alkenes react faster, consistent with prior alkene coordination. Thus, propene is also readily metallated by giving the allyl complex , which does not react further with propene.
All these studies gave an efficient access to a range of allyl-lanthanidocene complexes and, even more importantly, they explained why (R = H, alkyl) complexes make such poor propene polymerization catalysts.
Okuda et al. showed that the “constrained geometry” hydrido complex [(η5:η1-C5Me4SiMe2NtBu)Y(THF)(μ-H)]2 smoothly reacts with stoichiometric amounts of styrenes to give the corresponding allyl complexes (Scheme 26) [46]. Styrenes with no more than one ortho-substituent give 1-phenethyl complexes. An X-ray diffraction study of [Y(η5:η1- C5Me4SiMe2NtBu)(THF)(CHMeC6H4tBu-4)] confirmed that the insertion occurred in a 2,1-fashion and that the phenyl ring is engaged in an η3-coordination. Variable-temperature NMR spectroscopy further revealed fluxional behavior that includes THF dissociation, phenyl ring coordination, and rotation about the ipso- and α-carbon atom. When larger amounts of styrene (or dienes) were used, polymerization does not occur, reflecting the stability of these mono(insertion) products, possibly due to the presence of the coordinated THF molecule that cannot be displaced by styrene.
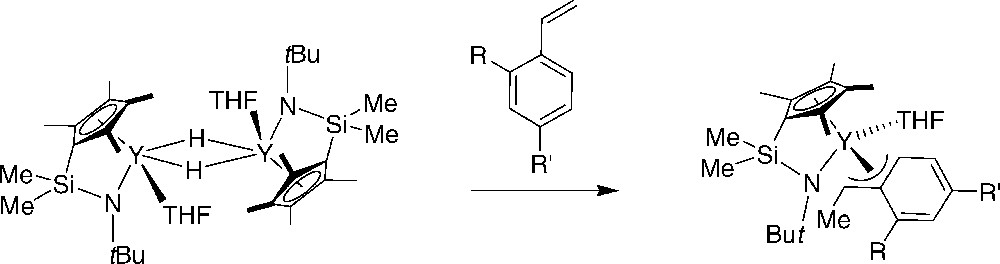
Insertion of styrenes in “constrained geometry” hydrido yttrium complexes [46].
Another effective entry to allyl-samarocenes relies on the reaction of alkenes with the divalent precursor [44]. In fact, this latter complex reacts rapidly and quantitatively with alkenes (propene, butene, and allylbenzene) in hexane or toluene to form the corresponding allyl complexes and alkane byproducts. Several possible routes were discussed to account for the formation of such allyl compounds. Based on the observed reactivity, notably towards styrene and dienes, it was concluded that allyl complexes form via pathways which do not involve free or intermediates. A bimetallic mechanism was eventually proposed (Scheme 27).
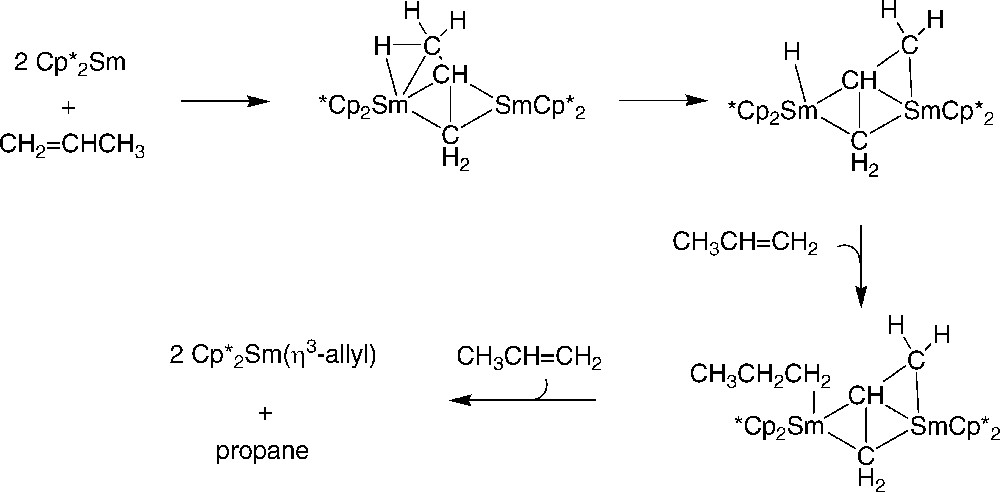
Possible reaction pathway for conversion of 3 mol of propene to propane and two allyl complexes within a tetracyclopentadienyl cavity generated from two units [44].
Relevant to diene polymerization catalysis, a structurally proven example of [allyl] → [alkyl] exchange reaction was reported by Evans et al. [47]. The addition of excess AlMe3 to the Cp-functionalized monoallyl-yttrocene complex quantitatively yields a dimeric aluminate, which was X-ray crystallographically characterized (Scheme 28). Bochmann et al. previously proposed on the basis of NMR spectroscopic investigations a similar allyl exchange reaction between lanthanide bis(allyl) complexes LnCl[1,3-C3H3(SiMe3)2]2(THF)x (Ln = La, x = 1; Ln = Y, x = 0) and triisobutylaluminum [21].
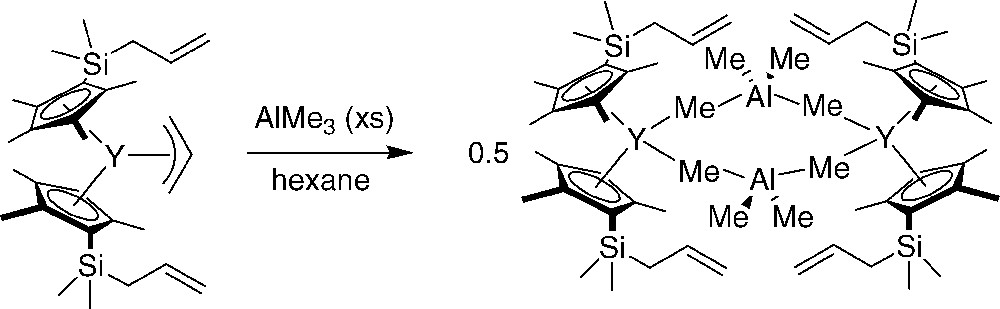
[allyl] → [alkyl] exchange reaction in a Cp-functionalized monoallyl-yttrocene complex [47].
Mono(allyl)-lanthanidocenes can react with protic compounds via a sigma-bond metathesis process, eventually releasing propene. For instance, Evans et al. showed that the mono(allyl)-lanthanidocene complexes (Ln = Ce, Sm, Y) react with pro-ligand 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidine (Hhpp) to form heteroallylic NCN metallocene complexes, , in which the (hpp)1− anion coordinates as a terminal bidentate ligand [48].
Evans et al. investigated also CO2 and CS2 insertion chemistry of mono(allyl)-samarocenes [49]. CO2 reacted with to form in > 90% yield (Scheme 29). CS2 reacted analogously with to form , which subsequently isomerized to (Scheme 29). The same reactivity was observed with COS.
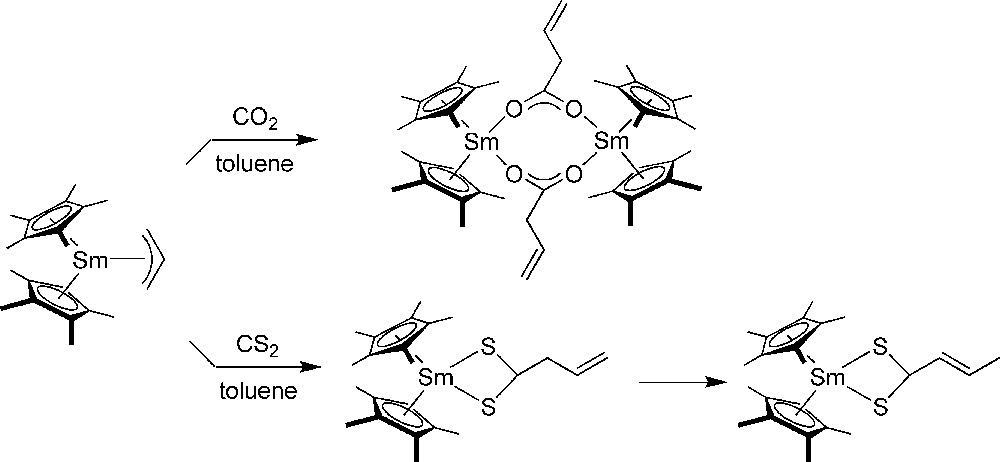
CO2 and CS2 insertion with [49].
Recently, the reactivity of allyl-lanthanidocenes (Ln = Y, La, Sm) with (trimethylsilyl)diazomethane, Me3SiCHN2, was studied [50]. This latter reagent is not metallated by the allyl-metallocene complexes but instead inserts to form the lanthanide hydrazonato complexes (R = CH2 = CHCH2) (Scheme 30). Although the La, Y, and Sm complexes are isomorphous, the double bond in the allyl substituent is oriented toward La and away from Y and Sm.
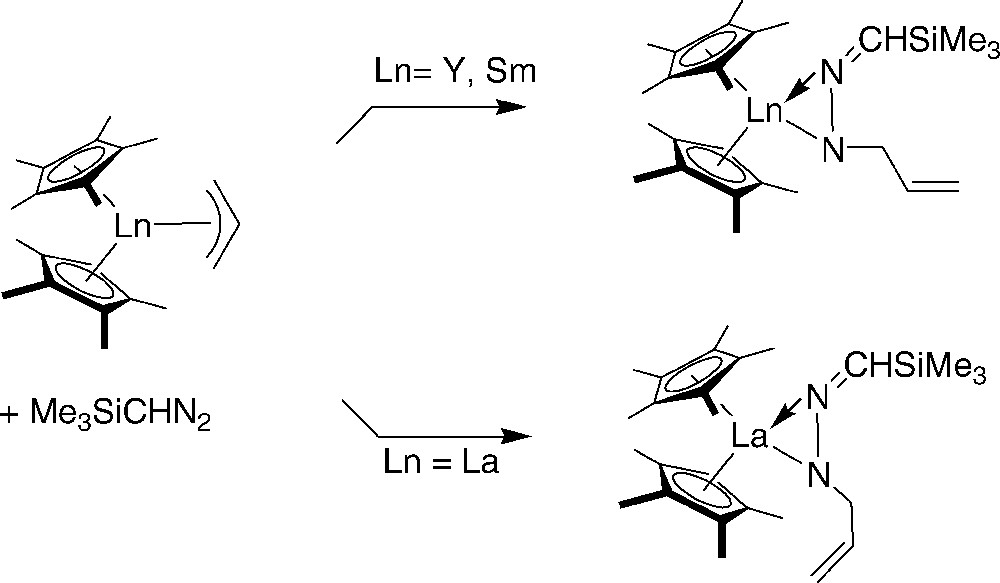
Reaction of allyl-lanthanidocenes (Ln = Y, La, Sm) with (trimethylsilyl)diazomethane [50].
A fundamental work about the fluxional behavior of allyl moieties in neutral allyl group 3 metallocenes was performed by Bercaw et al. using variable-temperature 1H NMR spectroscopy combined with spectral simulations and line shape analyses [51]. Spectroscopic data for a number of allyl-lanthanidocenes ((η5-Cp*)2Sc(η3-C3H5), meso-Me2Si(η5-3-tBu-C5H3)2Sc(η3-C3H5), meso-Me2Si[η5-2,4-(iPr)2-C5H2]2Sc(η3-C3H5), meso-Me2Si{η5-3-[2-(2-Me)-adamantyl]-C5H3}2Ln(η3-C3H5) (Ln = Sc, Y) and rac-Me2Si[η5-2,4-(iPr)2-C5H2]2Sc(η3-C3H5)) indicated an allyl rearrangement mechanism involving rate determining C = C dissociation from the metal center, i.e., an η3 to η1 change in coordination (Scheme 31). Donor solvents do not significantly affect the rate of olefin dissociation. A second rearrangement mechanism that involves 180° rotation of the η3-allyl moiety has been found to operate in those metallocenes whose ancillary ligand arrays adopt rigid meso geometries. Line shape analysis indicated that the rate of η3-allyl rotation is generally more than one order of magnitude faster than olefin dissociation for a given meso metallocene. On the other hand, no unambiguous assessments of the mechanism(s) were found for the fluxional behavior for the allyl derivatives of the racemic metallocenes.
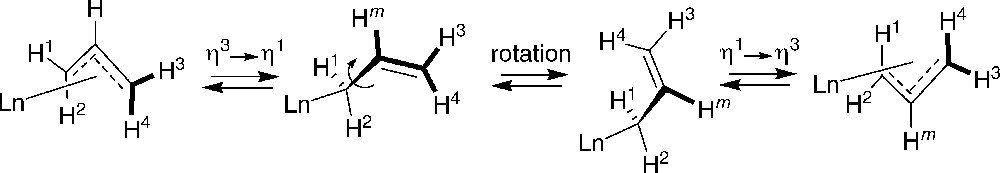
Syn ↔ anti interconversion in allyl-lanthanidocenes [51].
3.2 Mono- and dicationic half-sandwich allyl-lanthanide complexes
Quite recently, important contributions on cationic group 3 metal and lanthanide allyl complexes bearing mono(cyclopentadienyl) ligands have been reported independently by the groups of Xi and Hou [52] and Okuda [13]. This work was essentially motivated by the involvement of mono- and dicationic allyl complexes in diene polymerization catalysis evidenced in Taube's contributions (vide supra) and the topical excitement around cationic rare-earth-metal alkyl complexes.
Xi and Hou reported novel families of monocationic mono(cyclopentadienyl)scandium allyl complexes generated by the reactions of neutral di(allyl) precursors with various activators such as [PhNMe2H][B(C6F5)4], [Ph3C][B(C6F5)4], and [PhNMe2H][BPh4]. Well-defined cationic half-sandwich allyl complexes with various Lewis base ligands such as toluene, N,N-dimethylaniline, and THF were isolated under appropriate conditions (Scheme 32). Recrystallization of these compounds in THF yielded the corresponding THF separated ion pair complexes.
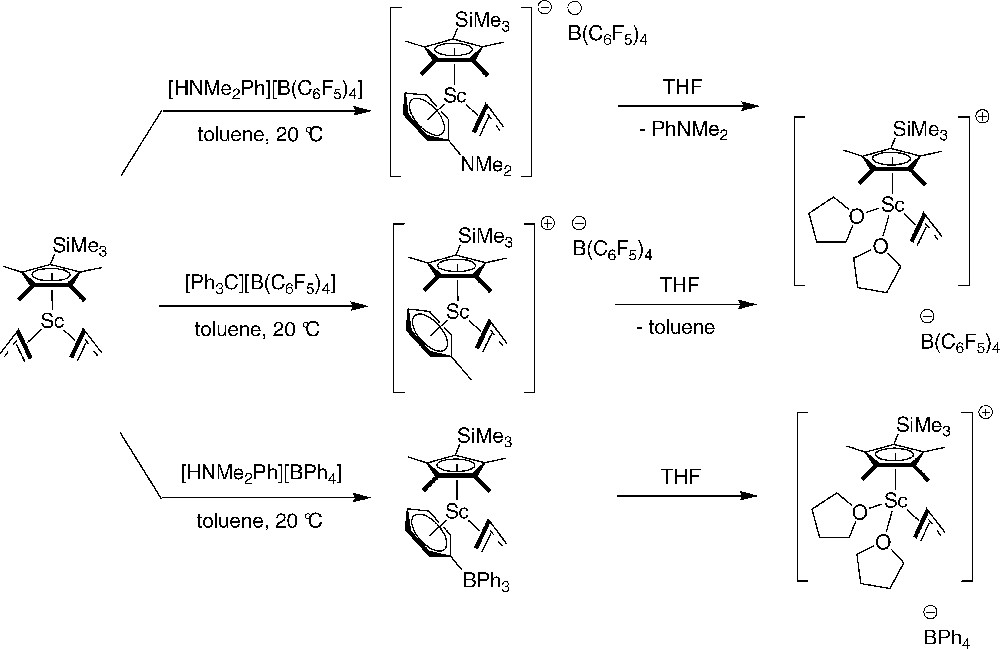
Monocationic half-sandwich allyl-scandium complexes [52].
These monocationic complexes were examined as catalysts for the polymerization and copolymerization of isoprene and norbornene, which demonstrated significant influence of the Lewis base ligands and the counteranions on the catalytic activity of the polymerization. Thus, [(η5-C5Me4SiMe3)Sc(η3-C3H5)(η6-PhNMe2)][B(C6F5)4] and [(η5-C5Me4SiMe3)Sc(η3-C3H5)(η6-toluene)][B(C6F5)4] are highly active, affording random copolymers previously unavailable with a broad range of isoprene content (33–86 mol%), while the tight ion pair [(η5-C5Me4SiMe3)Sc(η3-C3H5)(μ,η6-PhBPh3)] and the THF complexes are inactive.
The group of Okuda reported similar observations on corresponding yttrium and lanthanum complexes [13]. The monocationic half-sandwich complexes [Ln(η5-C5Me4SiMe3)(η3-C3H5)(THF)2][B(C6X5)4] (Ln = Y, La; X = H, F) were synthesized from the neutral precursors [Ln(η5-C5Me4SiMe3)(η3-C3H5)2(THF)] by protonolysis with the corresponding reagents.
4 Heteroleptic allyl-lanthanide complexes supported by non-Cp ancillaries
Surprisingly enough, there are few examples of well-characterized, allyl-lanthanide complexes supported by non-Cp ancillaries. Apparently, synthetic problems involving ligand transfer reactions and/or ligand redistribution processes have so far hampered the preparation of a large variety of such compounds.
Fryzuk et al. reported that the bis{amido(diphosphino)}-chloride complex [{N(SiMe2CH2PMe2)2}2YCl]2 reacts with allylmagnesium chloride or diallylmagnesium with elimination of one {N(SiMe2CH2PMe2)2}− ligand (and not of the Cl−) and formation of [{(η3-C3H5){N(SiMe2CH2PMe2)2}Y(μ-Cl)}2] (Scheme 33) [53]. On the other hand, the reaction of [{N(SiMe2CH2PMe2)2}YCl2] with diallylmagnesium occurred with elimination of MgCl2 and [(η3-C3H5)2{N(SiMe2CH2PMe2)2}Y] was obtained. This latter compound acts as a catalyst precursor for the polymerization of ethylene.
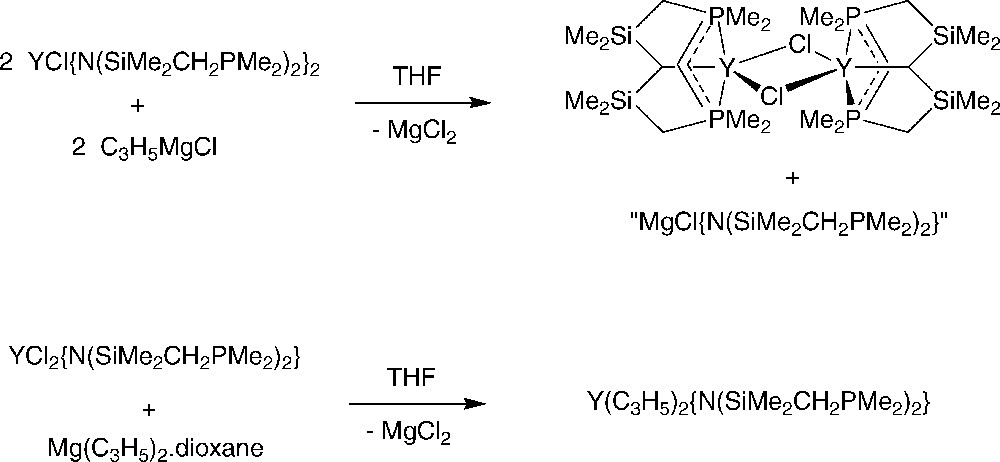
Transmetallation reactions between allyl Grignard and diallymagnesium and chloroyttrium complexes supported by amidodiphosphine ligands [53].
Also, Bochmann et al. investigated reactions of different magnesium/lanthanide allyl complexes with the weakly acidic diketimine pro-ligand BDI–H (BDI–H = 2-(2,6-diisopropylphenyl)amino-4-(2,6-diisopropylphenyl) imino-2-pentene). However, no lanthanide-containing product could be isolated. Instead, magnesium β-diketiminato complexes were formed as a result of ligand transfer reactions (Scheme 34) [54]. On the other hand, when the “ate” allyl-samarium complex [Mg(THF)6][Sm2(η3-C3H5)6(μ-η3: η2-C3H5)]2.toluene was treated with BDI-H, [Mg(THF)6][Sm(η3-C3H5)4]2.2THF, alongside Sm(η3-C3H5)2{η3-HC(MeCNC6H3iPr2-2,6)2} were produced [16].
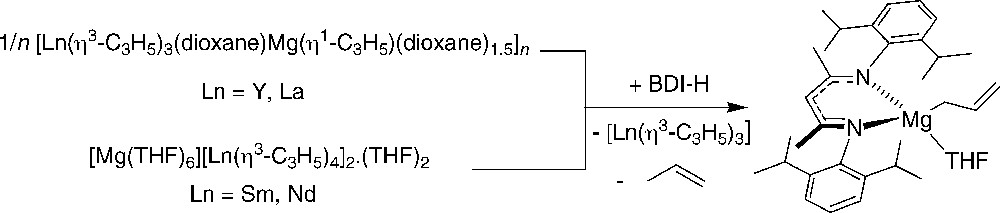
Reactions of a β-diketimine pro-ligand with mixed lanthanide-magnesium allyls [54].
Effective access to bis(allyl)(β-diketiminato)-lanthanide complexes was achieved through the reaction of [La(η3-C3H5)3(η1-dioxane)]2(μ-dioxane) and [Ln(η3-C3H5)3(η1-dioxane)]∞ (Ln = Y, Sm, Nd) with the proteo diketimine pro-ligand BDI-H in THF at 60 °C [55]. These reactions proceeded cleanly with propene elimination (Scheme 35). The solid-state structure of the samarium complex showed that the allyl fragment opposite the N atoms is η3-bonded, with three very similar Sm–C bond lengths, while bonding of the second allyl moiety is less symmetrical. All of these complexes are highly effective single-component catalysts for the ring-opening polymerization of ɛ-caprolactone and rac-lactide giving poly(ɛ-caprolactone) and atactic poly(lactide) with controlled molecular weights and narrow molecular weights distributions. Polymer end-group analysis shows that the polymerization process is initiated by allyl transfer to the monomer.
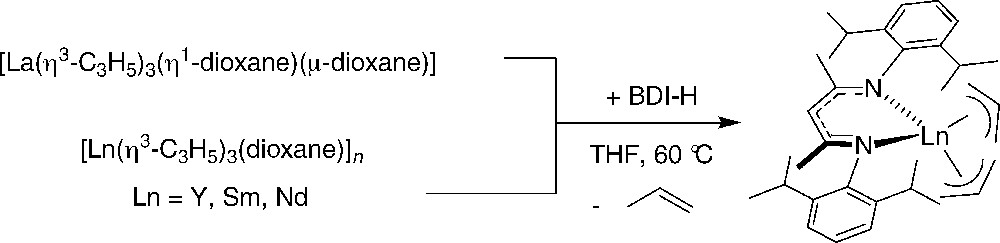
Synthesis of bis(allyl)(β-diketiminato)-lanthanide complexes [55].
5 Conclusions
Allyl complexes have attracted considerable interest over the past four decades. Efficient synthetic methodologies have allowed access to quite a large variety of compounds. The span of compounds now readily available includes “simple” homoleptic neutral, anionic and also cationic bis, tris and tetra(allyl) compounds and mono(allyl)-lanthanidocene complexes. The preparation of allyl complexes of group 3 metals and lanthanides supported by non-Cp ancillaries is much less mature. Considering the obvious interest of allyl-lanthanides in catalysis, notably in (co)polymerization of dienes and styrenics, one can expect that development of effective procedures for accessing new derivatives of such allyl postmetallocene complexes shall be the focus of future research.
1 Only well-defined allyl-lanthanide complexes are included in this review, which is not aimed to be fully comprehensive; accordingly, special focus is given onto structurally (or spectroscopically for the very first compounds) characterized complexes. Also, only regular allyl ligands are addressed. Thus, heteroallyl ligands such azaallyls or diphosphinomethanides are not included in this manuscript.
2 For general reviews in organolanthanide chemistry, with highlights on allyl-lanthanide complexes, see [1].
3 Attempts to isolate such neutral complex (or a related Lewis base adduct) were performed but the species could not be fully authenticated, see [34].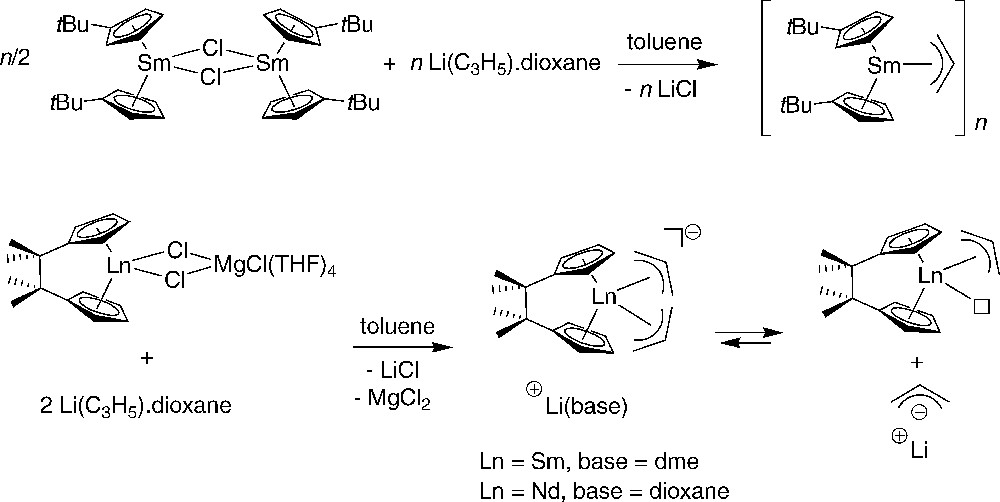
4 Syndiotactic oligostyrenes were also obtained by the in situ combination of the ansa-chloroneodymocene precursor [(CpCMe2Flu)Nd(μ-Cl)]2 with (allyl)2Mg or a regular dialkylmagnesium, see [36].