

1 Introduction
Cationic alkyl complexes of the rare-earth metals have recently been shown to participate in a number of C-C bond forming reactions, including catalytic polymerization of ethylene, 1-alkene, and 1,3-dienes [1–3]. Both monocationic and dicationic alkyl complexes have been implicated as the reactive intermediates. We have recently demonstrated that the dicationic complexes [LnMe(thf)n][BPh4]2 (Ln = Sc, Y, Lu) metalate the C-H bond of pyridine in the ortho-position to give a pyridyl complex (Scheme 1) [4]. Factors influencing this reaction are not yet well understood. The rates of reaction were found to be dependent on the metal radii, with the smaller metals being less reactive toward pyridine. C-H bond activation of pyridines by neutral rare-earth alkyls have been long known and demonstrated to proceed via σ-bond metathesis mechanism [6]. Compounds containing bulkier alkyl, aryl [5] [5a,b], or hydrido ligands [7] lead to side reactions, including C-C bond coupling [5] [5c,d,g,h]. Metal-pyridyl moieties are of synthetic value, as a number of unsaturated small molecules has been shown to insert into the Ln-C bond, yielding a variety of ortho-functionalized pyridines [6].
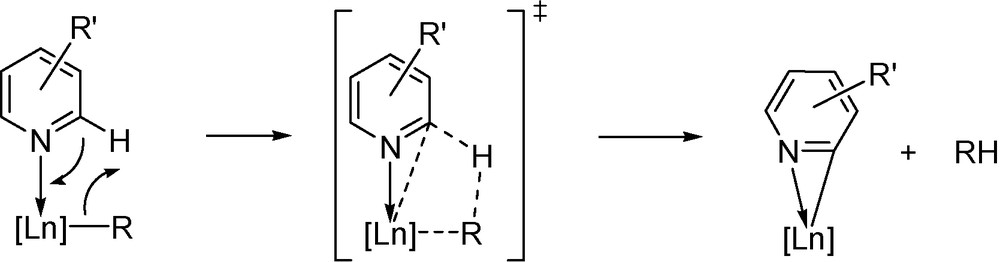
Schematic representation of the ortho-metalation of a pyridine derivative by a rare-earth alkyl moiety.
2 Results and discussion
In order to learn more about the C-H bond activation reactions between [YMe(thf)6][BPh4]2 (1) and pyridines [2], we have investigated the effects on the rate and reactivity, upon using para- or ortho-substituted pyridines. For the present study, the electron rich 4-(N,N-dimethyl-amino) pyridin (DMAP), the electron poor (4-trifluoromethyl)pyridine as well as 2-picoline were chosen (Chart 1).

Using DMAP has the advantage that unlike [YMe(py)6][BPh4]2 [4], the addition of at least 3 equiv of DMAP to 1 affords a thf soluble species such that the reactivity with small amounts of substrate can be followed. To maintain approximately constant DMAP concentrations throughout the reaction, an initial set of reactions between 1 and 25 equiv of DMAP was performed at 23, 35, 45, and 55 °C. 1H NMR spectroscopic monitoring of the reactions revealed pseudo first-order rates for the decomposition of 1. In comparison to the reaction of 1 with pyridine(-d5), the half-lives at all temperatures were found to be much smaller (Table 1). From the Eyring plot of the data sets, the activation parameters were found to be: ΔS‡ = −86.4 ± 12.0 J K−1 mol−1 and ΔH‡ = 71.6 ± 4 kJ mol−1. A negative entropy of activation is characteristic of an associative mechanism with a four-membered metallacyclic transition state, which is usually postulated for d0-transition metals undergoing this reaction type [5,8]. The activation energy Ea was determined to be 74.4 ± 4 kJ mol−1 from an Arrhenius plot. While a quantitative comparison to the C-H bond activation of compound 1 in pyridine is not possible due the large excess of pyridine or pyridine-d5 needed in this study [4], the reactions with DMAP have much shorter half-lives at the respective temperatures. This is due to the better electron donating ability of DMAP for the metal center, facilitating the coordination of DMAP and the Y-Me bond-breaking in the transition state. Although it is obvious that the number of pyridine molecules present affect the reaction rate [4], we assume that [YMe(DMAP)1+n(thf)5−n]2+ with n = 0 can be regarded as the crucial species. A similar approach, can be used to explain the different speeds of decomposition of the compounds [LnMe(thf)n][BPh4]2 in pyridine [4]. If mainly electrostatic interactions contribute to the bonding between rare-earth metals and the methyl group, as was found in our recent calculations [3h] and by others [9], a lower activation energy to reach the transition state is required for the larger metals due to weaker electrostatic bonding of the methyl group. In turn, this results in faster C-H bond activation. Faster rates, due to a greater acidity of the 2-proton on the DMAP ring, relative to pyridine can be ruled out. While no pKa data can be found for DMAP in the literature [10], electron-donating groups do not stabilize negative charges in the 2-position and therefore no increased acidity can be assumed [11].
Half-lives and rate constants for the reaction of 1 in pyridine-d5 and with 25 equiv DMAP in thf-d8.
| Reagent | T (°C) | τ1/2 (min) | k (s−1) |
| Pyridine-d5 (> 250 equiv)4 | 35 | 510 | 2.49 × l0−5 |
| 45 | 234 | 4.57 × l0−5 | |
| 55 | 69 | 1.62 × l0−4 | |
| 65 | 22 | 5.32 × l0−4 | |
| DMAP (25 equiv) | 23 | 269.9 | (4.28 ± 0.01) × l0−5 |
| 35 | 69.6 | (1.66 ± 0.01) × l0−4 | |
| 45 | 33.2 | (3.48 ± 0.03) × l0−4 | |
| 55 | 13.4 | (8.59 ± 0.3) × l0−4 |
When the reactions were repeated with 3 and 6 equiv of DMAP, compound 1 was also found to decompose in a pseudo first-order fashion, expressed by constant half-lives (Table 2). As expected for a fast substitution pre-equilibrium [4], the pseudo first-order decomposition amount significantly varies with unreacted DMAP during the course of the reaction. It appears that 1 and DMAP form a labile complex where DMAP undergoes fast exchange with thf-d8. Evidence for this is given by the difference in chemical shifts of the methyl group at the metal in the 1H NMR spectra of the kinetic studies. In the presence of 3 equiv of DMAP, the chemical shift of the methyl group is −0.41 ppm and it is at −0.20 ppm in the reaction with 6 equiv of DMAP. This suggests that at least one DMAP has to be coordinated at the metal to give [YMe(DMAP)1+n(thf)5−n]2+ which decomposes with first-order kinetics. For the reaction of 1 with 3 equiv of DMAP, it was found that ΔS‡ = −4.6 ± 7 J K−1mol−1 and ΔH‡ = 99.5 ± 3 kJ mol−1 and the reaction with 6 equiv of DMAP gave ΔS‡ = −42.9 ± 5 J K−1 mol1 and ΔH‡ = 85.0 ± 2 kJ mol−1. The activation energies Ea were determined to be 97.6 ± 2 kJ mol−1 and 84.4 ± 2 kJ mol−1.
Half-lives and rate constants for the decomposition of 1 with varying amounts of DMAP in thf-d8.
| eq. DMAP | T (°C) | τ1/2 (min) | k (s−1) |
| 3 | 23 | 988.9 | (1.17 ± 0.01)×l0−5 |
| 35 | 223.0 | (5.18 ± 0.03)×l0−5 | |
| 45 | 61.6 | (1.88 ± 0.03)×l0−4 | |
| 55 | 21.2 | (5.45 ± 0.05)×l0−4 | |
| 6 | 23 | 285.1 | (4.05 ± 0.02)×l0−5 |
| 35 | 75.6 | (1.53 ± 0.02)×l0−4 | |
| 45 | 27.9 | (4.14 ± 0.08)×l0−4 | |
| 55 | 9.8 | (1.17 ± 0.06)×l0−3 |
In the reverse situation, with less electron density on the yttrium center, slower rates of decomposition or in other words, a more stable complex towards C-H bond activation would be expected. Substituting the electron-donating NMe2 group for the electron-withdrawing CF3 group in the 4-position of the pyridine ring leads to a strong decrease of rates. Reactions between 1 and (4-trifluoromethyl)pyridine in thf-d8 could however not be measured, as 1 was insoluble even in the neat pyridine derivative. To enable the reaction to proceed [YMe(thf)6][B(C6F5)4]2 (1′) was prepared from the reaction of [Y(AlMe4)3] with two equiv [NPhMe2H][B(C6F5)4]. 1′ is soluble in thf and was mixed with 6 equiv of (4-trifluoromethyl)pyridine in thf-d8. However, even at 55 °C the half-life of the pseudo-first-order decomposition was found to be 1259.9 min, significantly higher than that of 1 in pyridine-d5 [4]. Cationic and neutral mixed iodo methyl compounds [YMeI(thf)5][BPh4] and [YMeI2(thf)3] [3] [3h] were recently found to be stable in pyridine-d5 at elevated temperatures for more than 1 d. This is in accordance with the above-suggested electronic influence of the ligands on the reactivity. The electronegative iodo ligands leave the metal center with decreased electron density, thus strengthening the electronic interactions with the methyl group.
Using the rates found for the reactions at 55 °C between 1 and 6 equiv DMAP, neat pyridine-d5 and 6 equiv (4-trifluoromethyl)pyridine, a Hammett plot was done (Fig. 1). The concentration difference for the reaction with neat pyridine-d5 is assumed to be insignificant as the coordination sphere of the yttrium center is saturated with six pyridine molecules [4]. The Hammett plot for the three reactions affords a nearly linear data set with a negative slope (ρ = −2.72). This suggests an increase in reaction rates for more electron-donating substituents in the para-position of the pyridine ring.
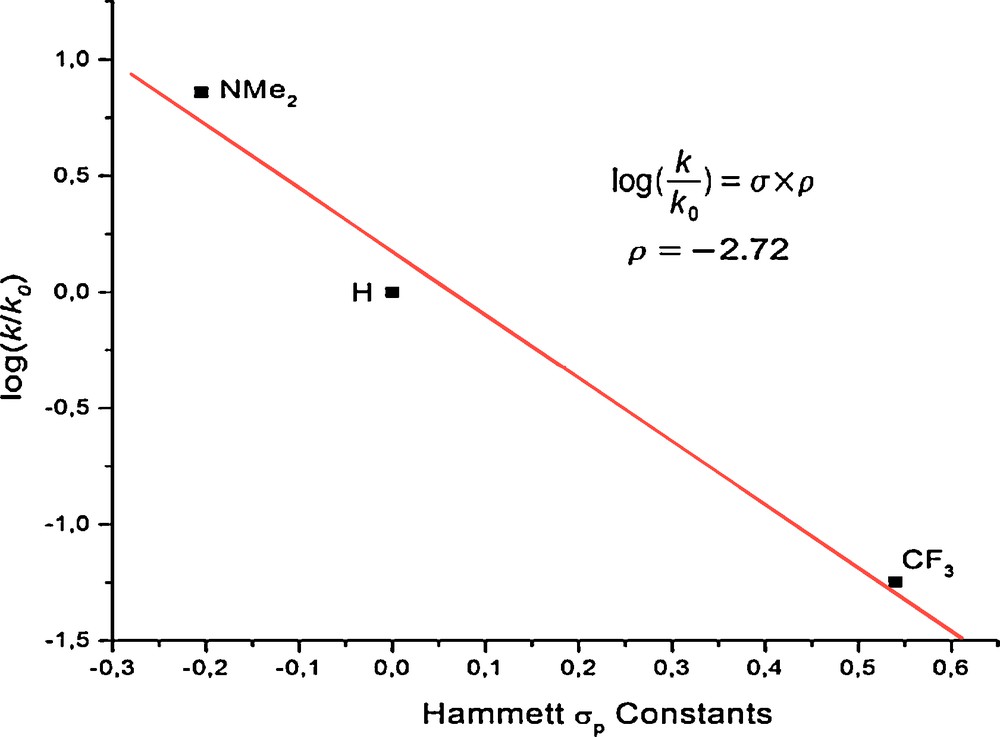
Hammett plot of the relative rates of C-H bond activation of NC5H4X by 1 or 1′ at 55 °C.
Previously Teuben et al. found that [{PhC(NSiMe3)2}2(Yμ-H)]2, [{PhC(NSiMe3)2}2YR] (R = CH(SiMe3)2, CH2Ph(thf)) and [{Me2Si(NCMe3)(OCMe3)}2YCH(SiMe3)2] react with 2-picoline by selective metalation of the methyl group [5] [5a,b]. In contrast to this, it was found that other complexes such as [Cp*2YMe(thf)] metalate the aromatic proton in the 6-position, rather than the 2-methyl group [12]. Upon reaction of [YMe(thf)6][B(C6F5)4]2 (1′) with 6 equiv of 2-picoline in thf-d8 exclusive metalation of the aromatic 6-H occurred. An isolation of the metalated product was not possible due to the slow reaction rate and concurrent decomposition of the product.
The product of the C-H bond activation reaction of 1 with 6 equiv of DMAP, [Y{η2-NC5H3(4-NMe2)}{NC5H4(4-NMe2)}5][BPh4]2 (2), can be isolated from a preparative scale reaction at 55 °C for 90 min. All thf molecules originating from 1 are displaced by DMAP molecules. In the 13C{1H} NMR spectrum of 2, the peak corresponding to the yttrium-bound carbon gives rise to a characteristic doublet at δ 214.1 ppm (1JYC = 26.9 Hz), similar in chemical shift and coupling constant to the corresponding pyridyl complex [4]. Complex 2 can in turn be protonated with [NEt3H][BPh4] to afford the trication [Y(DMAP)6(thf)][BPh4]3 (3). The thf-insoluble compound is comparable to an in situ generated tricationic lanthanum ion from the complete protonolysis of lanthanum tribenzyl [13].
3 Mechanistic discussion
It is commonly proposed that the metalation of pyridine by d0fx complexes proceeds via an associate mechanism with a four-membered metalacyclic transition state [4,6,7]. To investigate this more carefully, calculations at the DFT level were performed for the reaction between 1 and one equiv DMAP as well as on the reaction between [YMe2(thf)5][BPh4] and two equiv of DMAP. The energy profile for the reaction between 1 and DMAP is shown in Fig. 2.
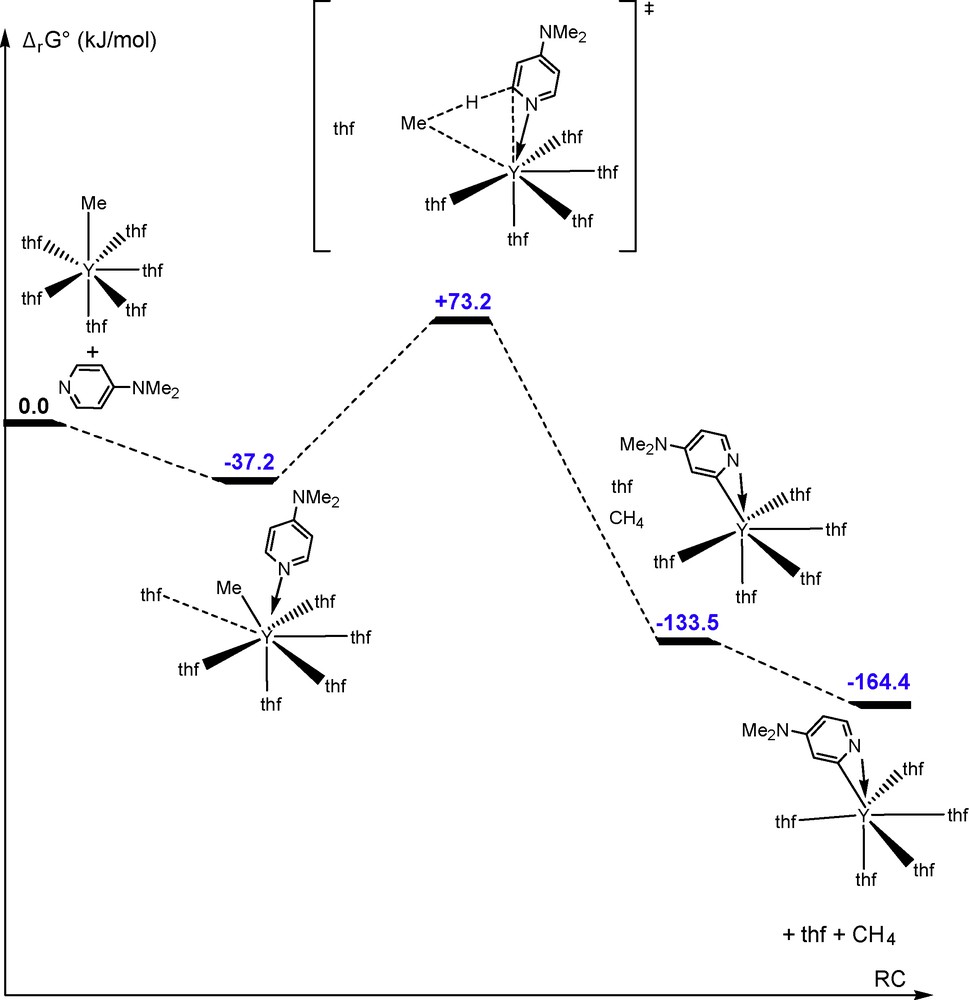
Energy profile of the reaction between 1 and one equiv DMAP.
Interestingly, unlike in the case with pyridine, the initial coordination of DMAP is exergonic. This eight-coordinate intermediate loses one molecule of thf and adopts a 4-membered metalacyclic transition state before eliminating methane. Re-ordering by displacing methane and thf out of the coordination sphere affords the final pyridyl product, which is 164.43 kJ mol−1 more stable than 1 and DMAP. The Ea of 110.5 kJ mol−1 is in excellent agreement with the experimental result, found for the reaction between 1 and 3 equiv DMAP (97.6 ± 2 kJ mol−1). This is in accordance with a decrease in Ea upon increasing the concentration of DMAP. In the final product, the pyridyl ligand is η2-(C,N)-coordinated.
A near-planar, four-center transition state was found, in which, the major atomic motions correspond to bond-breaking/bond-forming. The transition state corresponding to the reaction between 1 and DMAP is shown in Fig. 3. The geometry of the transition state deviates slightly from planarity, with the transferred proton being situated 0.171 Å away from the plane defined by Y7, C20 and C4. The molecule is oriented such that the C20-H52-C4 angle approaches linearity (166.19°) and the remaining methyl hydrogen atoms are pushed away from the approaching proton. The C3 axis of the methyl group is no longer pointing toward the metal center. It should be noted that there is no Y-H bond formation in the transition state, despite the close Y-H contact of 2.084 Å as the positive charge of the proton and the metal center prevent any bond formation. This has been verified by a full NBO analysis of the transition state and is in accordance to previous findings by some of us [14,15]. Contrary to these findings, earlier DFT calculations on the metalation of methane of [ScCp2Me] with Me-H claimed a full Sc-H bond in the transition state [16].
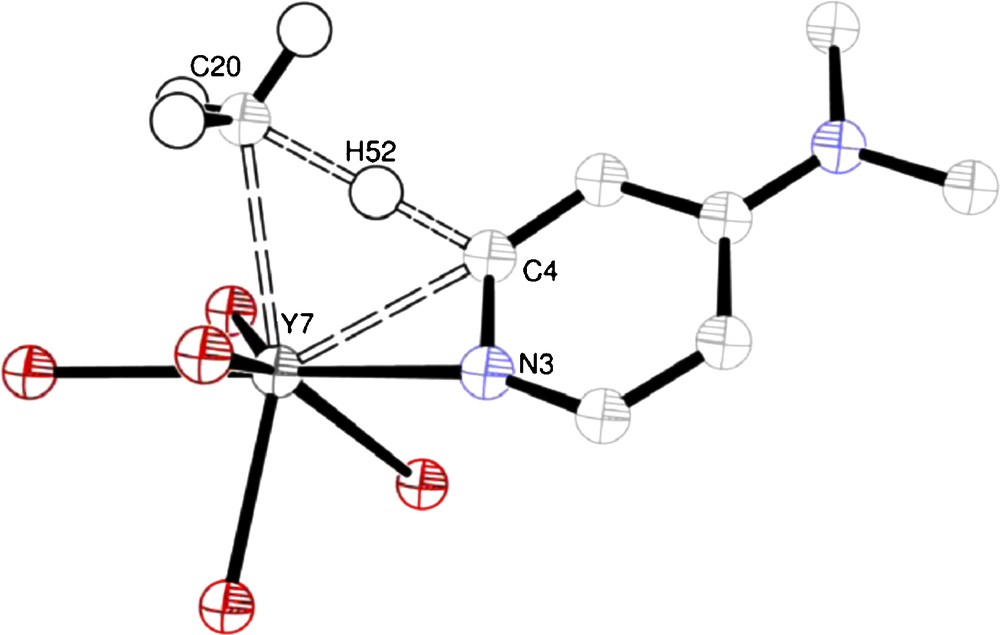
Transition state from DFT calculation for the reaction in Figure 2. Only selected atoms are shown for the sake of clarity. Selected bond lengths (Å) and angles (°): Y7-C20 2.586, C20-H52 2.084, H52-C4 1.335, C4-Y7 2.464; C20-Y7-C4 68.46, Y7-C20-H52 36.23, C20-H52-C4 166.19, H52-C4-Y7 57.76.
To gain further insight into the reactivity of such yttrium cations, the free energy profile of the reaction between the dimethyl monocation [YMe2(thf)5][BPh4] [2,3] [2b,3h] and two equiv of DMAP has been determined (Fig. 4). Two consecutives C-H bond activation of DMAP are kinetically and thermodynamically possible. The first activation barrier is predicted to be higher by 42 kJ mol−1 than the barrier for the dication. This is in line with the fact that the σ-bond metathesis activation barrier is mainly controlled by electrostatic effects rather than orbital overlaps for d0 elements (the orbitals are more contracted for the dication than the monocation and thus less accessible to reaction). It is confirmed by a NBO analysis, indicating a slightly more ionic transition state for the dicationic than the monocationic system (for instance Y: 2.04 vs 1.98, Me: −0.63 vs −0.60), and also by calculations on lanthanum complexes (more ionic and lower activation barrier). The second activation barrier is smaller than the first one by roughly 29.3 kJ mol−1, indicating that the pyridyl ligand has a stronger trans effect than the methyl group.
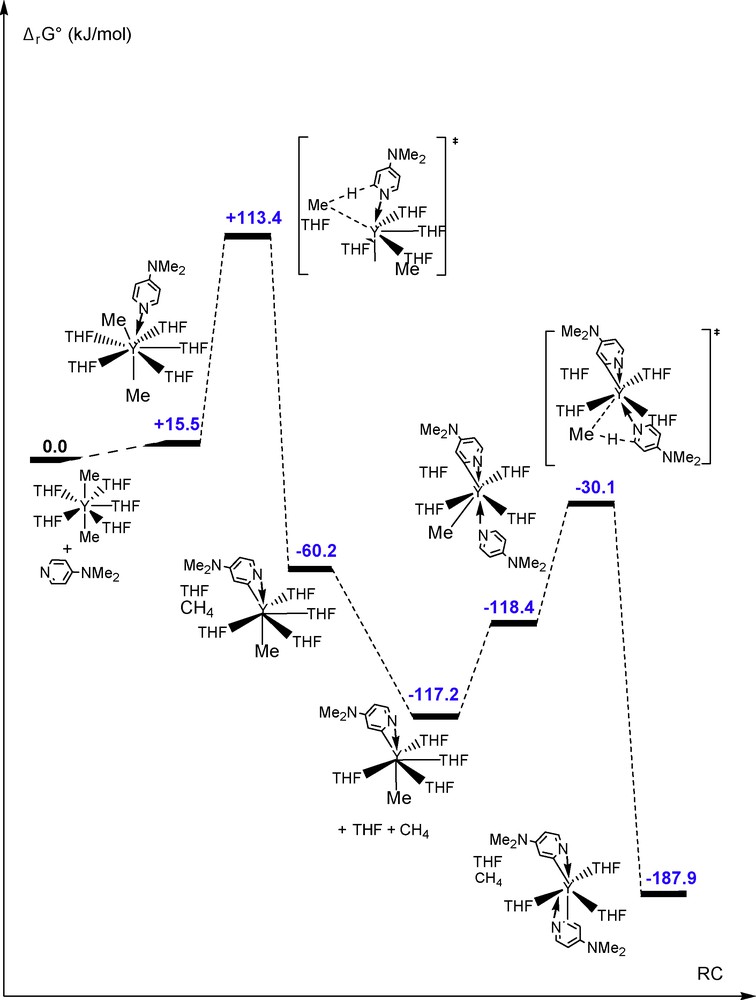
Energy profile of the reaction between [YMe2(thf)5]+ and 2 equiv of DMAP.
As for the monocation, for each C-H bond activation of a DMAP molecule, one thf molecule dissociates. The coordination number of the yttrium atom is kept to seven along the entire pathway. In the final product, the two pyridyls are also η2-(C,N)-coordinated and trans to each other. This final product is found to be more stable than the reactants by 187.9 kJ mol−1.
4 Conclusion
As the representative dicationic methyl complex of the rare-earth metals [LnMe(thf)n]2+, the yttrium complex [YMe(thf)6][BPh4]2 was reacted with substituted pyridines and found to undergo an ortho-metalation reaction. Electron donating properties of para-substituents of pyridines increase the rates of reaction, as indicated by the Hammett linear free energy relationship. DFT calculations confirmed the two-step reaction, consisting of a ligand substitution step and the rate-determining ortho-metalation step via σ-bond metathesis. DFT calculations also revealed for the C-H bond activation step an unusual transition state structure with a nearly linear methyl carbon-hydrogen-ortho-carbon arrangement.
5 Experimental
5.1 General considerations
All operations were performed under an inert atmosphere of argon using standard Schlenk-line or glovebox techniques. The thf was distilled from sodium benzophenone ketyl. Pentane was purified by distillation from sodium/triglyme benzophenone ketyl. Alternatively, the above solvents were dried using an MBraun SPS-800 solvent purification system. Compounds 1 [2,3] and [Y(AlMe4)3] [17] were prepared according to published literature procedures. All other chemicals were commercially available and were used after appropriate purification. Standard NMR spectra were recorded at room temperature on a Bruker DRX 400 spectrometer (1H, 400.1 MHz, 13C, 100.6 MHz, 11B, 128.4 MHz). Kinetic measurements were conducted on a Varian-Mercury 200 BB (1H, 200.0 MHz) at various temperatures. Chemical shifts for 1H and 13C NMR spectra are referenced internally using the residual solvent resonances and reported relative to tetramethylsilane. Thf-d8 was distilled from sodium and pyridine-d5 was distilled from CaH2. 11B NMR spectra are referenced externally to a 1 M solution of NaBH4 in D2O. Elemental analyses were performed by the Microanalytical Laboratory of the Organic Chemistry Department of RWTH Aachen University.
5.2 [YMe(thf)6][B(C6F5)4]2 (l′)
To a stirred solution of [Y(AlMe4)3] (200 mg, 0.571 mmol) in thf (5 mL) was added dropwise a solution of [NPhMe2H][B(C6F5)4] (915 mg, 1.142 mmol) in thf (8 mL). An immediate gas evolution was observed. Upon complete addition, the clear colorless solution was stirred for a further 20 min, then filtered, and dried under vacuum to afford a colorless oil. Repeated vigorous trituration and repeated washings with pentane (9 × 20 mL) gave 1′ as a colorless powder (1020 mg, 94%). 1H NMR (thf-d8): δ −0.44 (d, 2JYH = 2.3 Hz, 3H, YMe), 1.77 (m, 24H, β-thf), 3.62 (m, 24H, α-thf). 13C{1H} NMR (thf-d8): δ 26.1 (β-thf), 28.8 (d, 2JYC = 57.2 Hz, YMe), 68.2 (α-thf), 125.0 (br, Ph-1), 136.9 (d, 1JCF = 244.5 Hz, Ph-3), 138.9 (d, 1JCF = 244.4 Hz, Ph-4), 149.0 (d, 1JCF = 245.4 Hz, Ph-2). 11B{1H} NMR (thf-d8): δ −14.7. 19F{1H} NMR (thf-d8): δ −162.99, −159.38, −127.36. Anal. Calcd for C73H51BB2F40O6Y: C, 46.28; H, 2.71. Found: C, 46.29; H, 3.47.
5.3 [Y{η2-NC5H3(NMe2-4)}{NC5H4(NMe2-4)}5][BPh4]2 (2)
To a stirred suspension of 1 (1000 mg, 0.851 mmol) in thf (5 mL), solid DMAP (624 mg, 5.106 mmol) was added at once. All solids dissolved within 1 min and the colorless solution was heated to 50 °C for 1 h. The resulting reaction mixture was dried in vacuo and the resulting solid was washed with pentane, and dried to give 2 as an off-white solid (186 mg, 82%). 1H NMR (thf-d8): δ 2.77 (s, 30H, NMe2-DMAP), 2.98 (s, 6H, NC5H3NMe2), 6.32 (d, 3JHH = 6.5 Hz, 10H, 2,5-H DMAP), 6.56 (dd, 3JHH = 6.3 Hz, 4JHH = 2.8 Hz, 1H, 5-H-NC5H3NMe2), 6.67 (t, 3JHH = 7.0 Hz, 8H, 4-Ph), 6.77 (t, 3JHH = 7.5 Hz, 16H, 3-Ph) 7.13 (d, 4JHH = 2.5 Hz, 1H, 3-H-NC5H3NMe2), 7.24 (m, 16H, Ph-2), 7.83 (d, 3JHH = 6.3 Hz, 10H, 2,6-H-DMAP), 8.11 (d, 3JHH = 6.3 Hz, 1H, 6-H-NC5H3NMe2). 13C{1H} NMR (thf-d8): δ 39.1 (NMe2-DMAP), 39.3 (NC5H3NMe2), 107.8 (3,5-DMAP), 108.3 (C5-NC5H3NMe2), 110.8 (C3-NC5H3NMe2), 122.0 (4-Ph), 125.9 (q, 3JBC = 2.6 Hz, 3-Ph), 137.2 (m, 2-Ph), 143.2 (C6-NC5H3NMe2), 149.5 (C2,C6-DMAP), 153.9 (C4-NC5H3NMe2), 155.9 (C4-DMAP), 165.1 (q, 1JBC = 49.4 Hz, 1-Ph), 214.1 (d, 1JYC = 26.9 Hz, C2-NC5H3NMe2). 11B{1H} NMR (thf-d8): δ −6.53. Anal. Calcd for C90H99B2BN12Y: C, 74.07; H, 6.84; N, 11.51. Found: C, 73.05; H, 6.99; N, 11.75.
5.4 [Y(NC5H4{NMe2-4})6(thf)][BPh4]3 (3)
To a stirred solution of 2 (450 mg, 0.308 mmol) in thf (4 mL), a solution of [NEt3H][BPh4] (130 mg, 0.308 mmol) in thf (2 mL) was added dropwise. After complete addition, a light-yellow oil was formed. The solution was decanted from the oil and after repeated trituration with thf and pentane, an off-white solid had formed. Drying under reduced pressure afforded 3 (400 mg, 70%). 1H NMR (py-d5, 400 MHz): δ 1.63 (m, 4H, β-CH2, thf), 2.68 (s, 36H, NMe2-DMAP), 3.66 (m, 4H, α-CH2, thf), 6.49 (d, 3JHH = 7.3 Hz, 12H, 2,5-H DMAP), 7.08 (t, 3JHH = 7.3 Hz, 12H, 4-Ph), 7.22 (t, 3JHH = 7.5 Hz, 24H, 3-Ph), 7.94 (m, 24H, Ph-2), 8.06 (d, 3JHH = 6.8 Hz, 12H, 2,6-H-DMAP). Anal. Calcd for C118H128BB3N12OY: C, 76.54; H, 6.97; N, 9.08. Found: C, 75.07; H, 7.93; N, 8.81.
5.5 Kinetic measurements for the decomposition of 1 with DMAP
A J. Young's NMR tube was charged with a solid mixture of 1 (70 mg) and either 6 (44 mg) or 3 (22 mg) equiv of DMAP. The thf-d8 (0.5 mL) was added to the reaction mixture and the sample was shaken vigorously. The samples were either measured at room temperature (23 °C) or heated to 35, 45, or 55 °C in the NMR spectrometer. 1H NMR spectra were recorded at regular intervals (room temperature and 35 °C: every 15 min for 14 h; 45 °C: every 10 min for 12 h, 55 °C: every 10 min for 10 h) with t1 = 3.0 s, nt = 10. Pseudo first order rate constants were determined by plotting ln(I0/I) against t (I = integration of YMe protons). The Ph-2 signals of the BPh4 anions were used as an internal standard to calculate the extent of conversion from 1 to 2.
Acknowledgements
Financial support of this work by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie (Kekulé Scholarship to M.U.K.) is gratefully acknowledged. L.M. and A.Y. thank the CINES and CALMIP for generous grant of computing time. L.M. is member of the Institut Universitaire de France.