

1 Introduction
Nowadays, actinide chemistry is an important field of both academic and industrial research, notably due to environmental problems as well as the matter of nuclear waste reprocessing. However, the experimental studies are mainly focused on thorium (Th) and uranium (U) species [1–7]. Indeed, doing experiments with transuranium elements like neptunium (Np) or plutonium (Pu) is a very difficult task which requires high security requirements. In that context, theoretical studies can be very useful. However, for actinide systems, calculations are not easy mainly because of the importance of relativistic effects [8,9] and electronic correlation effects due to the presence of 5f unpaired electrons in the ground state [10,11]. Density functional theory (DFT) methods have to be used with caution because the ground state wave function is not necessarily represented by only one Slater determinant. Thus, elaborate theoretical studies have been mainly limited to small molecules, such as actinyl derivatives [8,12–31], which are very common and interesting species for experimentalists and nuclear waste reprocessing. Concerning these systems, geometries and frequencies calculated with DFT compare well with experiments but it is not the case for the energies. For example, work by Vallet et al. [8] demonstrated that DFT was not reliable to represent the reduction of the uranyl ion by water.
Much less is known about organoactinide complexes [32–35]. Barros et al. [36] have proposed a comprehensive study of the reactivity of organo-uranium complexes (Cp2UO and Cp2UNMe) using DFT methods and the results compare very well with experiment. In particular, it has been demonstrated that DFT studies can be safely used in that case by comparing the geometry and the DFT wave function with CASSCF ones, which include static electronic correlation. Moreover, our group has also realized CASSCF calculations on other organoactinide complexes (Cp2An(IV)Me2 for An = U, Np and Pu) which have shown that the ground state wave function can be actually described by a single Slater determinant [37]. Thus, it seems that DFT methods can be safely used for this type of organoactinide complex.
Recently, Kiplinger and co-workers observed a C-H activation of pyridine N-oxide by uranium (IV) bis-alkyl complexes, (C5Me5)2U(CH2R)2 (with R = H, Ph) [38]. Initially, the experiment aimed at synthetizing a uranium (VI) alkylidene complex, (C5Me5)2U(=O)(=CHR) by an oxygen transfer followed by an α-hydrogen abstraction. Indeed, pyridine N-oxide is usually used for oxygen atom transfer properties in the high-valent oxo complexes [39–41]. The C-H activation of pyridine N-oxide seems to be the most favourable reaction pathway [42] leading to a stable η2-(O,C) orthometalated complex, which does not then evolve.
In this article, the predictive formation of a formaldehyde complex from reaction between actinide bis(methyl) complexes Cp2An(IV)Me2 (with Cp = C5H5 and An = U, Np, Pu) and the pyridine N-oxide is reported. This reaction can occur for An = U and An = Pu, whereas it is less obvious with An = Np, and the difference of behaviour between Np and the two other actinides is underlined. Indeed, the formation of a transient carbene intermediate is less favourable for An = Np than for An = U and An = Pu. This difference can be explained by a Natural Bond Orbital (NBO) analysis in parallel with the use of a large core pseudopotential (LC-ECP).
2 Computational details
Explicit 5f electron treatments for U, Np and Pu were performed using the very small core Stuttgart-Dresden pseudopotential in combination with their adapted basis set [43,44]. This leads to computing a triplet spin state for U(IV), a quartet for Np(IV) and a quintet for Pu(IV). In all cases, no spin contamination has been found and the <S2> is 3.0, 3.75 and 6.0 for U, Np and Pu, respectively. All the calculations were carried out with the very small core Relativistic Effective Core Potential (RECPs) except when explicitly stated. The newly extracted f-in-core, adapted to a given oxidation state, was also used for U, Np and Pu [45,46]. No spin-orbit effects were accounted since the reactions are of the metathesis-type (no change in oxidation state). Carbon (C), nitrogen (N), oxygen (O) and hydrogen (H) atoms have been described with a 6-31G(d,p) double-ζ basis set [47]. Calculations were carried out at the unrestricted DFT level of theory using the hybrid functional B3PW91 [48,49]. Geometry optimizations were carried out without any symmetry restrictions, and the nature of the extrema (minima and transition states) was verified with analytical frequency calculations. Gibbs free energies were obtained at T = 298.15 K within the harmonic approximation. Intrinsic Reaction Coordinate (IRC) calculations were performed to check the connections of the optimized transition states. The calculations were carried out with the Gaussian03 [50]. Finally, the electronic density (at the DFT level) has been analyzed using the Natural Bond Orbital (NBO) technique [51].
3 Results and discussion
The nature of the bonding in the reactants Cp2AnMe2(1) will first be discussed. NBO analyses indicate that the bonding is covalent in the case of uranium with a participation of both the 5f and 6d atomic orbitals. The participation of the 5f is around 30% and of the 6d around 49%. For Np, the bonding is more covalent, that is the 5f and 6d orbitals are more involved (30% and 51%, respectively). The situation is even more pronounced for Pu where the 5f participation is 40% and of the 6d is 46%. It is thus clear that the bonding is becoming more covalent while increasing the nuclear charge within the actinide series. The presence of the Cp ligands changes the properties of the metal centre, in terms of covalence, as it is admitted that the actinides are becoming more lanthanide-like (more ionic in bonding) while increasing the nuclear charge. Indeed, the presence of the Cp ligands reduces the symmetry of the complexes allowing a greater mixture of the 5f and 6d orbitals than e.g. in the actinyl ions.
The initial step of the formation of the formaldehyde complex will be first introduced. The C-H activation of pyridine N-oxide by Cp2An(IV)Me2 (1) (with releasing of methane and formation of a cyclometalated pyridine N-oxide complex [4]) has been investigated (Fig. 1). The reaction begins with the formation of a pyridine N-oxide adduct, found as slightly endergonic. The coordination is rather strong as it overcomes the loss of translational entropy [52]. The strength of the adduct is associated with the coordination of the negatively charged O to the positive metal centre. The reaction is kinetically accessible for the three actinides, with a higher barrier for An = U (+19.9 kcal.mol−1) than for An = Np (+15.4 kcal mol−1) and An = Pu (+14.3 kcal.mol−1). The structure of the transition state is a 6-member ring with an almost ideal charge distribution [42]. The nature of the transition state has been discussed in details in a previous study [42]. It should be noticed that the barrier is lowered by the nucleophilic assistance of the O atom.

Calculated free energy profile of C-H activation of pyridine N-oxide catalyzed by Cp2An(IV)Me2 with An = U, Np, Pu. The free energy is given in kcal.mol−1.
Thermodynamically, the reaction is calculated to be favourable for the three actinides (exergonic by 29.2 to 33.2 kcal mol−1). Two isomers of the cyclometalated products can be obtained that differ from the orientation of the C with respect to the methyl group, namely a η2-(O,C) (4) and a η2-(C,O) (5) (Fig. 2). A small energy difference between them leads to the possible formation of both.

Calculated free energy of two isomers of cyclometalated products (4) and (5).
Moreover, the geometry of the transition state (3) (Figs. 3 and 4) allows formations of both (4) or (5). Indeed, the pyridine N-oxide is not lying in the same plane as the methyl groups and An. The ring is nearly perpendicular to the equatorial plane. So, the pyridine N-oxide ring can easily turn around the U-C bond to point toward or opposite to the methyl group and an equilibrium between the two isomers is possible. Thus, formations of (4) and (5) are equivalent as the only difference is a rotation around the U-C bond (more details are available on a precedent work [53]). In conclusion for this first part, the C-H activation reaction of pyridine N-oxide catalyzed by (1) is favourable for the three actinides.

C-H activation transition states geometries for An = U, Np and Pu.
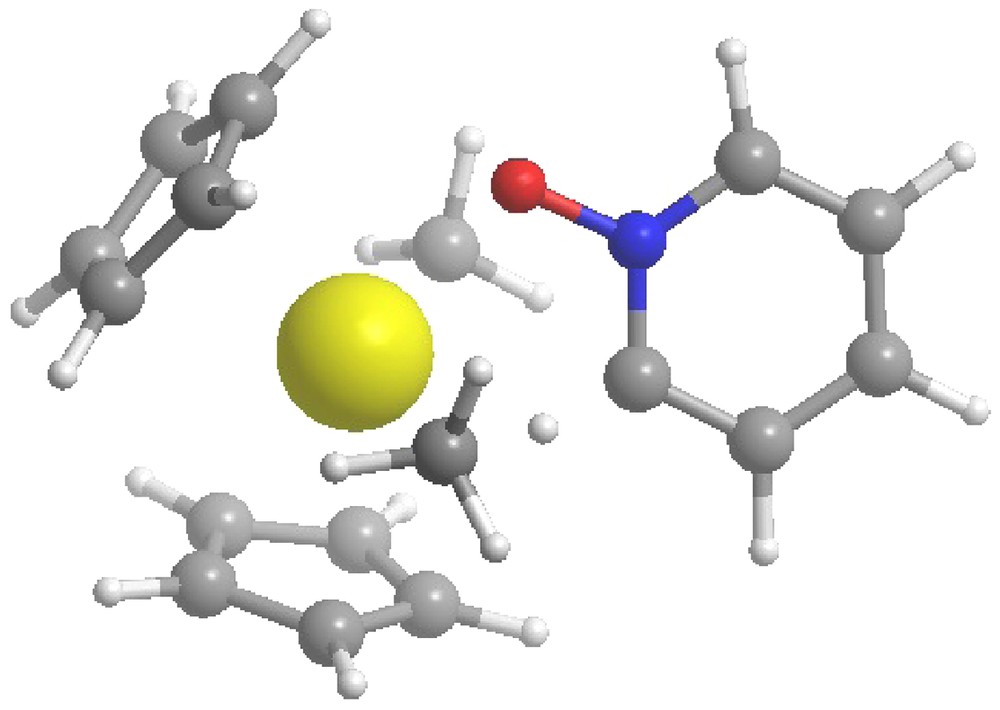
C-H activation transition state geometry for An = Np.
Is there any further possible reactivity from complex (5)? From the experimental point of view, the Kiplinger's group has not reported any further reactivity using Cp* ligand but recently a theoretical study proposed further reactivity using Cp or CpTMS ligands [53]. The proposed reaction occurs in two steps. First, an α-hydrogen abstraction from the methyl group by the activated C of pyridine N-oxide (6) has been investigated. This reaction yields an adduct of pyridine N-oxide to an actinide (IV) carbene complex (7). Then, this transient carbene complex (7) inserts into the An-O bond (8) yielding the actinide (IV) formaldehyde complex (9) with pyridine releasing. The complete reaction profile for the three actinides has been computed (Fig. 5).
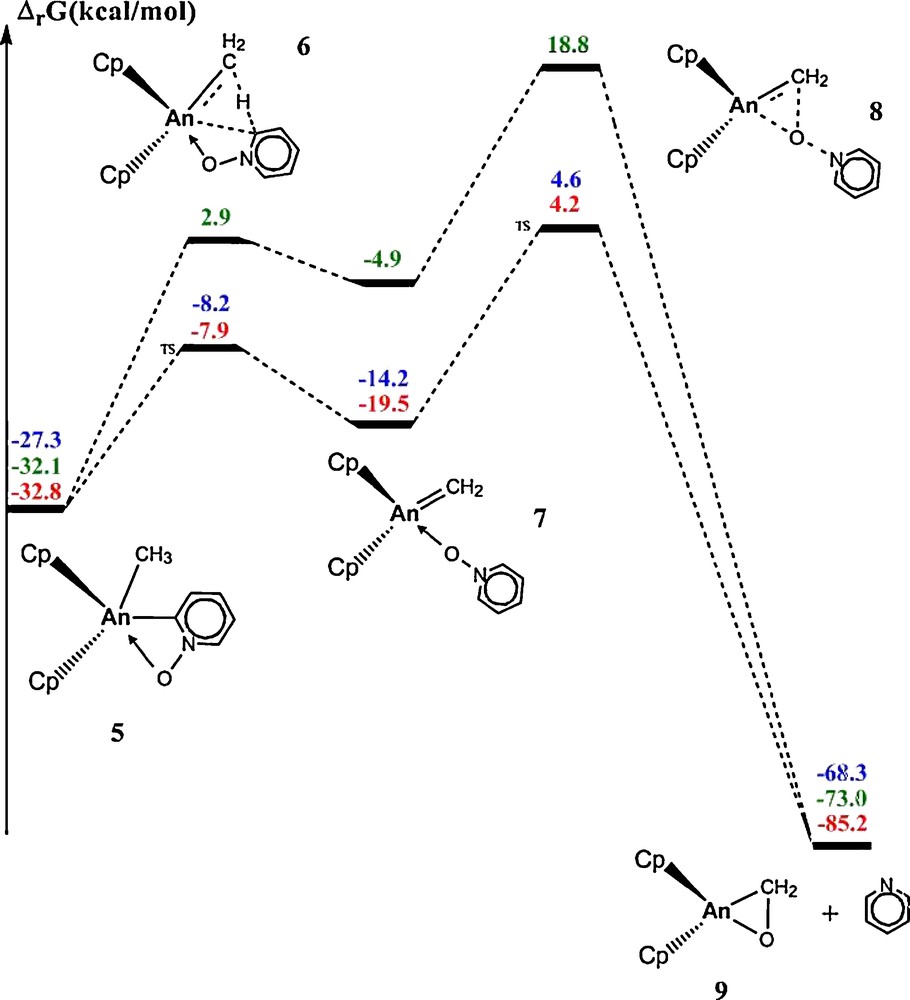
Calculated free energy profile for the α-hydrogen abstraction followed by the formation of the aldehyde complex.
The α-hydrogen abstraction reactions are kinetically accessible for An = U and An = Pu (−8.2 kcal mol−1 for An = U and −7.9 kcal mol−1 for An = Pu with respect to the entrance channel), whereas it is less obvious for An = Np (+2.9 kcal.mol−1 with respect to the entrance channel). Indeed, the relative barrier (with respect to complex 5) is around 20 kcal.mol−1 for U and Pu and 35 kcal.mol−1 for Np. At the transition state (6), the carbene CH2 is almost already formed as well as the C-H bond in the pyridine N-oxide, for An = U and An = Np, whereas the transition state is predicted to be “early” for An = Pu. Indeed, the An-CH2 bonds are fully formed for U and Np (around 2.16–2.17 Å) but not fully formed for Pu (2.42 Å). At the same time, the CH2…H distances are longer for U and Np than for Pu (Table 1). Thus, we can notice that the geometry of (6) is different for An = Pu while the activation barrier is different for An = Np. In order to understand this difference, a NBO analysis was performed. Special attention was paid to the nature of the interaction between the metal and the carbene. For An = U, a single bond character is found, with a lone pair on the carbene (charges of +2.40 and −0.86 for U and CH2, respectively). No significant interaction between the lone pair of CH2 and the metal centre is found at the second order donor–acceptor perturbation theory. The bonding is thus described as a single bond with a strong electrostatic component. Such a bonding situation was reported by Barros et al. on Cp2UO [36]. For An = Np, a double bond character is found (charges of +2.20 and −0.77). Thus, Np(IV) is more covalent than U(IV). Such behaviour was already found for the Cp2AnMe2. For An = Pu, a single bond character is found (charges of +2.19 and −0.53) but with less electrostatic component. Indeed, a residual bond lies between Pu and the ortho carbon of the pyridine N-oxide. The transferred H shares one electron with CH2 and one electron with the C of the pyridine N-oxide. Thus, for Pu, H is not fully transferred yet, and bonds present in (5) still exist. This NBO analysis for An = Pu is in agreement with the geometry of a slightly earlier transition state than for U and Np.
Bond distances in the transition state of α-hydrogen abstraction structure (6).
| D (Ǻ) | U | Np | Pu |
| An-CH2 | 2.17 | 2.16 | 2.42 |
| CH2-H | 1.66 | 1.65 | 1.44 |
| C(ortho)-H | 1.27 | 1.27 | 1.37 |
Thus, as expected, the presence of a more covalent character in the bond between Np and the carbene increases the activation barrier. This is in line with the fact that ionic σ-bond metathesis transition states are more favourable than covalent ones [54]. This covalence is not present for An = U, which displays an ionic single bond character, or for An = Pu. Electronic reorganization of Pu is easy to achieve in order to gain a better stability. It is related to the electronic repulsion between the unpaired f electrons and the lone pairs of the carbene, which has to be minimized. Without this skill, the transition state for An = Pu might be more covalent than for An = Np and exhibits a higher activation barrier. It seems that Np complex cannot reorganize its electronic density and is unable to do longer bonds than it currently does. Indeed, any attempt to locate a Transition state (TS) similar to Pu for Np (and also U) failed. To ensure this statement, the activation barriers of this reaction with LC-ECP were computed for the three systems. Using LC-ECP ensures to keep the same oxidation state (+IV) and so the same electronic situation. In this case, the three barriers are equivalent (+13.8 kcal mol−1 for An = U, +11.2 kcal mol−1 for An = Np and +11.6 kcal mol−1 for An = Pu). It confirms the fact that the difference of values is due to a difference of electronic reorganization at the transition state.
The carbene intermediate (7) and the following transition state (8) for An = Np are shifted by the same amount of energy than the transition state of α-hydrogen abstraction (6) (Fig. 3). With respect to the adduct, the free energy data (activation barrier and reaction energy) are similar for An = Np as for An = U and An = Pu. From a thermodynamic point of view, formation of (7) is endergonic because cyclometalated products (5) are more stable than complexes (7), which can be described as Shrock-type (or dianionic) carbenes in interaction with the actinide (IV). The dianionic character of the carbene is confirmed by a NBO analysis with the lack of first order bonds between actinides and the carbene group. On the contrary, the electronic density is mainly localized on the carbene (a lone pair is indeed present on the carbene group). Thus, (7) is not very stable and has to be considered as a transient carbene.
Thermodynamically, the product (9) of this last reaction is found very stable for all actinides (−68.3 kcal mol−1 for An = U, −73.0 kcal mol−1 for An = Np and −85.2 kcal mol−1 for An = Pu). This product is described as a dianionic formaldehyde complex. The product for An = Pu is more stable than the two others. This is due to a geometrical difference that exists between An = Pu on one hand and An = U or Np on the other hand (Table 2). As for the α-abstraction TS, the An-CH2 bond is longer for Pu than for U and Np. At the same time, the O-CH2 bond is shorter for Pu than for U and Np. This is due to the repulsion between the unpaired f electrons and the lone pairs of the dianionic formaldehyde, which is maximal for Pu in the considered series (four unpaired electrons). Thus, the product looks different for Pu than for U and Np. This is further confirmed by the NBO analysis.
Bond distances in the formaldehyde complexes (9).
| D (Ǻ) | U | Np | Pu |
| An-CH2 | 2.34 | 2.35 | 2.64 |
| An-O | 2.04 | 2.06 | 2.19 |
| O-CH2 | 1.43 | 1.41 | 1.33 |
The NBO analysis shows that one bond O-CH2 and one bond An-CH2 are formed in the case of An = U and Np, with an electrostatic interaction An+-O−. It is the classical description of a dianionic formaldehyde adduct. It is different for An = Pu. Indeed, the double bond O-CH2 is almost formed, but interactions at the second order donor–acceptor level still exist between CH2 and Pu or O and Pu. It can be described as a monoanionic adduct of formaldehyde with Pu at the oxidation state +III. This is consistent with the fact that Pu is more electronegative than U and Np. Thus, we understand the shorter bond O-CH2 and the longer bonds Pu-CH2 and Pu-O. These differences of geometrical and electronic configurations lead to a product more stable than for An = U and Np.
From a kinetic point of view, the barriers are found to be accessible (+4.6 kcal mol−1 for U, +4.2 kca mol−1 for Pu and +18.8 for Np). Relative to the transient carbene (7), the barriers are similar for all three atoms (around 20 kcal mol−1 as for the α-abstraction TS). Concerning the kinetic of the carbene insertion into the An-O bond, there is no difference between the three metals because the transition state (8) of An = Np is just shifted.
4 Conclusion
We have shown that the first step of ortho C-H activation of pyridine N-oxide catalyzed by Cp2An(IV)Me2 can occur with the three actinides (U, Np and Pu) and that they can form η2-(C,O) orthometalated complexes. The low symmetry of the metallocene complexes allows a strong mixture between the 5f and 6d orbitals, changing the properties of the bonding. Indeed, an increase of the covalency is observed in the actinide series, whereas the reverse is observed for the free ions or the actinyl ions. We have also concluded that for An = U and An = Pu, it is possible to lead to very stable actinide (IV) formaldehyde complexes going through actinide (IV) Schrock-type carbene intermediates. We have to notice that this reaction is calculated to be possible with a cyclopentadienyl ligand Cp. As shown in a precedent work [53], the reaction with (C5Me5) ligand is unfavourable due to a high barrier at the H2C-O coupling transition state (8). On the other hand, we think that it can be realized with a CpTMS ligand due to reduced steric effects. The formation of the formaldehyde complex is less obvious for An = Np because of a higher activation barrier for the transition state of formation of the transient carbene intermediate. It can be explained by a more covalent bond between Np and the carbene than for the two other actinides. Indeed, ionic metathesis transition states are more stabilized than covalent ones. This study is a new step to gain knowledge on the chemical behaviour of organoactinide complexes.
Acknowledgements
We are grateful to the CEA, CNRS and UPS for financial support of this work. LM is grateful to Institut universitaire de France. CalMip (CNRS, Toulouse, France), CINES (CNRS, Montpellier, France) and CCRT (CEA, France) are acknowledged for calculation facilities.