

1 Introduction
The history of actinide chemistry has been greatly influenced by the nuclear industry and many research programs were supervised in specialized laboratories having the necessary equipment in an interdisciplinary framework. This development was however much uneven, in terms of times, space and scope, because rapid answers must be given to the most urgent problems. Major attention was first turned to the synthesis of volatile compounds of uranium, for isotopic enrichment and fabrication of the atomic bomb, while recent efforts were devoted to the separation and elimination of wastes coming from nuclear power plants and destruction of weapons. These works obviously induced significant advances in actinide chemistry but large domains of this discipline had been neglected and were much less explored than those of other metals. It is clear today that this underdevelopment was not only the result of a chemical necessity but reflected a disfavored historical background. This situation is well illustrated with the advances in organometallic chemistry of actinides, in particular uranium.
The first organouranium compound, Cp3UCl (Cp = η-C5H5), was synthesized by Reynolds and Wilkinson in 1956, a few years after the discovery of ferrocene [1]. Fisher and Hristidu prepared the tetracyclopentadienyl complexes Cp4An (An = U, Th) in 1962 [2]. The German workers extended the series of cyclopentadienyl compounds to transuranium elements [3] and isolated the first organometallic complex of uranium(III), Cp3U, in 1970 [4]. A milestone in organometallic chemistry was the preparation in 1968 by Streitwieser and Mueller-Westerhoff of the so-called uranocene (COT)2U (COT = η-C8H8), the first representative of a new class of π-bonded cyclooctatetraenyl sandwich complexes [5]. Lugli et al. reported in 1974 on the stereospecific polymerization of butadiene catalyzed by uranium allyl complexes [6]. These initial results revealed the unique properties of uranium compounds, the novelty of their structures determined by the size of the ions and the participation of the f orbitals in metal-ligand bonding, and their remarkable performances in catalysis. But following a period of relative stagnation, it is only in the 1980's that organoactinide chemistry received a more regular and sustained attention and since the beginning of the 21st century, this discipline is witnessing a speeding up of its historical process which can be measured quantitatively by the increasing number of publications and characterized compounds and, very importantly, by the emergence of a new generation of young enthusiastic chemists [7]. In addition to the wish to find applications and give solutions to the topical problems of environmental remediation, these advances were clearly motivated by the revealed existence of fundamental and fascinating aspects of f elements chemistry. Organouranium complexes became much more attractive in view of their structures which were unsuspected or reputed non accessible, the multiplicity of their oxidation states exploited in redox reactions, their efficiency in the activation of small molecules and their peculiar magnetic properties. The remarkable improvements in computational investigations now permit to support an increasing number of experimental structural and thermodynamic data by a detailed theoretical analysis, giving a clear description of the bonding and the role of the f electrons.
This history of organoactinide chemistry, with its initial exciting discoveries followed by a rather long period of stagnation and its recent blooming, is well illustrated by the development of the uranium carbene complexes. By comparison with the considerable amount of work devoted to the carbene complexes of d transition metals due to their fundamental aspects and their extensive applications in organic synthesis and catalysis [8], the chemistry of such compounds with f elements remains largely unexplored. This was related to the strong ionic character of the metal-ligand bonding and the weak stabilization of the carbenic centre by π-back-donation from the metal fragment. The phosphoylide uranium compounds Cp3UCHP(Me)RR’, reported by Gilje et al. in 1981, were the first actinide carbenes with significant metal–carbon multiple bond character to be structurally characterized. However, nearly three decades passed before such new compounds came out again with the use of bis-phosphorus(V) stabilized carbon dianions as ligands. Meanwhile, uranium carbenoid species were detected in matrix isolation experiments and evidenced in McMurry type reactions; the only isolated carbene complexes were N-heterocyclic carbene (NHC) compounds which are in fact simple Lewis base adducts without significant M–C double bond character. Here we give a complete description of these successive steps in the development of uranium carbene complexes, in which we have been involved since 1997.
2 The first uranium carbene complexes
The uranium complexes Cp3UCHPMeRR’ (R = R’ = Me, Ph; R = Me and R’ = Ph) were synthesized by reaction of Cp3UCl with the lithium salt of the phosphoylide anion Li(CH2)(CH2)RR’ (Scheme 1) [9]. The crystal structures of Cp3UCHPMe2Ph and Cp3UCHPMe3 determined by X-ray or neutron diffraction analysis [9,10], showed that the uranium-carbon bond is the shortest yet observed, with a distance very close to 2.29 Å, and the UCP angle is slightly larger than 140°. These geometrical parameters clearly revealed the multiple bond character of the UC bond. The concept of multiple bond in Cp3UCHPR3 was supported by extended Hückel molecular orbital calculations which show covalency with a significant π component for the UCα bond [11]. The multiple uranium-carbon bonding can be formulated in terms of several resonance structures (A–C) with the hybrid D of these structures, as given in Scheme 1.
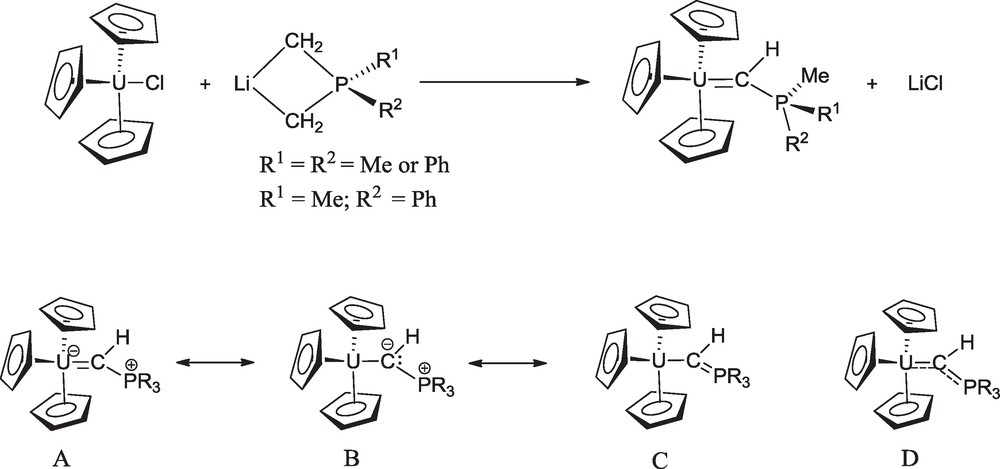
Synthesis and resonance forms of the phosphoylide complexes (C5H5)3UCHPR3.
Cramer, Gilje et al. outlined that these complexes represent, after the so called Fisher carbene compounds and the Shrock alkylidene derivatives, a third class of complexes with a metal carbon double bond in which a negative charge on the α carbon is stabilized by an electron withdrawing heteroatom substituent [12]. These authors also predicted that such bonds probably will be the most common type of metal-carbon double bonds in compounds of high-valent, largely ionic metals, where effective ligand to metal charge delocalization cannot occur.
In contrast to both Fisher carbene complexes and Shrock alkylidene complexes, the uranium phosphoylides were inert in the presence of internal monoalkenes and alkynes. However, consistent with the polar nature of the UC bond and the nucleophilic character of the α carbon, they react with the electrophile MeI, the weak acids Ph2NH and PhCCH, and the polar unsaturated molecules CO, MeCN, C6H11NC and PhNCO [13], according to Scheme 2. Thus, products from the insertion of small molecules into a metal-carbon multiple bond were structurally characterized for the first time.
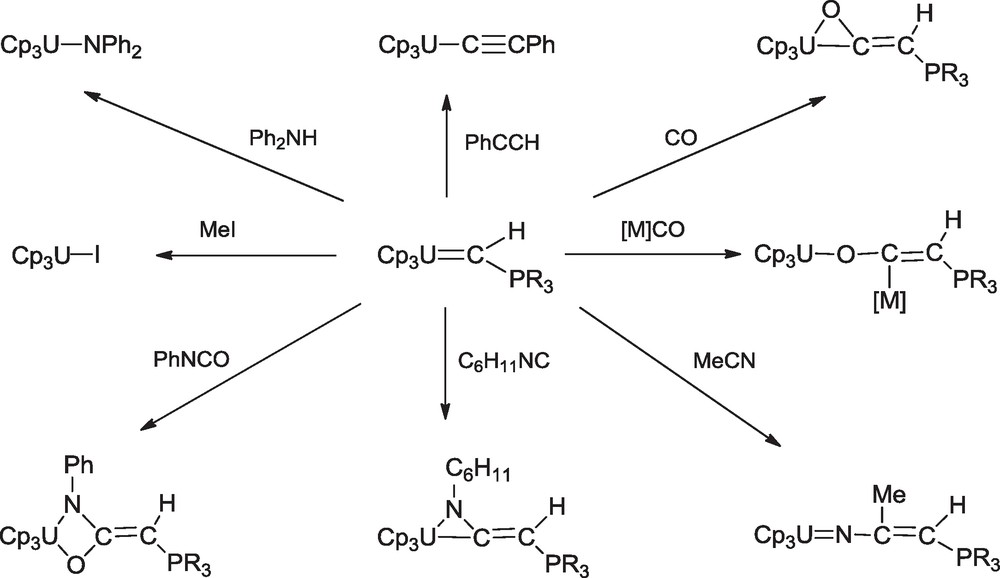
Reactions of the phosphoylide complexes (C5H5)3UCHPR3.
The carbene complexes Cp3UCHPMePhR also react with coordinated CO in carbonyl compounds [M](CO) [M = CpMn(CO)2, W(CO)5, CpCo(CO)] to give bimetallic derivatives of general formula MC(OUCp3)CHPMePhR (Scheme 2) [13c,e]. The last paper on these series of uranium carbenes was published in 1990, which described the neutron diffraction crystal structure of Cp3UCHPMe3 [10], and no study on such compounds was reported until 2009, with the use of geminal dianions (see chapter 5).
It should be noted here that a novel route to a uranium phosphoylide complex was designed in 2011 by the group of Hayton (Scheme 3). Treatment of U(NR2)3 (R = SiMe3) with 1 equivalent of the Wittig reagent Ph3PCH2 gave the U(III)-ylide adduct U(CH2PPh3)(NR2)3 which underwent in diethyl ether a one-electron oxidation concomitant with formal loss of H radical from the coordinated Wittig ligand to give the uranium(IV) carbene U(CHPPh3)(NR2)3. This latter was found to be in equilibrium with the metallacycle U(CH2SiMe2NR)(NR2)2 and the free Wittig reagent, an equilibrium which is shifted toward the formation of the carbene in diethyl ether at low temperature, thus permitting its synthesis under these conditions [14].
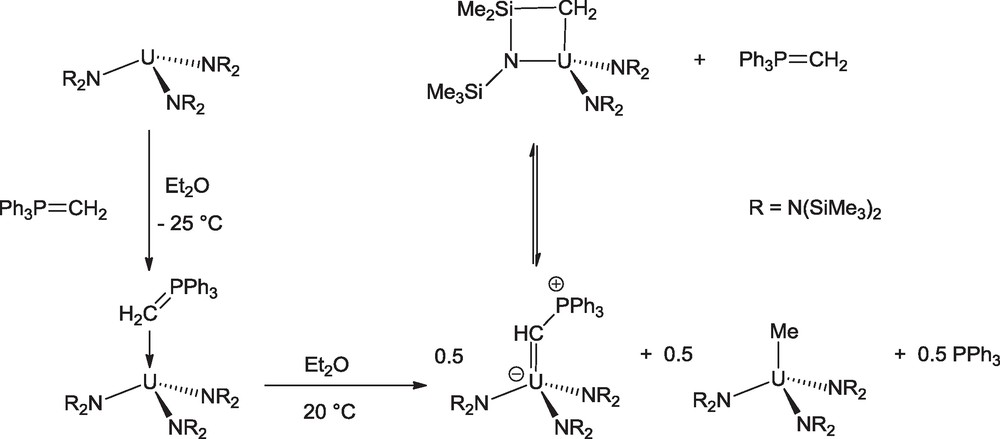
Synthesis of the phosphoylide complex U(CHPPh3)(NR2)3 (R = SiMe3).
3 Matrix preparation of actinide methylidene complexes and evidence for uranium carbenoid species in McMurry type reactions
3.1 Actinide methylidene complexes
The methylidene complexes H2CAnHX (X = F, Cl, Br, I) and H2CAnH2 (An = Th, U) have been prepared in 2006 by Andrews et al. by reactions of excited thorium and uranium atoms with methyl halides and methane in solid argon at 7 K (Scheme 4) [15].
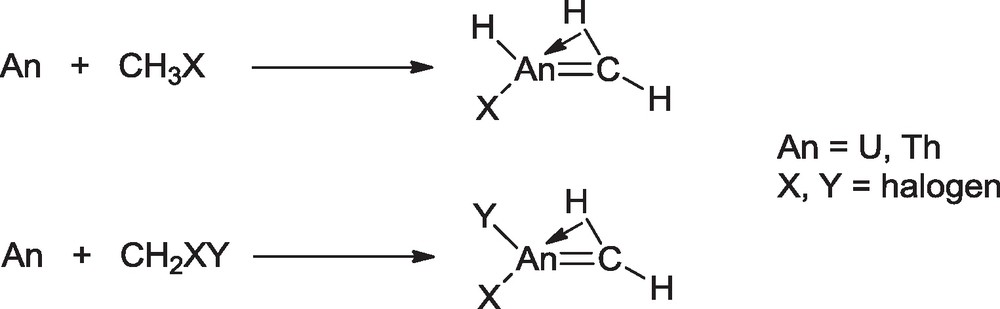
Synthesis of actinide methylidene complexes.
These simple organometallic complexes were identified by matrix infrared spectra through isotopic substitution and by comparison with vibrational characteristics calculated by DFT. Similar reactions of laser-ablated uranium atoms with methylene halides CH2XY (XY = F2, FCl and Cl2) gave the methylidene molecules H2CUF2, H2CUFCl, and H2CUCl2 as the major products [16]. These complexes exhibit highly distorted agostic structures with agostic HCU angles of ca 89°, in contrast with those of analogous transition-metal complexes such as H2CWF2, where a nonagostic ethylene-like symmetric structure is computed. Electronic structure calculations reveal that these U(IV) molecules all have strong UC double bonds in the triplet ground states with 5f2 configurations. The calculated bond lengths and bond energies indicate that the CU double bonds are slightly weaker in the fluoride species (2.066 Å, 109 kcal.mol−1) than in the chloride species (2.049 Å, 113 kcal.mol−1) because of the radial contraction of the U (6d) orbitals by the inductive effect of the fluorine substituent.
3.2 Uranium carbenoid species in McMurry type reactions
Our first encounter with uranium carbenes occurred in 1997 during our studies on the mechanism of the McMurry reaction, which is the reductive coupling of carbonyl compounds into alkenes upon treatment with low-valent titanium reagents [17]. Uranium and titanium complexes exhibit strong similarities in structure and reactivity, and uranium metal powder is effective in the reductive coupling of aromatic ketones. Moreover, uranium compounds have some advantages over their titanium counterparts: they can be easily detected by their highly shifted paramagnetic NMR signals and they often crystallize with less difficulty. Therefore, the chances of isolating and characterizing intermediates are greater. Indeed, the first metallopinacolate complexes isolated in a McMurry reaction were obtained at 25 °C by treating benzophenone or acetone with the UCl4-Na(Hg) system, and were transformed into the corresponding pinacol upon hydrolysis or the tetrasubstituted alkenes Ph2CCPh2 and Me2CCMe2 after further treatment with sodium amalgam at higher temperature [18,19]. These results were in agreement with the generally accepted mechanism of the McMurry reaction which involves the intermediacy of a metallopinacolate resulting from dimerization of two ketyl radicals.
Reaction of diisopropyl ketone was found to be quite different from that of acetone since the only coupling product, formed at 25 °C, was tetraisopropylethylene. In fact, formation of the corresponding pinacol was never observed in any McMurry reaction of diisopropyl ketone. Moreover, it was demonstrated that iPr2CCiPr2 could not be obtained by deoxygenation of the pinacolate Cl3M-OCiPr2CiPr2O-MCl3 which, in the case of MTi, was not stable towards rupture of the pinacolic CC bond and was readily transformed into TiCl3 and iPr2CO. The facile cleavage of the pinacolates and the absence of pinacol in the product mixtures indicated that reductive coupling of iPr2CO would not proceed by dimerization of ketyl radicals. A more careful analysis of the products showed that the McMurry reactions of iPr2CO afforded an important quantity of 2,4-dimethyl-2-pentene resulting from deoxygenation of the ketone. This observation, which was overlooked so far, revealed that carbenoid species were likely intermediates. These latter would be formed by reduction and deoxygenation of the ketyl radicals; their reaction with another molecule of ketone would give iPr2CCiPr2, presumably via a metallaoxetane complex, and their rearrangement by H migration would afford Me2CC(H)iPr (Scheme 5) [20].
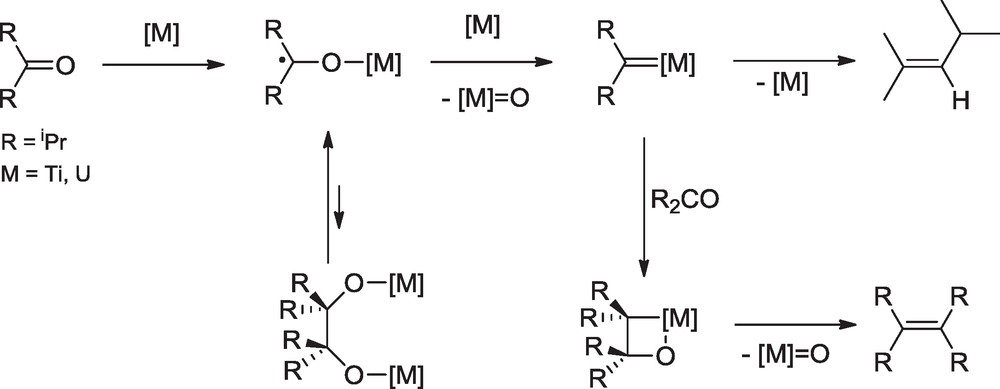
Involvement of carbenoid species in the reductive coupling of diisopropyl ketone with the MCl4-Li(Hg) system (M = Ti, U).
These results revealed the dual nature of the mechanism of the McMurry type reactions. Contrary to the generally accepted mechanism, metallopinacols are not the only precursors to the alkene; if the ketyl radicals can be effectively coupled into pinacolate intermediates, they can also be reduced and deoxygenated into carbenoid species which provide the alkene after further reaction with the ketone. The course of the reaction, via the metallopinacol or the carbenoid intermediates, is largely determined by the steric hindrance of the ketone; the most hindered ketones would follow the carbenoid route because of the difficult coupling of the ketyl radicals and the reversible cleavage of the pinacolic CC bond [21]. The involvement of carbenoid species in the McMurry reaction of sterically hindered ketones was further demonstrated by analysis of the products resulting from the reduction of ditertiobutyl ketone. In that case, H migration within the carbenoid species [M]CtBu2 gave the expected cyclopropane compound in 2% yield with M = Ti. The major product was tBu2CH2 (40 and 50% yield for M = Ti and U, respectively) which was liberated after hydrolysis of the reaction mixture; deuterolysis experiments confirmed that this alkane was formed by successive addition of H(D) atoms to the carbenoid species [M]CtBu2. The latter were thus much more stable than [M]CiPr2 and could also be trapped with aldehydes RCHO to give the cross-coupling products tBu2CC(R)H (R = Me, tBu) (Scheme 6) [21].
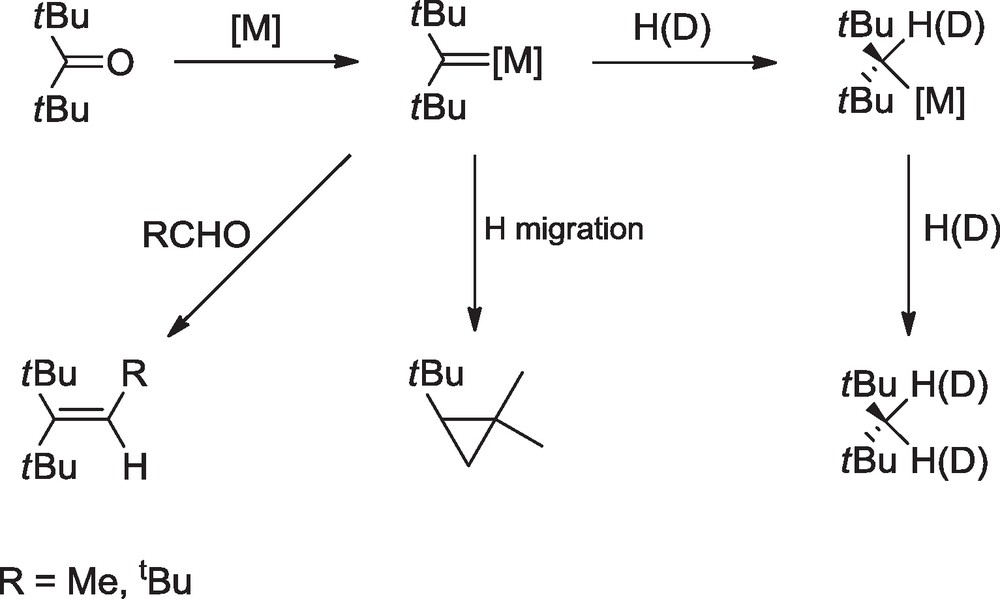
Involvement of carbenoid species in the reaction of ditertiobutyl ketone with the MCl4-Li(Hg) system (M = Ti, U).
4 Uranium complexes with N-heterocyclic carbene ligands
N-heterocyclic carbene (NHC) ligands are most well-known as soft, two-electron σ-donors and are used widely as strongly basic phosphine analogues, to support late transition metal complexes [22]. In contrast to alkyl phosphines, NHC are effective ligands for high-valent metal complexes [23]. The first NHC compounds of an f element, Cp*2Sm(C{NMeCMe}2)2 and (tBu2acac)3Eu(C{NHCMe}2) (tBu2acac = tBuC(O)CHC(O)tBu) were isolated by Arduengo et al. in 1994 [24], and such carbenes were coordinated to uranium after 2000 [25]. To the best of our knowledge, no NHC complex of thorium was structurally characterized. Monodentate NHC adducts have been reported for tri-, tetra- and hexavalent uranium, as illustrated by ({Me3Si}2N)3U(C{NMeCMe}2) [26], Cp*2UO(C{NMeCMe}2) [27] and UO2Cl2(C{NMesCH}2)2 (Mes = mesityl) [28], represented in Scheme 7.
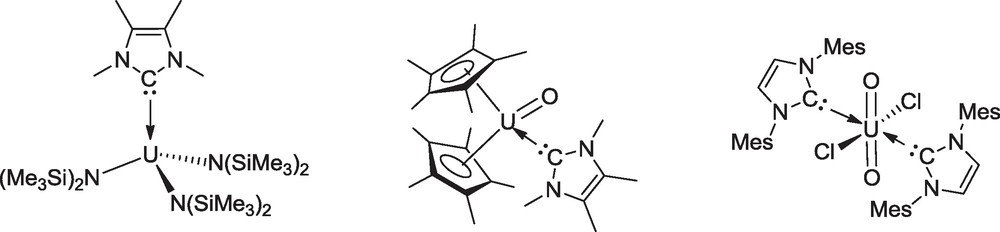
NHC complexes of uranium in the +3, +4 and +6 oxidation states.
Functionalized NHC ligands were designed to stabilize electropositive metal cations. Reaction of UCl4 with the pincer dicarbene ligand CNC gave the adduct (CNC)UCl4 [CNC = 2,6-bis(arylimidazol-2-ylidene)pyridine with aryl = 2,6-iPr2C6H3] [29]. The UO22+ ion was complexed by the alkoxide- and amido-NHC ligands OCMe2CH2[1-C(NCHCHNiPr)] (Lo) [30] and tBuNCH2CH2[1-C(NCHCHNtBu)] [31]. The alkoxide-NHC ligand Lo was also attached to the U4+ ion in the complexes U(Lo)3I and U(Lo)4; in this latter, one of the four carbene groups remains uncoordinated, being in fast exchange in solution with the other bound NHC ligands, and was trapped by BH3 and a range of 16-valence-electron metal carbonyl fragments [32].
We have found a novel aspect of the NHC ligand's chemistry, which is their high capacity to discriminate between trivalent lanthanide (Ln) and actinide (An) ions. This separation, which is a difficult task in view of the similar properties of the ions, represents a challenging problem in both its fundamental aspects and potential applications, especially the reprocessing of spent nuclear fuels. Selective complexation of An3+ over Ln3+ ions reflects the slightly less hard character of the former, leading to a better affinity towards soft nitrogen donors and to the formation of bonds with a larger degree of covalency. A usual way to probe the extent of covalent interactions in actinide complexes is to compare bond distances in analogous uranium(III) and lanthanide(III) compounds [33]. The crystal structures of the two pairs of isomorphous U(III) and Ce(III) complexes (C5H4tBu)3M(C{NMeCMe}2) and Cp*2MI(C{NMeCMe}2) (M = U, Ce) (Fig. 1) show that the uranium-carbene distances are shorter by ca 0.03 Å than the cerium-carbene distances while the ionic radius of uranium(III) is 0.02 Å longer than that of cerium(III) [34]. Such shortening of the uranium-ligand distances from a purely ionic bonding model is accounted for by the presence of a stronger, more covalent interaction between the actinide and the soft ligand.
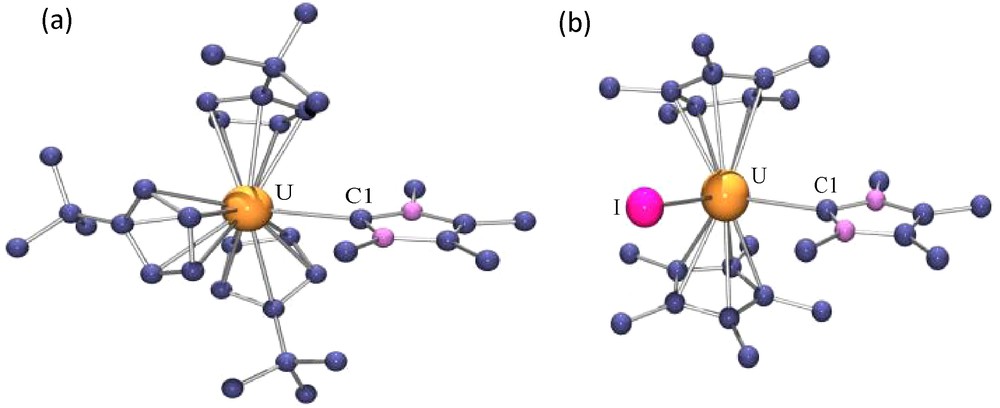
X-ray crystal structures of (C5H4tBu)3U(C{NMeCMe}2) (a) and Cp*2UI(C{NMeCMe}2) (b).
The much stronger affinity of the NHC molecule for the 5f than the 4f ion was confirmed by competition reactions (Scheme 8). Addition of the NHC molecule to 1 mol equivalent of both the analogous trivalent uranium and cerium complexes in THF gave the uranium and cerium carbene complexes in a molar ratio of 80:20 at 23 °C, and 90:10 at –60 °C. By comparison with the other molecules which have been considered for the differentiation of the (C5H4R)3U and (C5H4R)3Ce complexes (i.e. phosphine, phosphite, isonitrile and azine ligands), the NHC is by far the most efficient, presenting the best affinity and the best selectivity for the U(III) compound [34]. The selective complexation of the uranium over the cerium metallocenes by C3Me4N2 in THF can only be compared with that of UX3 over CeX3 (X = I, OTf) by 2,6-bis(5,6-dialkyl-1,2,4-triazin-3-yl)pyridine (Rbtp) molecules in pyridine [33a]. Currently, these terdentate nitrogen ligands are among the most promising extractants available for the hydrometallurgical reprocessing of nuclear waste.
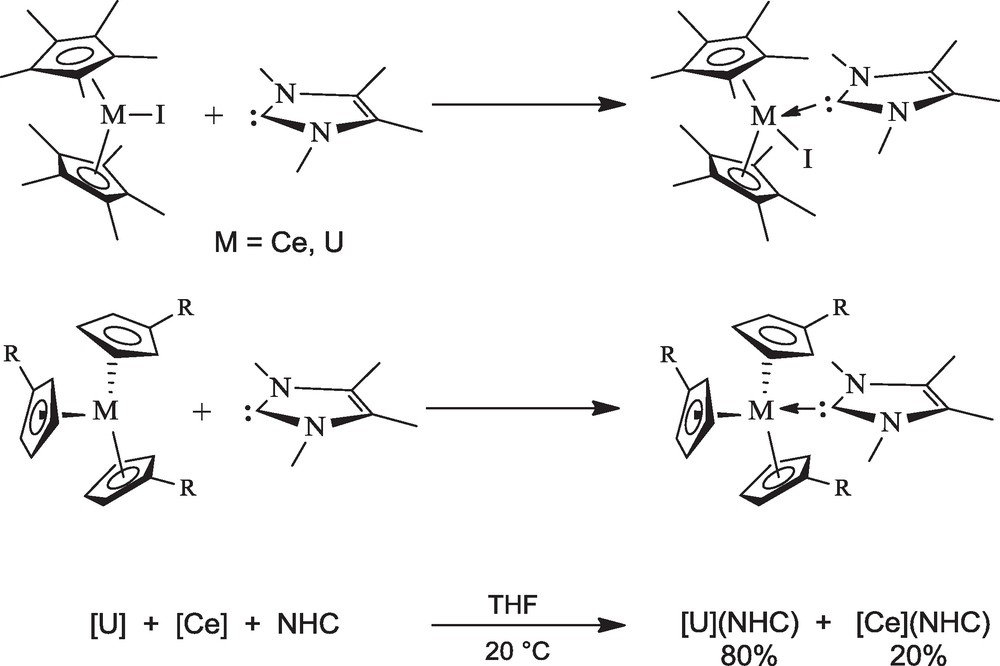
Efficiency of NHC in cerium(III)/uranium(III) differentiation.
5 Bis-phosphorus stabilized actinide carbene complexes
5.1 Synthesis and crystal structures of the actinide(IV) complexes
A novel synthetic route to carbene complexes was opened with the design and use of geminal carbon dianions stabilized by phosphorous(V) substituents [35]. The real beginning of this new development dates back from 1999, when the groups of Stephan and Cavell reported independently the high yield preparation of the bis(iminophosphoranyl) methanediide ligand [Ph2P(NR)]2C2−(R = SiMe3) [36,37]. Other such bis(iminophosphorane) derivatives with various R substituents, named (RNCN)2− thereafter, were synthesized during the 2000–2006 period, while Le Floch et al. reported in 2004 the quantitative preparation of the bis(thiophosphinoyl)methanediide [Ph2P(S)]2C2−, named (SCS)2− thereafter [38]. The most accurate Lewis structures to describe the electronic structure of the germinal dianions (RNCN)2− and (SCS)2− are presented in Scheme 9.
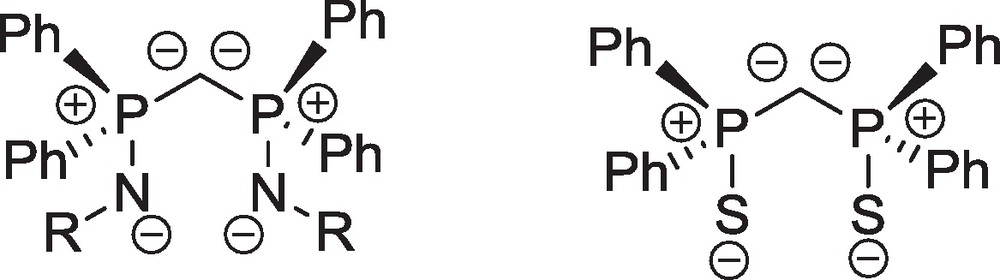
Most accurate Lewis structures to describe the electronic structures of the germinal dianions (RNCN)2− and (SCS)2−.
In the new class of RNCN and SCS complexes, and like in the phosphoylide complexes isolated by Gilje et al., the four electrons of the formal MC double bond are provided by the sole ligand, in contrast to the Fischer and Schrock type carbenes, where both the carbenic and metal fragments bring two electrons to the MC interaction (Scheme 10). The bis-phosphorus stabilized carbene compounds could therefore be constructed from any electron deficient metal center and, most notably, from oxidized f-element ions, where stabilization of the carbenic centre by π-back donation from the metal fragment is very unlikely. Similarly to Schrock alkylidenes, these carbene complexes exhibit a nucleophilic character.
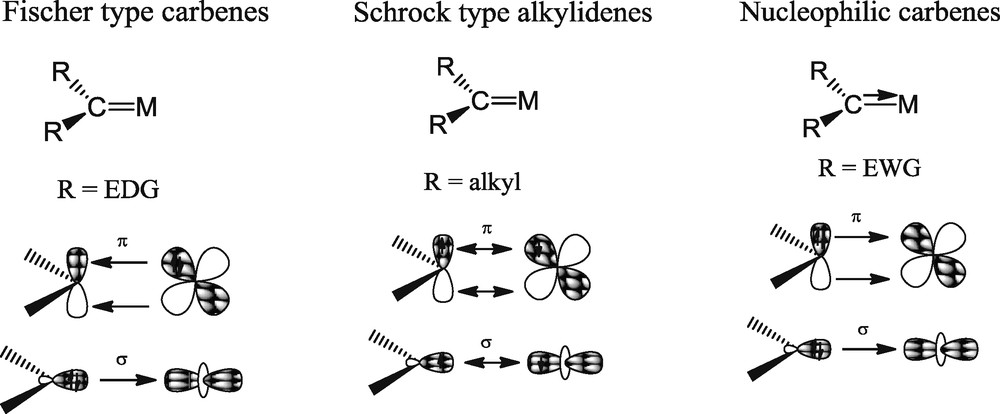
Bonding modes in the different types of metal carbene complexes.
We introduced the SCS ligand in 5f element chemistry with the reactions of the lithium salt of the germinal dianion and uranium tetrachloride [39,40] or uranium tetraborohydride [41]. Treatment of UCl4 with 1, 2 or 3 molar equivalents of Li2SCS gave respectively the mono-carbene “ate” complex Li(THF)2U(SCS)Cl3(THF), the bis-carbene complex U(SCS)2(THF)2 and the unique example of a homoleptic tris-carbene complex, Li2(OEt2)2U(SCS)3, in very good yields (Scheme 11). Without going into details of experimental conditions, it is necessary to note the importance of the nature of the solvent and the order of introduction of the reagents, in particular for avoiding the detrimental protonation of the (SCS)2− dianion into the (SCHS)− monoanion in protic solvents. The thorium analogues Th(SCS)2(DME) and [Li2(DME)Th(SCS)3]n were synthesized by Fang et al. from ThCl4(DME)2 [42]. The uranium bis-carbene complex was also obtained in almost quantitative yield from a 1:2 mixture of uranium tetrachloride and the tris-carbene in THF. Under the same conditions, the comproportionation reaction between uranium chloride and the bis-carbene in a 1:1 molar ratio led to the quantitative formation of the neutral mono-carbene compound U(SCS)Cl2(THF)2 [39,40].
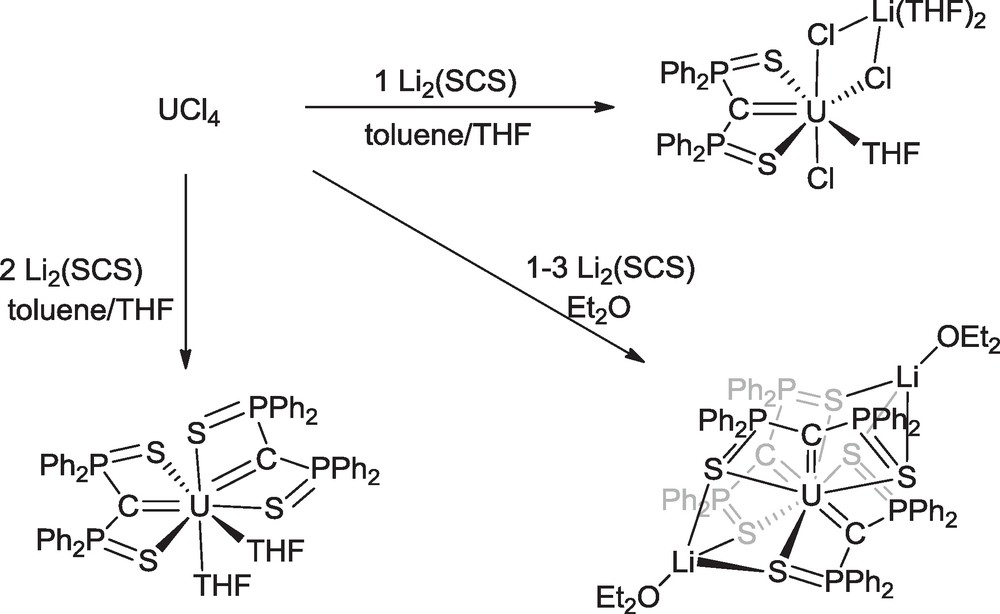
Synthesis of uranium carbene complexes from UCl4.
The tris-carbene complex was also obtained by using the uranium tetraborohydride as precursor. Reaction of U(BH4)4 with 1 equivalent of Li2SCS in toluene afforded the trinuclear complex U(μ-SCS)3[U(BH4)3]2 which was transformed in THF into the mononuclear derivative U(SCS)(BH4)2(THF)2 (Scheme 12) [41].
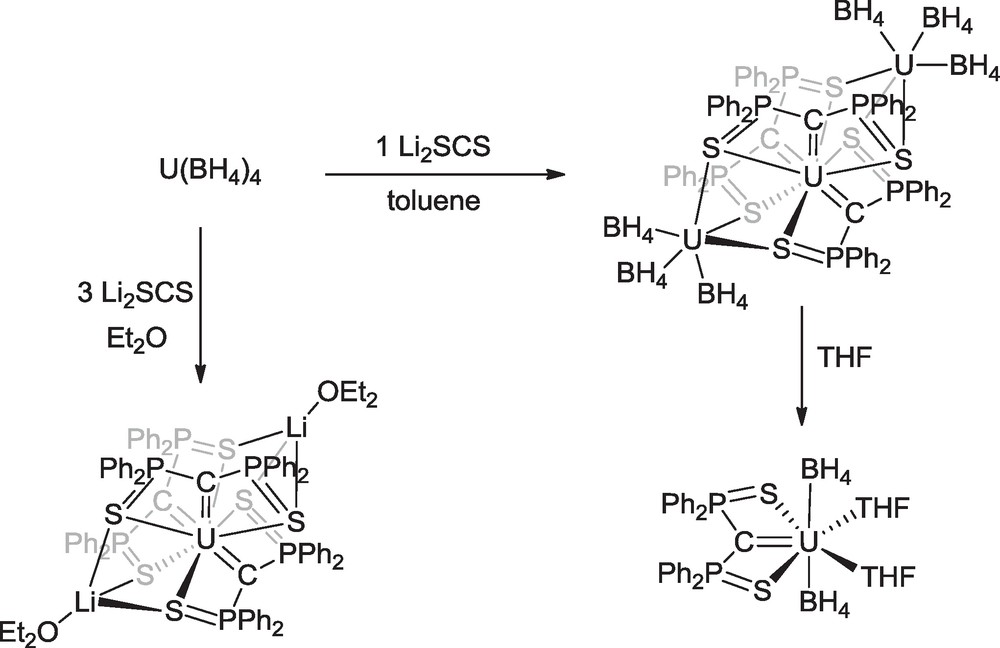
Synthesis of uranium carbene complexes from U(BH4)4.
The uranium amides U(NEt2)4 and [U(NEt2)3][BPh4] were found to be also good precursors of carbene complexes by protonolysis reactions with the neutral molecule [Ph2P(S)]2CH2 (SCH2S). Thus were obtained U(SCS)(SCHS)(NEt2), the first mixed alkyl-carbene compound of an actinide with both methanide and methanediide ligands, and [U(SCS)(NEt2)(THF)3][BPh4], the first example of a cationic uranium carbene complex (Scheme 13) [40]. The crystal structures of the chloro-, tetrahydroborato and amido-carbene complexes are shown in Fig. 2.
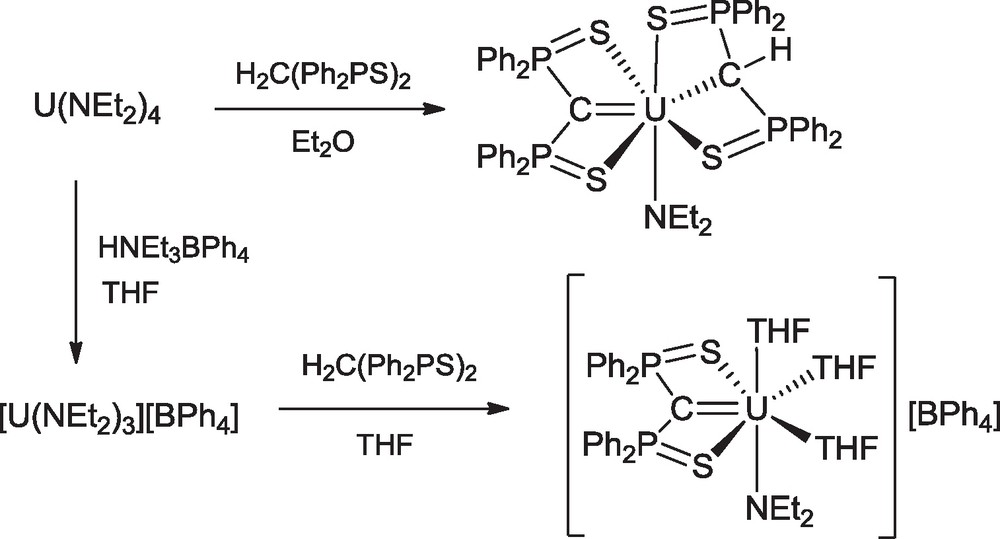
Synthesis of uranium carbene complexes from U(NEt2)4.
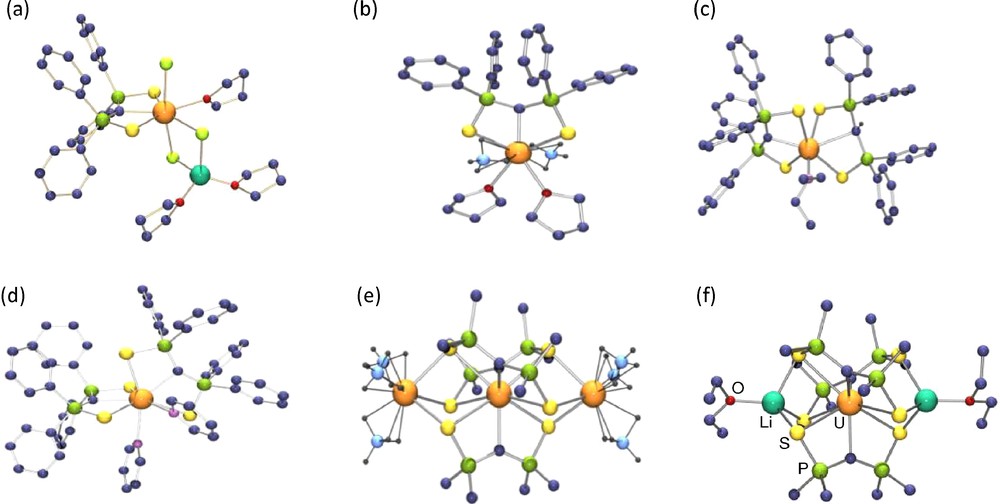
X-ray crystal structures of Li(THF)2U(SCS)Cl3(THF) (a), U(SCS)(BH4)2(THF)2 (b), U(SCS)(SCHS)(NEt2) (c), U(SCS)2(py)2 (d), U(SCS)3[U(BH4)3]2 (e), Li2(OEt2)2U(SCS)3 (f).
Liddle et al. have used the (MesNCN)2− ligand to prepare the homoleptic bis-carbene complex U(MesNCN)2 [43] while Cavell et al. synthesized the mono-carbenes An(™SNCN)Cl2 (An = U, Th) which were transformed into the cyclopentadienyl and the pyrazolyl borate derivatives Cp2An(™SNCN) and TpAn(™SNCN)Cl [44].
The series of mixed uranium SCS carbene complexes was extended to π-organometallic compounds. The anionic, neutral and cationic mono-carbenes were used as precursors for the synthesis of cyclopentadienyl and cyclooctatetraenyl derivatives (Schemes 14 and 15) [40]. Treatment of the “ate” complex with 1 molar equivalent of freshly sublimed TlCp in THF gave the trinuclear compound Tl[CpU(SCS)]2(μ-Cl)3 which can be seen as the assemblage of the expected monocyclopentadienyl uranium carbene CpU(SCS)Cl with half the quantity of the by-product thallium chloride. Such compounds where thallium(I) coordinates to metal-bound halide ions to form soluble coordination compounds are quite uncommon and have been obtained by serendipity. The same reaction with 2 molar equivalents of TlCp gave the biscyclopentadienyl complex Cp2U(SCS). In contrast, the bis(pentamethylcyclopentadienyl) analogue could not be obtained from the “ate” complex and was synthesized by reaction of Cp*2UCl2 and Li2SCS in diethyl ether (Scheme 14).
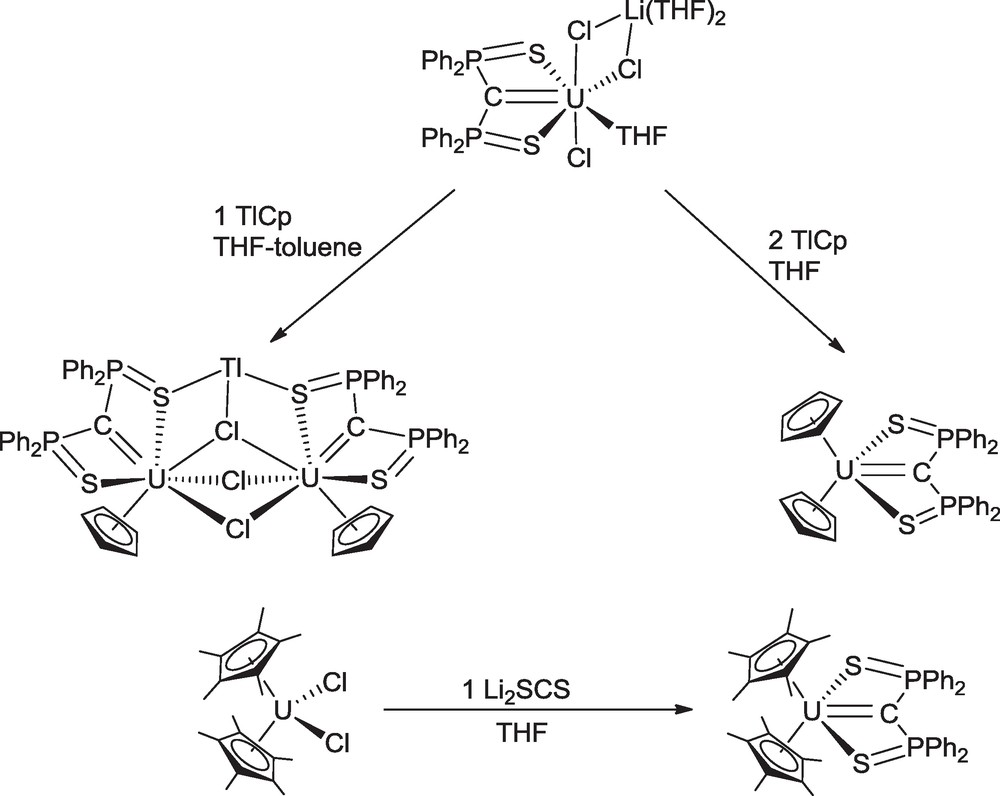
Synthesis of cyclopentadienyl uranium carbene complexes.
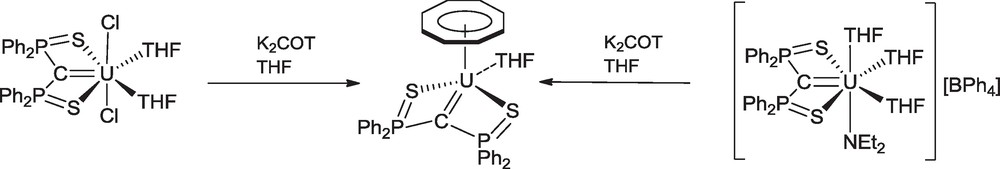
Synthesis of the cyclooctatetraenyl uranium carbene complex (COT)U(SCS)(THF).
Although the chemistry of the monocyclooctatetraenyl uranium compounds has been significantly developed during the last years, such complexes with uranium-carbon bonds remain very rare. The monocyclooctatetraenyl uranium carbene (COT)U(SCS)(THF) was synthesized by metathesis reaction of U(SCS)Cl2(THF)2 and K2COT in THF. This complex was alternatively prepared in almost quantitative yield from the cationic precursor [U(SCS)(NEt2)(THF)3][BPh4] (Scheme 15) [40]. The crystal structures of the cyclopentadienyl and cyclooctatetraenyl carbene complexe are shown in Fig. 3.
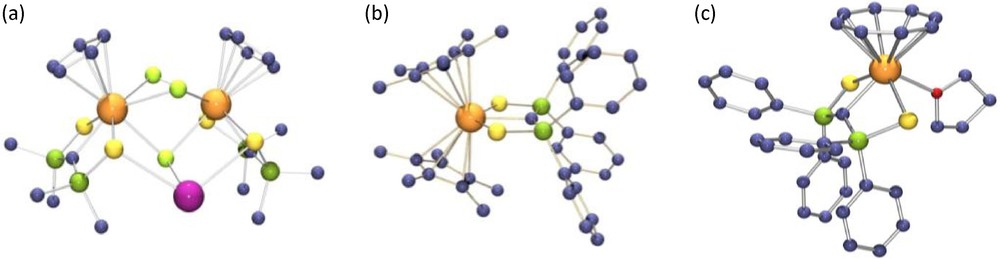
X-ray crystal structures of Tl[CpU(SCS)]2(μ-Cl)3 (a), Cp*2U(SCS) (b) and (COT)U(SCS)(THF) (c).
The crystal structures of the uranium SCS compounds show that, with the exception of the trinuclear compounds M2U(SCS)3 [M = U(BH4)3 or Li(OEt2)] where the central uranium atom is sterically encumbered and very electron rich, the UC bond distances vary from of 2.327(3) Å in U(SCS)(BH4)2(THF)2 to 2.396(4) Å in Cp*2U(SCS) derivative. These short distances are similar to those measured in the phosphoylide complexes isolated by Gilje et al. They are smaller than the σ UC bond lengths of uranium(IV) alkyl compounds, which are comprised between 2.4 and 2.6 Å, and significantly smaller than those found in uranium(IV) complexes with N-heterocyclic carbene ligands, by more than 0.2 Å. In all the complexes, the P–C distances with an average value of 1.676 Å are short, suggesting that the two lone pairs at the carbon atom are stabilized by negative hyperconjugation into the Ph2PS arms. The U–S–P–C–P–S cores are planar, except in the monocyclooctatetraenyl compound which adopts a classical four legged piano stool configuration. The short U–C distances and the planarity at the carbene carbon atoms show the donation of both lone pairs from the dianionic fragment to the metal centre and the likely formation of a UC double bond.
Similar short U–C distances were found in the uranium RNCN complexes, as shown by the values of 2.336(6) and 2.351(2) Å for Cp2U(SCS) [40] and Cp2U(™SNCN) [44], respectively. However, the RNCN ligands generally adopt an “open book” conformation, in contrast to the planar SCS ligands. This difference likely reflects the much greater steric constraints due to UN and PN bonds much shorter than US and PS bonds and to the presence of bulky substituents on the coordinated nitrogen atoms.
Comparison of the crystal structures of analogous uranium and thorium carbene complexes Cp2An(™SNCN) (An = U, Th) [44], TpAn(™SNCN)Cl (An = U, Th) [44], U(SCS)2(py)2 [40] and Th(SCS)2(DME) [42], Li2(OEt2)2U(SCS)3 [40] and [Li2(DME)Th(SCS)3]n [42] indicates that the differences of 0.07–0.09 Å between the Th–C and U–C distances are larger than the value of 0.05 Å expected from the variation in the radii of the Th4+ and U4+ ions [45]. These features suggest that the double bond character of the MC bond is less pronounced for M = Th than for M = U.
5.2 Electronic structures of the actinide(IV) complexes
The electronic structure of the actinide carbenes has been investigated using DFT approaches. The optimized structure of the model complex U(C{PH2(S)}2)(BH4)2(THF)2 is in perfect agreement with the X-ray diffraction data obtained for U(SCS)(BH4)2(THF)2. The interaction between the uranium(IV) ion and the dianionic ligand is described by the HOMOs (Fig. 4). In particular, the HOMO–6 clearly describes the UC σ-bond which results from the donation of the carbon sp2 lone pair to a vacant hybrid orbital on U while the HOMO–2 mainly corresponds to a UC π-bond, polarized on the C atom [41].
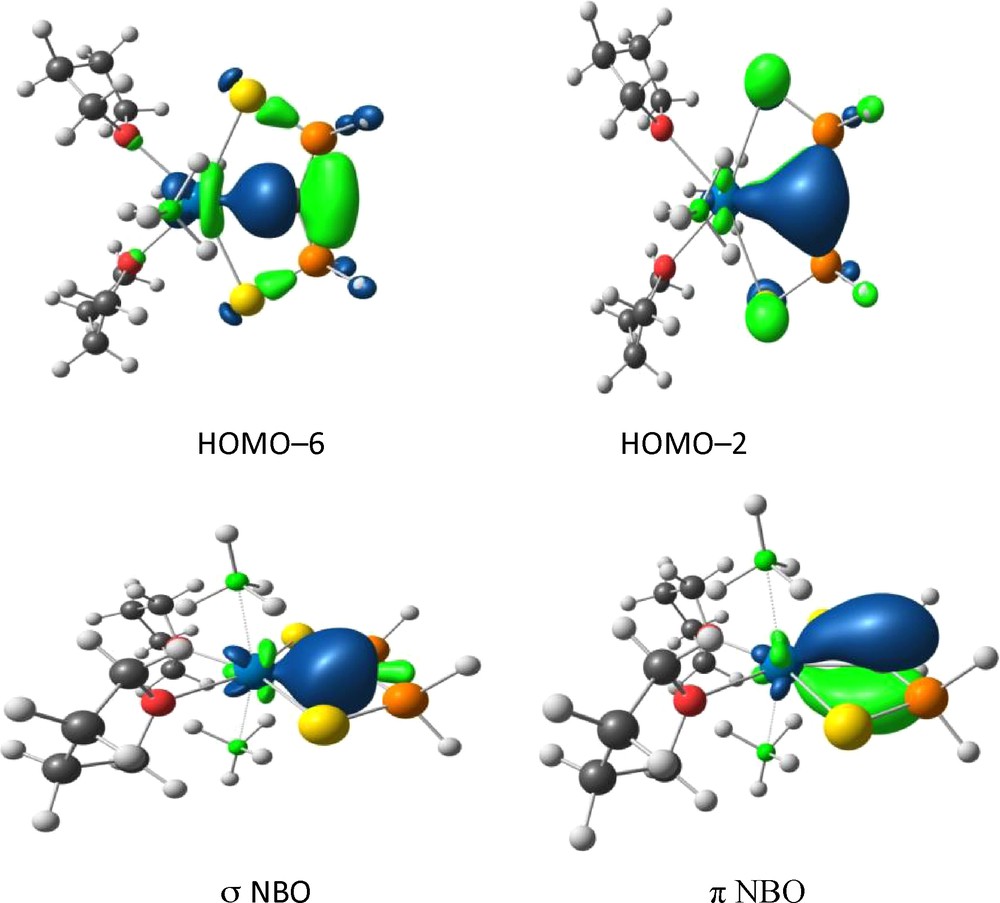
Plots of the HOMO and NBO for U(C{PH2(S)}2)(BH4)2(THF)2 describing the UC double bond.
The Natural Bond Orbital analysis gives a more localized and chemically relevant picture of the UC interaction. The NBO describing the two UC σ and π-bonds are represented in Fig. 4. The UC σ-bond comes from the interaction of the C sp2 lone pair (80.7%) with a uranium hybrid orbital (19.3%) of 52.6% 5f and 37.0% 6d character. The UC π-bond is made of a carbon 2p pure lone pair (82.9%) and a metal hybrid orbital (17.1%) of 59.0% 5f and 40.9% 6d character. This bonding scheme shows that the UC double bond is polarized towards the carbon atom, in agreement with the Mulliken analysis. The polarization is not only due to the greater electronegativity of carbon compared to uranium but also to the presence of two thiophosphinoyl groups attached to the carbene centre. The two lone pairs on the carbon atom are indeed stabilized by negative hyperconjugation in the free SCS2− ligand. The same occurs also, but to a lower extent, in the complex. Upon coordination to the metal fragment, the charge of the carbon atom drops from –1.90 in the free ligand to –1.52 in the complex. This is diagnostic of a significant electron transfer from the two carbon lone pairs to the U(IV) metal centre that establishes the UC double bond. The NBO analysis also confirms the important involvement of the uranium 5f orbitals in stabilizing the UC double bond. Overall, the contribution of the uranium 5f orbitals is somewhat higher than that of the 6d orbitals in the UC interaction. Though the 5f orbitals are more radially contracted than the 6d AOs, the 5f AOs are indeed lower in energy in uranium and can lead to greater angular overlaps where symmetry constraints are high (such as in HOMO–2) [41].
DFT calculations on the whole molecule of U(MesNCN)2 [43] and parent models of Cp2An(™SNCN) and TpAn(™SNCN)Cl (An = U, Th) [44] also showed that the components of the MC double bond involve the 5f and 6d orbitals of the actinide metal and the carbene 2p orbitals, as both σ bond and π bond components. The formation of the formal MC double bonds is not disfavoured by the ‘open book’ conformation of the RNCN carbene ligands. Wiberg bond indices indicated that the UC bond is stronger than the ThC bond, which is consistent with the relative U–C and Th–C distances in Cp2An(™SNCN). The double σ + π donation toward the metal atom was demonstrated in the thorium complexes Th(SCS)2(DME) and [Li2(DME)Th(SCS)3]n where the ThC double bonds were described as single bonds with a lone pair localized on the carbene carbon atom [42].
Theoretical calculations revealed that the bonding scheme in uranium(IV) phosphoylides U(CHPPh3)(NR2)3 and Cp3UCHPMe2Ph is quite identical to that determined in SCS and RNCN carbene compounds, the U–C interaction being highly polarized with modest π character [14].
For comparing the electronic structure of the uranium(IV) carbene complexes with transition metal analogues and explaining the distinct influence of the coordination environment around the M4+ ion (M = Zr, U) on the MC double bond, the electronic structure of parent models of the complexes M(SCS)Cl2(py)2 and Cp2M(SCS) (M = Zr, U) has been investigated [40]. Together with crystal data, theoretical calculations show that the UC multiple bond presents a covalent character similar to the bonding situation in transition metal complexes. In addition, the vacant 5f atomic orbitals on the actinide ion engage in covalent interactions with the carbene centre, so as to better stabilize the carbene valence orbitals. This effect is noticeable in the Cp2M(SCS) complexes for which the repulsive interaction between the SCS dianion and the Cp anions is observed for MZr while the 5f orbitals play a “buffer” role in the uranium compound, leading to a metal-carbon bond elongation in the zirconium complex only (Scheme 16). As a result, and in contrast with transition metal complexes, changes in the coordination sphere of the uranium(IV) centre have little influence on the UC bond.
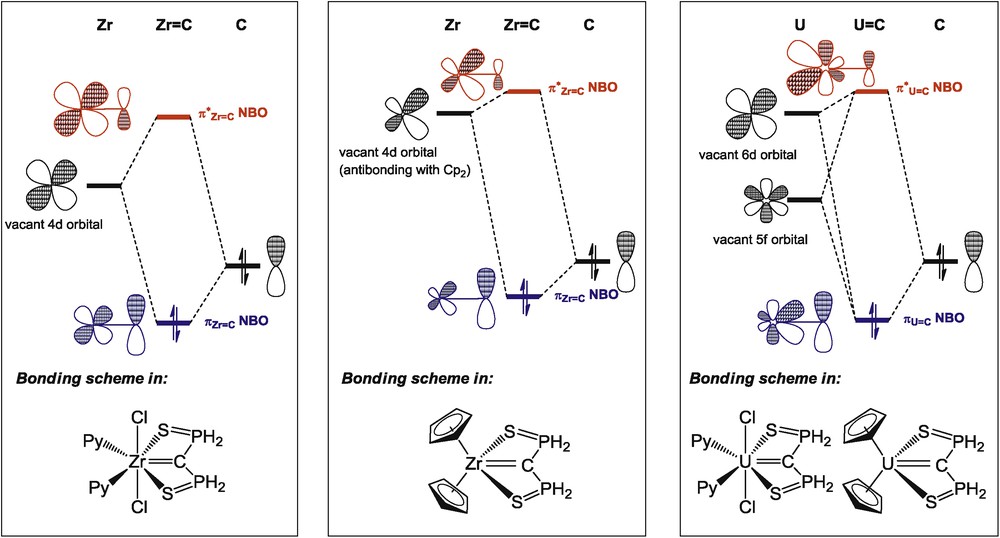
Representation of the metal–carbon double bond in zirconium and uranium carbene complexes.
5.3 Uranium carbene complexes in the +5 and +6 oxidation states
The SCS and RNCN ligands were found to also stabilize uranium compounds in their highest oxidation states. The group of Liddle recently reported on the oxidation of the uranium(IV) “ate” complex Li(THF)2U(™SNCN)Cl3(THF) with iodine and 4-morpholine N-oxide which led to the formation of the uranium(V) compound U(™SNCN)Cl2I [46] and the uranium(VI) oxo derivative U(™SNCN)Cl2(O) [47], respectively (Scheme 17). The U–C distances decrease with increasing oxidation state, with values of 2.310(4), 2.268(10) and 2.183(4) Å.
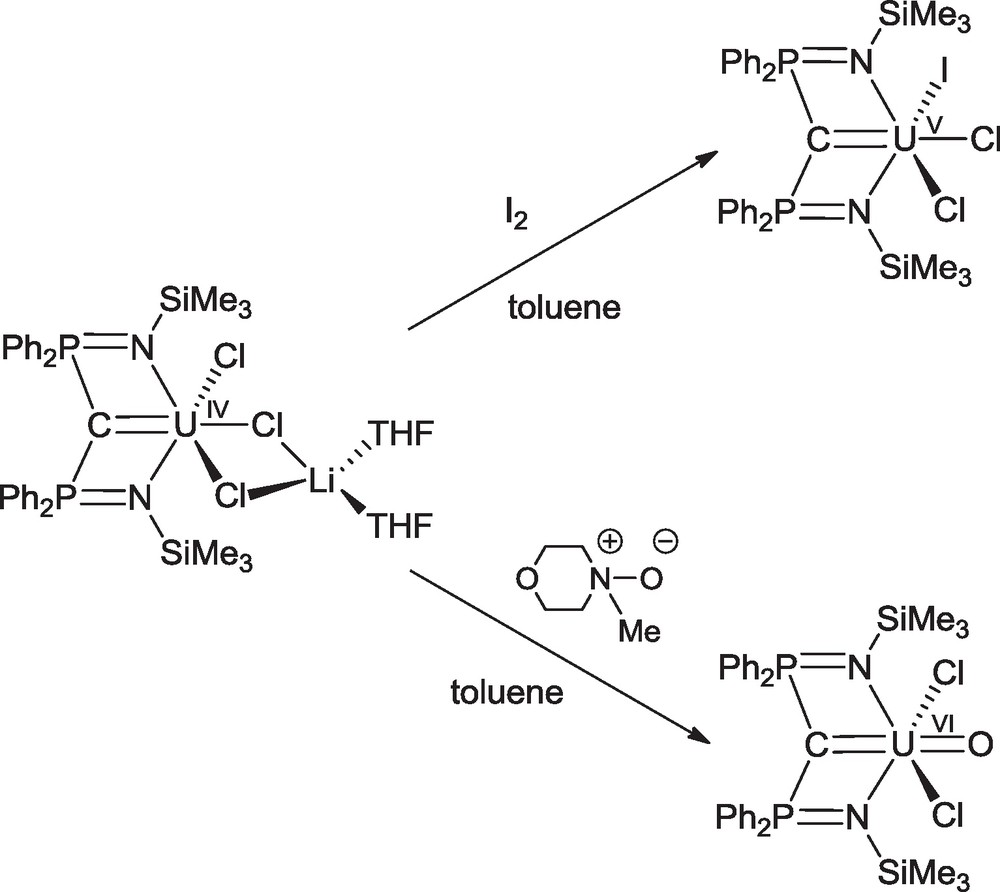
Synthesis of U(V) and U(VI) carbene complexes.
Natural Bond Order analyses indicate that upon oxidation from uranium(IV) to (V) to (VI) the uranium contribution to the UC σ-bond can increase from ca. 18 to 32%, while for the corresponding UC π-components, the uranium contribution increases from ca. 18 to 26% but then decreases to ca. 24%. The calculations suggest that as a function of increasing oxidation state of uranium, the radial contraction of the valence 5f and 6d orbitals may outweigh the increased polarizing power of uranium in U(™SNCN)Cl2(O) compared to U(™SNCN)Cl2I [47].
The synthesis of uranyl compounds with metal–carbon bonds remains a challenging goal because of the reduction of U(VI) to U(V) by the alkyl ligand. As such, only two uranyl complexes with alkyl or π-organometallic ligands have been observed to date: the cyclopentadienyl compound [NEt4]2[UO2(Cp*)(CN)3] [48] and the bis-iminophosphoranyl complex [UO2(™SNCHN)Cl]2 and its derivatives [49]. Interestingly, the cyclopentadienyl uranyl complex was not synthesized by treatment of a UO2X2 precursor with the [C5Me5]− anion, a reaction which provides in fact a convenient route to the corresponding pentavalent UO2X derivative [50], but was isolated from the reaction of the linear metallocene [NEt4]3[U(Cp*)2(CN)5] and pyridine N-oxide. The uranyl carbene complex UO2(SCS) was synthesized in 95% yield by treatment of uranyl triflate with Li2SCS in pyridine or by protonolysis of UO2(N{SiMe3}2)2(THF)2 with SCH2S (Scheme 18) [51]. The crystal structure shows the planar carbene ligand (Fig. 5). The U–C distance of 2.430(6) is 0.2 Å smaller than those measured in the uranyl methanide complexes UO2(SCHS)(OTf)(Et2O) and UO2(SCHS)2, which were isolated from reactions of UO2(OTf)2 with LiSCHS [51]. However, this distance is 0.1 Å larger than those found in mononuclear uranium(IV) carbenes, while the radius of the UO22+ ion is 0.2 Å smaller than that of U4+. These features suggest that the multiple bond character of the UC bond is less pronounced in the uranyl than in the uranium(IV) compounds.
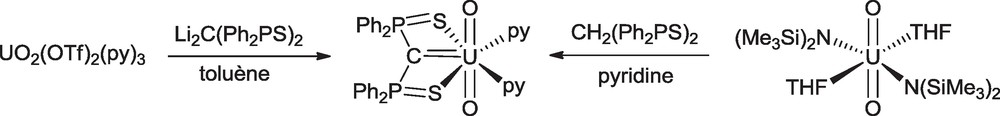
Synthesis of the uranyl carbene complex UO2(SCS)(py)2.
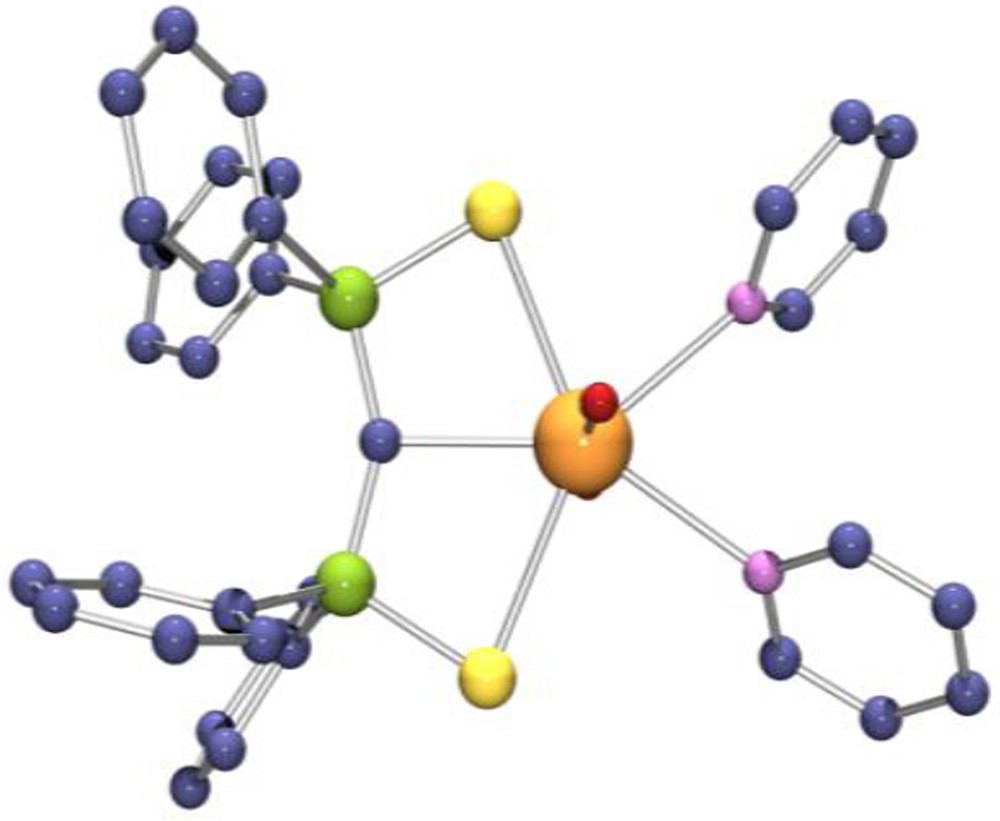
X-ray crystal structure of UO2(SCS)(py)2.
DFT calculations showed that the UC interaction in the uranyl carbene is properly described by two distinct NBO, as previously found for uranium(IV) and zirconium(IV) complexes (Fig. 6). The formation of the UC double bond upon conversion of the methanide to carbene complex is clearly reflected by the change in the uranium–carbon bond Wiberg bond order which increases from 0.43 in the alkyl complex to 0.91 in the carbene. However, the bond polarization is more marked than in uranium(IV) complexes. This result is surprising considering the greater electrophilicity of U(VI) compared to U(IV) which was expected to maximize the UC interaction and therefore electron donation from the ligand to uranium. However, inspection of the UO Wiberg bond indexes and qO charges in the alkyl and carbene complexes show that the uranium-oxo interactions are only slightly perturbed by the formation of the UC double bond. In other words, among the three double bonds present in the uranium coordination sphere, the metal ion preferentially accommodates the oxo ligands, leaving a nucleophilic carbene center.
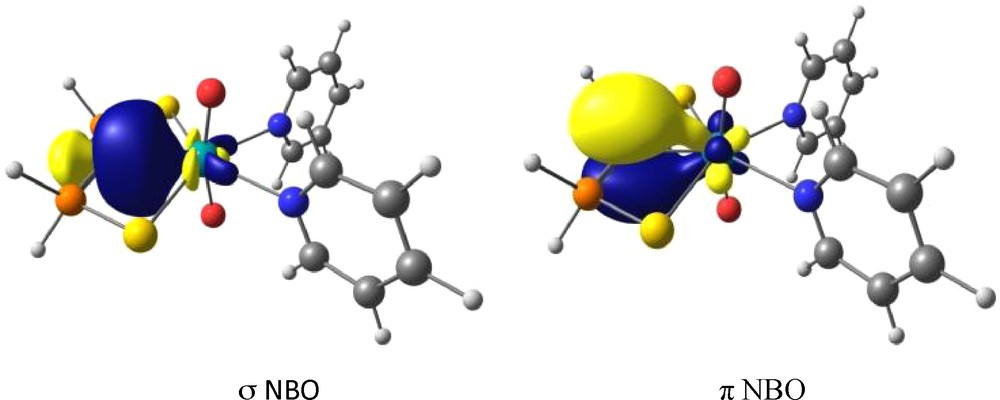
Plots of the NBO for UO2(C{PH2(S)}2)(py)2 describing the UC double bond.
5.4 Reactions of the actinide carbene complexes
While reactions of the complexes Cp3UCHPR3 were much explored by Gilje et al. (Scheme 2), limited studies were devoted to the more recently synthesized SCS and RNCN compounds. The nucleophilic character of the U(IV), Th(IV), U(V) and U(VI) carbene complexes was illustrated by metallo-Wittig reactivity with 9-anthracene carboxaldehyde, benzaldehyde or benzophenone to afford the alkene (Ph2PNR)2CCR1R2 or (Ph2PS)2CCR1R2 (R1 = 9-anthracene or Ph and R2 = H; R1 = R2 = Ph) [40–42,47]. A novel behaviour of the UC double bond was revealed by the reaction of U(™SNCN)Cl2 with MeCN or PhCN in which the CN triple bond of the nitrile suffered a 1,2-cycloaddition to the carbon-metal bond to form a new CC bond and build a tetradentate ligand with three imine centers which coordinate to the uranium atom while maintaining a single UC bond. The newly formed complex dimerizes with one equivalent of unconverted U(™SNCN)Cl2 using two chlorides and the imine group derived from the nitrile as three connecting bridges (Scheme 19) [44].
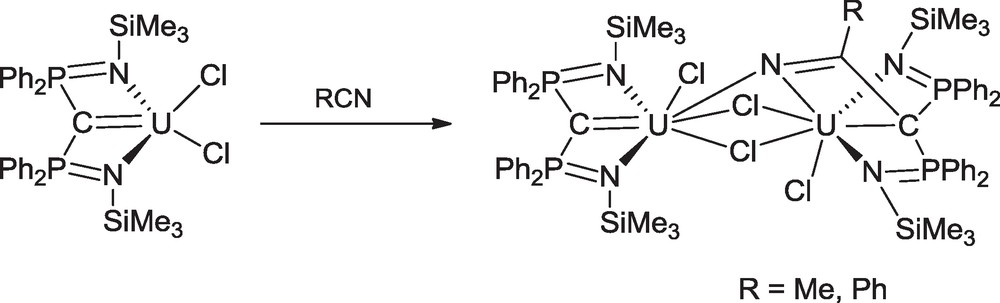
Reaction of U(™SNCN)Cl2 with RCN.
6 Conclusion
After the initial discovery of the phosphoylide complexes Cp3UCHPR3, the detection of actinide methylidene compounds in solid argon and the evidence for carbenoid uranium species in McMurry type reactions, the spectacular recent advances in uranium carbene chemistry have to be related to the use of geminal carbon dianions stabilized by phosphorous(V) substituents. A variety of uranium compounds in the +4, +5 and +6 oxidation states were thus isolated and fully characterized. At the same time, the progress of theoretical calculations led to a good description of the UC double bond that demonstrates a double σ and π donation toward the metal atom with the involvement of the 5f orbitals. Reactions of these complexes were not much developed, being limited to reactions with aldehydes and ketones, and confirmed the polarization of the UC bond toward the nucleophilic carbene carbon atom. Further attention should be paid to the characterization of thorium carbene complexes, which remain very rare, and their reactions, for comparison with those of the uranium analogues, in order to determine the influence of the f electrons on their structure and reactivity.