

1 Introduction
For a long time, organophosphorus chemistry has been built on tricoordinate to hexacoordinate phosphorus compounds [1a]. In group 14, the organosilicon (or –germanium) chemistry has involved mainly tetracoordinate silicon and germanium. Whereas doubly-bonded compounds of the lightest elements of groups 14 and 15, such as alkenes or imines, have been very well studied and have proved to be major building blocks in organic chemistry owing to the reactivity of the double bond, the stable examples of their heavier counterparts were unknown. The first reports claiming the formation of such compounds (for example diphosphene PhP = PPh [1b], silene Ph2Si = CH2 [1c]) appeared to be wrong. Thus, the “double bond rule”, which states that elements of the third or fourth row are unable to form stable double bonds has been postulated.
The success in the almost simultaneous synthesis of the first stable diphosphene Mes*P = PMes* by Yoshifuji et al. (Mes* = 2,4,6-tri-tert-butylphenyl) [2], disilene Mes2Si = SiMes2 by West et al. (Mes = 2,4,6-trimethylphenyl) [3] and silene (Me3Si)2Si = C(t-Bu)OSiMe3 by Brook et al. [4] has demonstrated that the stabilization of such derivatives was possible by using sterically demanding organic substituents around the double bond preventing the approach of two molecules and thus their dimerization. Spectacular progress in this field of low coordinate derivatives has been achieved from the mid 1980s and now almost all combinations E14 = E14 [5], E14 = E15 [5a–b,5e,6–8] and E15 = E15 [5a,5i,9] (E14 = Si, Ge, Sn; E15 = N, P, As) have been obtained. In the field of compounds with a E = C double bond, following the synthesis of heavier analogues of alkenes, >E14 = C<, and imines, –E15 = C<, another challenge has been the synthesis of heavier analogues of allenes E = C = E′; such derivatives are particularly interesting due to the presence of two cumulative double bonds but seemed more difficult to stabilize due to the two unsaturations.
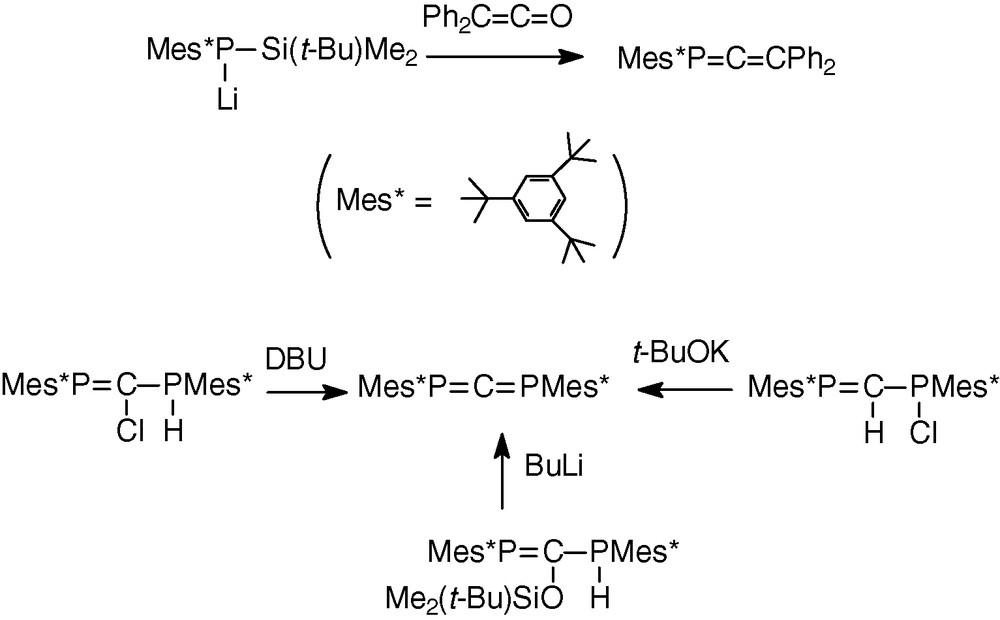
The first heteroallenes were the phosphaallene Mes*P = C = CPh2 [10] synthesized by Yoshifuji et al. in 1984 and the diphosphaallene Mes*P = C = PMes* [11] obtained the same year simultaneously by Appel, Yoshifuji and Karsch by different methods (Scheme 1). Since this date, other allenic compounds of phosphorus of the type –P = C = E15– (E15 = N, As) or –P = C = E16 (E16 = O, S) have been successfully isolated or characterized [12–17].
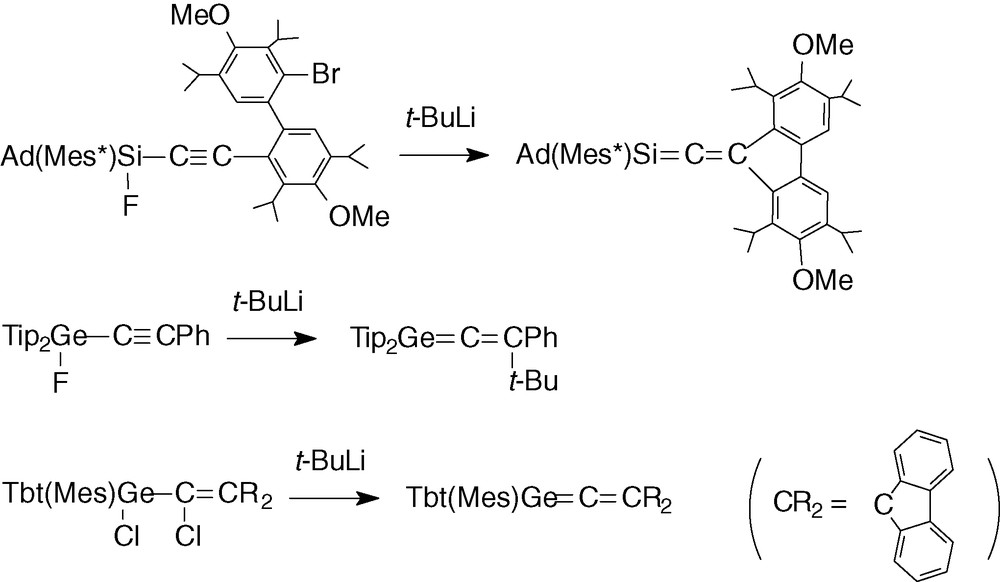
In the field of heavier group 14 elements, some silaallenes >Si = C = C< [12–18] and two germaallenes >Ge = C = C < [12–17,19] have been isolated and structurally characterized (Scheme 2) for the synthesis of the first representatives.
Owing to our experience on doubly-bonded compounds of phosphorus and germanium such as phosphagermenes –P = Ge< [20] and germenes >Ge = C< [21] and on synthetic equivalent of silenes >Si = C< [22] we have been particularly interested in studying heteroallenic compounds featuring double bonds on phosphorus and silicon or germanium of the types >Si = C = P– and >Ge = C = P–. Such compounds appear interesting from an academic point of view: many questions deserve an answer, such as the determination of the geometry (is it close to that of an allene or completely different, is the P = C = Si (or Ge) angle of 180° and the dihedral angle around the double bond of 90°), the charges (are the unsaturations polarized Pδ+ = Cδ– and Si (or Ge)δ+ = Cδ– or is there a reverse polarity), the electronic configuration, the types of substituents (electron releasing or withdrawing) able to stabilize the double bonds, and on which element (E14 or E15) they are the most efficient. The study of their reactivity is also very interesting: does only one double bond react preferentially, and which one (even if the E14 = C double bond is suspected to be more reactive than the E15 = C double bond) or do both double bonds react? It is also possible to imagine the reactivity of the lone pair at phosphorus and the cleavage of the Si = C (or Ge = C) double bond to silylene >Si (or germylene >Ge) and C = P–. Thus, such compounds are promising as building blocks in organophosphorus and organometallic chemistry, as precursors of polymers and as ligands.
This short review describes theoretical studies, synthesis, physicochemical characterizations and the reactivity of phosphasilaallenes >Si = C = P– and phosphagermaallenes >Ge = C = P–.
2 Results and discussion
2.1 Theoretical calculations
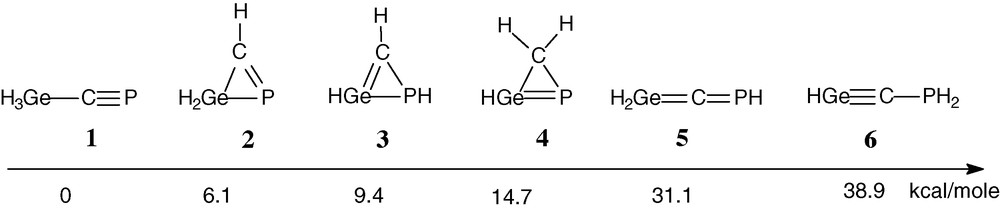
Calculations (B3LYP/6-31G(d,p)) have been carried out on the parent compound H2Ge = C = PH and its isomers of H3CGeP formula [23] (Scheme 3). Among the structures with a PCGe linkage 1–6, the most stable isomer is, as expected, the triple bond PC compound 1 (many phosphaalkynes have been isolated [24,25]). Three phosphagermirenes 2, 3 and 4 are more stable than phosphagermaallene 5, which lies 31.1 kcal/mol higher than phosphaalkyne 1. Note that the three-membered ring compound 7 (Fig. 1), analogous to 2, has been isolated by reaction between a germylene and a phosphaalkyne [26]. The triple bond GeC derivative 6 is the least stable, which is not surprising since no stable germyne has been reported until now.
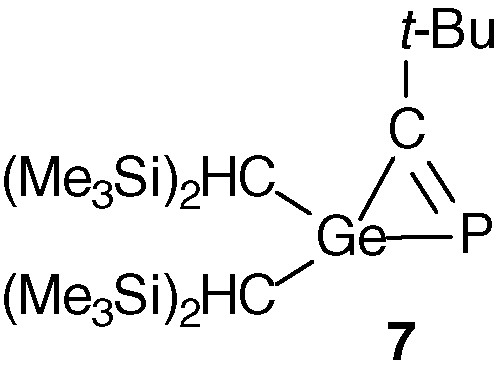
Structure of compound 7.
The main geometrical parameters (Ge = C and P = C bond lengths and GeCP bond angle) have been calculated at two levels of theory: CCSD/6-31G(d,p) Ge = C = 1.784 Å, P = C = 1.648 Å, GeCP = 164.04° and MP4(SDQ)/6-31G(d,p) Ge = C = 1.789 Å, P = C = 1.650 Å, GeCP = 162.56° [23]. They are in good agreement with the parameters recently determined by X-ray: Ge = C = 1.761(2) Å, P = C = 1.625(2) Å, GeCP = 166.55(14)° (unpublished results).
From this study it can be supposed that the stabilization of a phosphagermaallene >Ge = C = P– is not obvious; like in the case of other heteroallenes, the choice of the substituents at the germanium and phosphorus atoms is probably extremely important for this stabilization.
2.2 Transient phosphagermaallene and phosphasilaallene
2.2.1 General procedure
From our experience on silenes and germenes >E = C< (E = Si, Ge) [21,22] and the results of the literature [10,11] it appeared that the E = C double bond should be probably more reactive than the P = C double bond. Thus, the best strategy to synthesize a phosphagerma- (or -sila) allene was to create first the P = C double bond since the simultaneous formation of both double bonds seems unlikely. Thus, the general procedure is presented in Scheme 4.
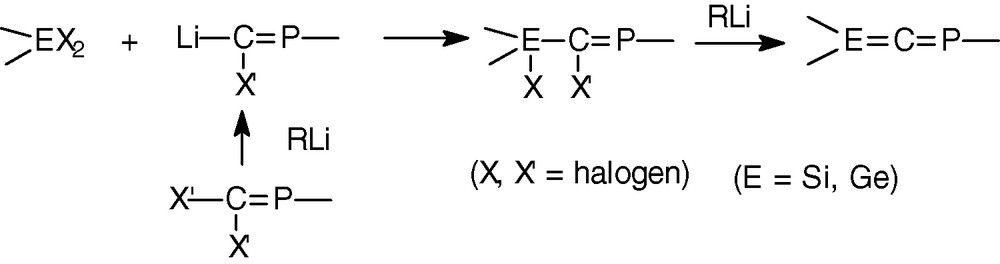
2.2.2 Phosphagermaallene 8
The 2,4,6-tri-tert-butylphenyl group, called supermesityl (Mes*), widely used to stabilize doubly bonded phosphorus compounds, was chosen on the phosphorus atom. As mesityl groups (Mes = 2,4,6 trimethylphenyl) were very efficient to stabilize the germene Mes2Ge = CR2 (CR2 = fluorenylidene) [21a–b] they were chosen on the germanium atom.
In order to avoid side reactions, particularly reduction of the Ge-X bond or alkylation (by the lithium compound), fluorine was preferred to chlorine as halogen on the germanium atom. Moreover the 19F NMR allowed us to monitor the reaction.
Phosphagermapropene 9 [27a], precursor of phosphagermaallene 8, was synthesized by a coupling reaction between the phosphacarbenoid Mes*P = C(Br)Li 10 [28] and difluorodimesitylgermane in the form of only one geometrical isomer (δ31P = 321.3 ppm, 3JPF = 29.8 Hz). We postulate it to be the Z isomer due to the selective lithium/bromine exchange of the bromine atom in Mes*P = CBr2 that is in trans in relation to the huge Mes* group as previously reported in the literature on similar compounds [28].
Addition of t-BuLi to an Et2O solution of 9 led to the immediate formation of the lithium compound 11 (δ31P = 397.4 ppm, 3JPF = 16.9 Hz) [27a]. Elimination of LiF was observed at −60 °C leading to an orange solution of the expected phosphagermaallene 8 in about 70% yield [27a] (Scheme 5).
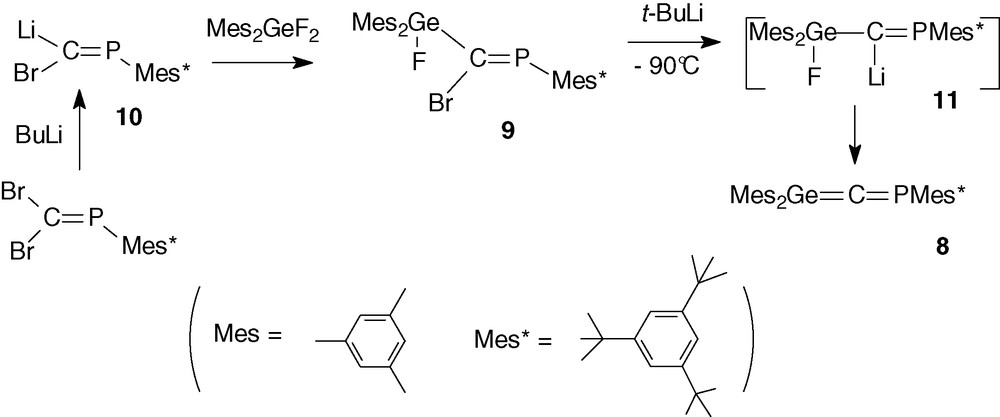
Phosphagermaallene 8 was characterized by a low-field shift in 31P NMR (240.0 ppm) and particularly in 13C NMR (280.9 ppm, 1JCP = 54.3 Hz for the sp carbon atom) [27a]. Similar low-field chemical shifts have been reported for the sp carbon atom in other heteroallenes: Mes*P = C = AsMes* (299.5 ppm) [29], Tip2Ge = C = C(Ph)t-Bu (235.1 ppm) [19a] (Tip = 2,4,6-triisopropylphenyl) and Tbt(Mes)Ge = C = CR2 (243.5 ppm) [19b] (Tbt = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl, CR2 = fluorenylidene).
Heteroallene 8 was stable only below −40 °C and warming to room temperature led to dimers (section 2.2.4).
2.2.3 Phosphasilaallene 12
Using the same mesityl groups on silicon failed to afford the phosphasilaallene analogue. The coupling reaction of Mes2SiF2 with the phosphacarbenoid Mes*P = C(Br)Li 10 occurred with a poor yield due to the too large steric congestion hindering the approach of the two reagents. Thus, Tip (2,4,6-triisopropylphenyl) and Ph groups were used, the presence of a small group like phenyl allowing the coupling to form the phosphasilapropene Tip(Ph)Si(F)-C(Br) = PMes*. However, even in this case, the coupling was difficult, the main product being the phosphaalkyne Mes*CP resulting from the decomposition around −80 °C of 10, before its reaction with the difluorosilane, followed by a rearrangement. Thus, its much more stable chlorine analogue Mes*P = C(Cl)Li 13 [30] was used.
Phosphasilaallene 12 [31] was obtained by dechlorination of phosphasilapropene 14 by tert-butyllithium at −90 °C in a mixture toluene/Et2O 90/10 (Scheme 6). The intermediate formation of the lithium compound 15 was evidenced in 31P NMR by the very low-field shift (417.6 ppm) characteristic of the presence of two electropositive elements (Si and Li) on the sp2 carbon atom and by hydrolysis leading to Tip(Ph)(Cl)Si-C(H) = PMes* [31].
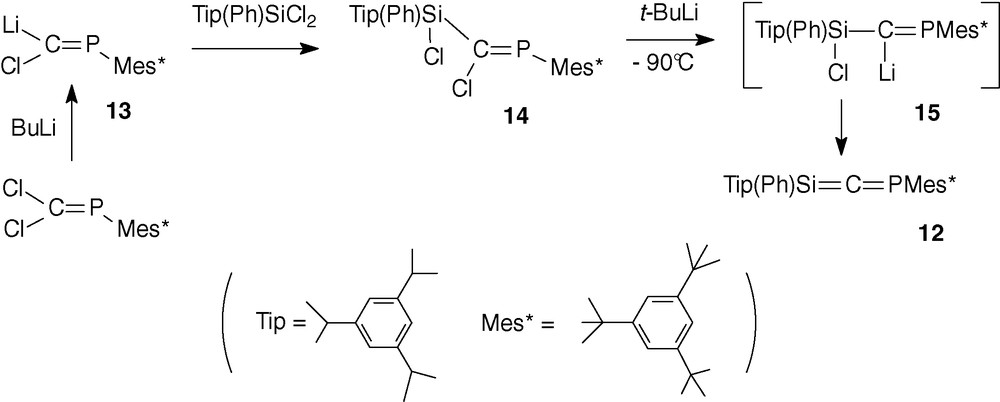
The formation of phosphasilaallene 12 was proved by its low-field shift in 31P (288.7 ppm), 13C (269.1 ppm, 1JCP = 45.8 Hz) and 29Si NMR (75.7 ppm, 2JSiP = 18.3 Hz). 13C and 29Si chemical shifts are comparable to those observed in silaallenes Ad(Mes*)Si = C = CR2 (CR2 = substituted fluorenylidene) (225.7 and 48.4 ppm) [18a] and t-Bu(Mes*)Si = C = C(Ph)t-Bu (216.3 and 55.1 ppm) [18b].
Like its germanium analogue, phosphasilaallene 12 was stable only below −30 °C and dimerized above this temperature (section 2.2.4).
2.2.4 Reactivity
Heteroallenes 8 and 12 reacted nearly quantitatively with methanol and phosphagermaallene 8 with methyllithium. These additions occurred exclusively onto the E = C double bond, according to the expected Eδ+ = Cδ− polarity and led to the sole E-phosphaalkenes (Scheme 7).
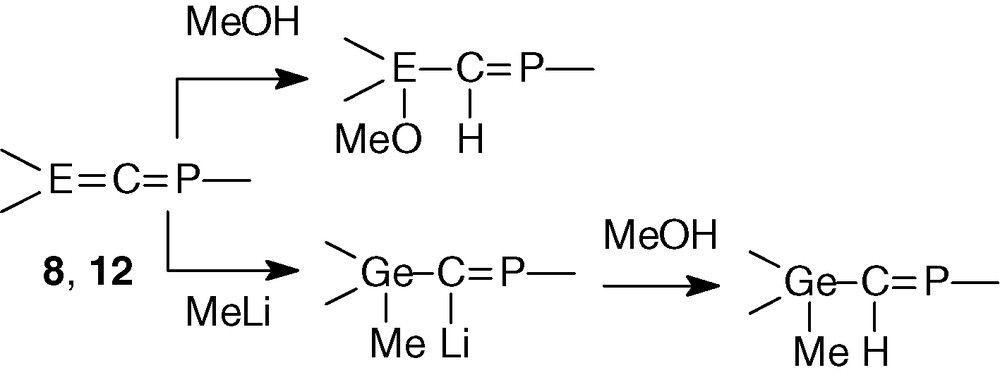
In the absence of trapping reagent, phosphasilaallene 12 and phosphagermaallene 8 give two types of dimers: the expected head-to-tail dimers 16 and 17 and the unsymmetrical dimer 18 [27a,31] (Scheme 8). 1,3-Dimetallacyclobutanes 16 and 17, containing two exocyclic P = C double bonds, are obtained by dimerization involving two E = C double bonds; a dynamic equilibrium is observed between the dimers 16 and 17; according to the NMR analysis, the major one is 17 with the two Mes* groups in cis disposition in relation to the P = C⋯C = P axis; this assumption was proved in the case of the silicon compound 17a by an X-ray analysis which displays a nearly planar four-membered ring [31]. Very low-field chemical shifts in 31P NMR are observed for 16 and 17 particularly for the silicon compounds (trans 431.9 and cis 425.4 ppm) due to the presence of two silicon atoms onto the sp2 hybridized carbon atom [31].
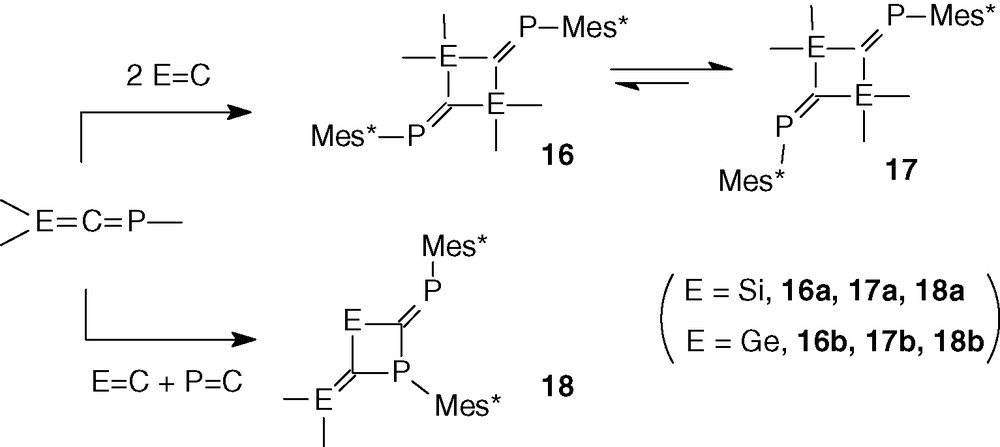
Dimers 18 were formed by a cycloaddition between E = C and P = C double bonds and are the major ones (E = Si: 18a 60%, 16a/17a 40% [31], E = Ge: 18b 88%, 16b/17b 12% [27a]). They were characterized by two doublets in 31P NMR: 18a = 294.9 and 86.9 ppm (2JPP = 224 Hz); 18b = 269.5 and 69.3 ppm (2JPP = 225.4 Hz). According to the large PP coupling constant, it seems that the isomer obtained has the lone pair on the λ3σ2 P in trans disposition in relation to the λ3σ3 P atom since the literature reports that 2JPP is 15–30 Hz in cis structures but much larger (more than 100 Hz) in trans structures [32].
The unsymmetrical dimerization seems a priori surprising but is not so unexpected since it has been reported to occur in other phosphaallenic derivatives such as phosphatioketene Mes*P = C = S [33]. Both types of dimerization occur in phosphaazaallenes Mes*P = C = NPh depending on the experimental conditions [34] (Scheme 9).

To have a rough idea about the relative stability of the dimers of parent compound H2Ge = C = PH, calculations have been performed [27a] (restricted Hartree-Fock calculations using Gaussian 92 package: for C, P and Ge atoms, effective core potentials were used [27b]; for all atoms the valence atomic basis sets consist of four Gaussian functions contracted to a double-ζ level and augmented by a polarization function; the geometries have been optimized by using a gradient technique). As expected, due to the greater stability of the P = C than Ge = C double bond, the dimers between two P = C double bonds 19 and 20 lie 25 kcal/mol above dimers 21 and 22 formed by Ge = C and P = C cycloadditions (Scheme 10). Dimers 23 and 24 formed from two Ge = C double bonds are normally more favourable by 18 kcal/mol [27a]. Only analogous of 21 and 23 have been experimentally observed because the formation of 22 and 24 is unfavorable due to the steric hindrance caused by the Mes and Mes* groups.
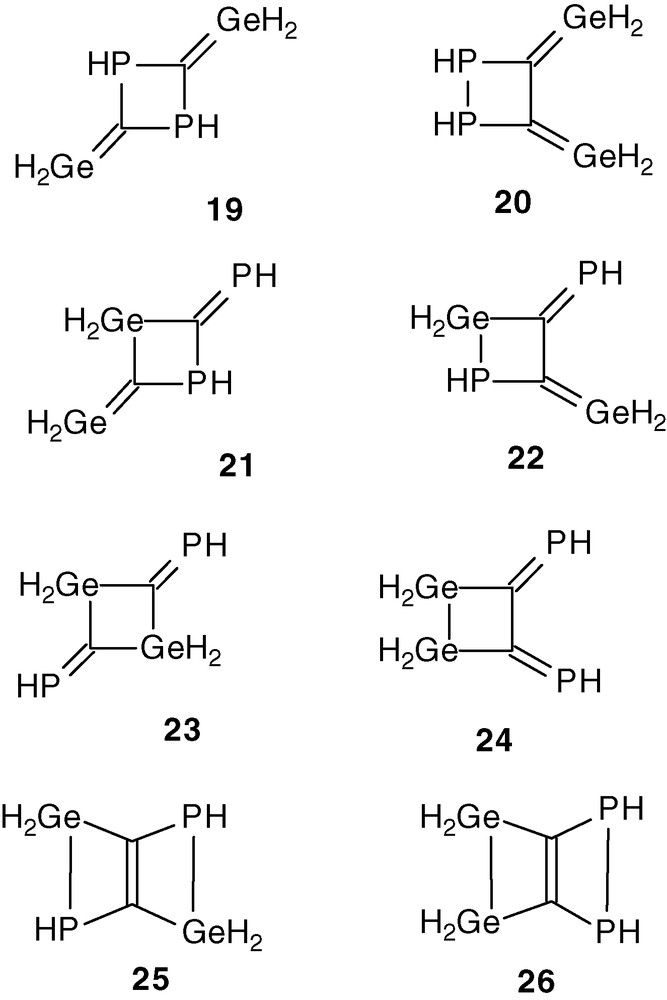
The most stable dimers are the butterfly structures 25 and 26. These bicyclic derivatives were calculated to lie 15 kcal/mol below 23 and 24. However, the formation of such structures, arising from 22 and 24 by a second cycloaddition between one Ge = C and one P = C or two P = C double bonds, are not possible with large groups at the germanium and phosphorus atoms.
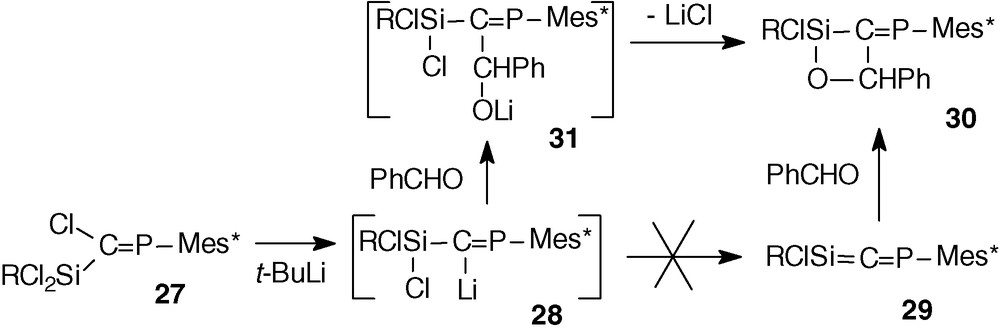
2.3 Chlorosilylphosphaethenyllithium
In order to increase the synthetic utility of phosphasilaallene, we tried to attach at the silicon atom a chlorine atom which could allow functionalisation of the Si = C = P system. The other group at Si was a 9-methylfluorenyl group intended to provide extrasteric protection for the Si = C double bond. The trichlorophosphasilapropene 27 [35] was prepared by the same routes as dichlorophosphasilapropene 14 by coupling phosphacarbenoid 13 with dichloro(9-methylfluorenyl)silane (Scheme 11).
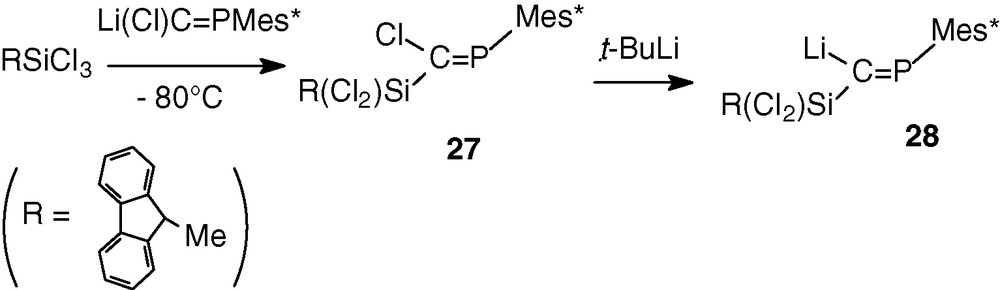
The 31P NMR analysis displayed, as previously, the formation of only one isomer (δ = 328.1 ppm). The X-ray analysis confirmed the formation of the Z isomer due to the selective lithium/chlorine exchange of the chlorine atom in trans in relation to the huge Mes* group.
Treatment of 27 with tert-butyllithium resulted in the formation of the expected lithium compound 28 which was evidenced by a very low-field chemical shift in 31P NMR (441.4 ppm) and by hydrolysis with one equivalent of water leading to RCl2SiC(H) = PMes* [35] (Scheme 11).
Surprisingly, whereas the elimination of lithium halide occurred at low temperature in the case of 15 and 11 substituted at Si and Ge by aryl groups, no elimination was observed in the case of 28.
Calculations on the [Cl3-nHnSi-C = PH]− anion at the RHF/6-31 + G** level show that the increasing number of chlorine atoms at the silicon increases the negative charge (natural charges) on the central carbon from −1.224 (n = 3) to −1.393 (n = 0) [35]. This is in favour of form a whereas form b should be necessary to form the double bond (Scheme 12), according to a study of Wiberg on compounds of the type Si(X)-C(Li) [36].
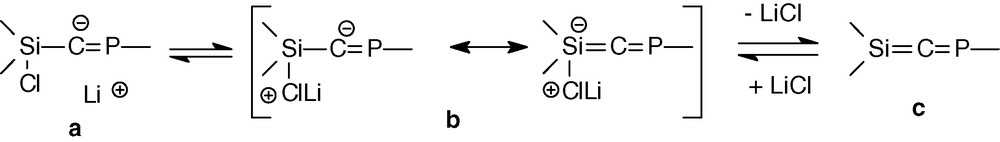
To have a better understanding about the non elimination of LiCl, molecular orbital calculations (RHF/3-21G* level) have been carried out to determine the relative stability of the conformers of the simplified system Cp′Cl2Si-C(Li) = PMes* (Cp′ = 1-methylcyclopentadienyl). The most stable isomer shows a strong interaction between the lithium atom and the π systems of Mes* and Cp′ with short distances between Li and the centroid of these aromatic systems. This lithium-π interaction also favours form a.
However, the chlorosilyllithium 28 can be considered as a synthetic equivalent of the phosphasilaallene 29; for example, reaction with benzaldehyde afforded the oxasiletane 30 probably via the intermediate 31 followed by cyclization (Scheme 13); reaction between the phosphasilaallene 29 and benzaldehyde should presumably give the same four-membered ring compound 30 by a [2 + 2] cycloaddition between the Si = C and C = O double bonds as in the case of a stable phosphagermaallene and benzaldehyde (section 2.5).
2.4 2,4-Diphosphinylidene-1,3-digermacyclobutanes
The replacement on the germanium atom of the two mesityl groups by two t-Bu groups which present a steric hindrance more important close to the germanium atom than Mes groups could theoretically prevent the dimerization of the Ge = C double bond.
In fact, addition of t-BuLi to the phosphagermapropene 32 [37] (formed from difluorogermane 33 and phosphacarbenoid Mes*P = C(Cl)Li), did not lead to the expected phosphagermaallene t-Bu2Ge = C = PMes*, but to its formal dimers, the 2,4-diphosphinylidene-1,3-digermacyclobutanes 34 and 35 as two isomers with exocyclic P = C double bonds (δ31P = 367.4 and 367.7 ppm for the cis and trans isomers in relation to the P-C⋯C-P axis) [37] (Scheme 14). Owing to their different solubilities (the cis isomer 34 has a non-zero dipole moment and thus a better solubility in polar solvents) both isomers have been purified by crystallization and characterized by an X-ray structure.
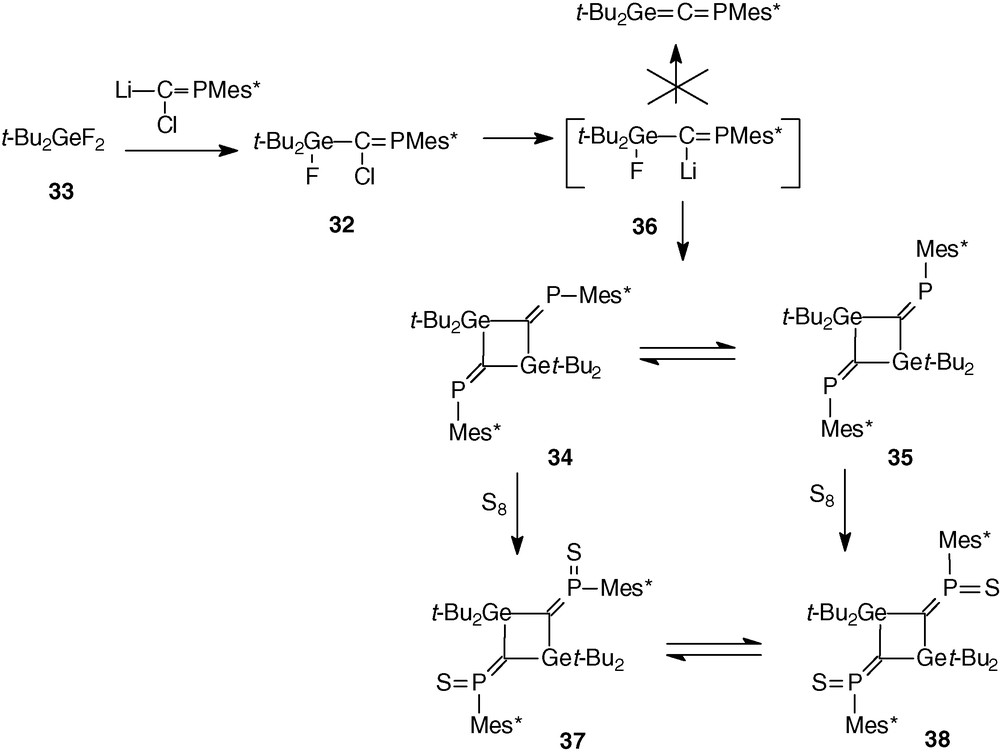
Monitoring the reaction by 31P NMR from −80 °C to 20 °C did not evidence the expected phosphagermaallene t-Bu2Ge = C = PMes*, but an intermediate (δ31P = 286.7 and 443.8 ppm, JPP = 38.5 Hz) probably resulting from an intermolecular condensation between two molecules of 36 with loss of LiF instead of the expected intramolecular elimination to give the heteroallene.
Thus, this reaction is completely different of that observed in the case of 11 and 8 (Mes groups on Ge) and it seems that at least one aryl group on the metal atom is necessary to cause the intramolecular elimination of lithium halide to take place and to form the heteroallene.
Note that reaction of 34 and 35 with an excess of sulphur afforded the first bis(methylenethioxo)phosphoranes 37 and 38 with two C = P(S)Mes* units [37] (Scheme 14).
2.5 Stable phosphagermaallene 39
2.5.1 Synthesis and physicochemical data
From the experience gained in the previous attempts, it seemed that a combination of one aromatic group (Tip) and one alkyl group (t-Bu), which presents the advantage of a large steric hindrance at long distance (Tip) and short distance (t-Bu) of the germanium atom, could be successful to stabilize a phosphagermaallene. Difluorogermane 40 [38] was obtained from TipGeCl3 [39] via the methoxy compounds TipGe(OMe)3 and Tip(t-Bu)Ge(OMe)2. Reaction of 40 with the phosphacarbenoid 13 led to 41, the expected precursor of the phosphagermaallene. The great steric hindrance in 41 was clearly evidenced in the 1H NMR spectrum by the presence of two signals for the o-t-Bu groups of Mes* and a very large one for the methyls of the o-iPr groups due to their slow rotation on the NMR time scale. Only one isomer was observed, probably the Z-isomer as demonstrated previously in similar reactions. Addition of one equivalent of tert-butyllithium to an Et2O solution of 41 at −90 °C led to the nearly quantitative formation of phosphagermaallene 39 [38] (Scheme 15).
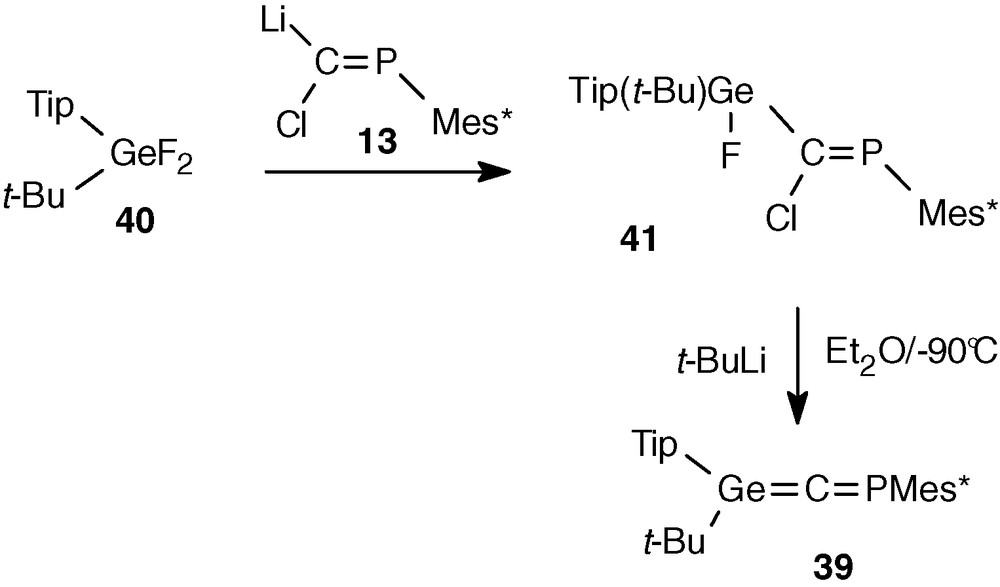
Red-brown solutions of 39 are extremely air and moisture sensitive, but no change was observed for months in an inert atmosphere. Crystallization in a sealed tube afforded orange crystals.
39 is the first stable heteroallenic compound with two heavy group 14 and 15 elements. Thus, a rather small change in the substitution at the germanium atom (Tip and t-Bu instead of two mesityl groups) changed drastically the stability of the heteroallene.
The structure of 39 was proved by a low-field shift in 31P NMR (250 ppm) and particularly in 13C NMR for the central sp carbon atom (280.4 ppm, JPC = 60.9 Hz) [38] comparable to that previously reported for transient phosphagermaallene 8 and phosphasilaallene 12.
2.5.2 Reactivity
The structure of 39 was also proved by its chemical reactivity. A regiospecific addition of water on the Ge = C double bond occurred, with the hydroxy moiety bonded to the germanium atom because of the Geδ+-Cδ− polarity of the double bond [38] (Scheme 16). The addition is also stereoselective, but it was impossible from the 2JPH coupling constant (25.0 Hz) to determine the configuration of the reaction product (in the similar C-germylphosphaalkene Me3GeC(H) = PMes* [28a], the 2JPH constants are 26.4 and 23.5 Hz for the E and Z isomers, respectively).
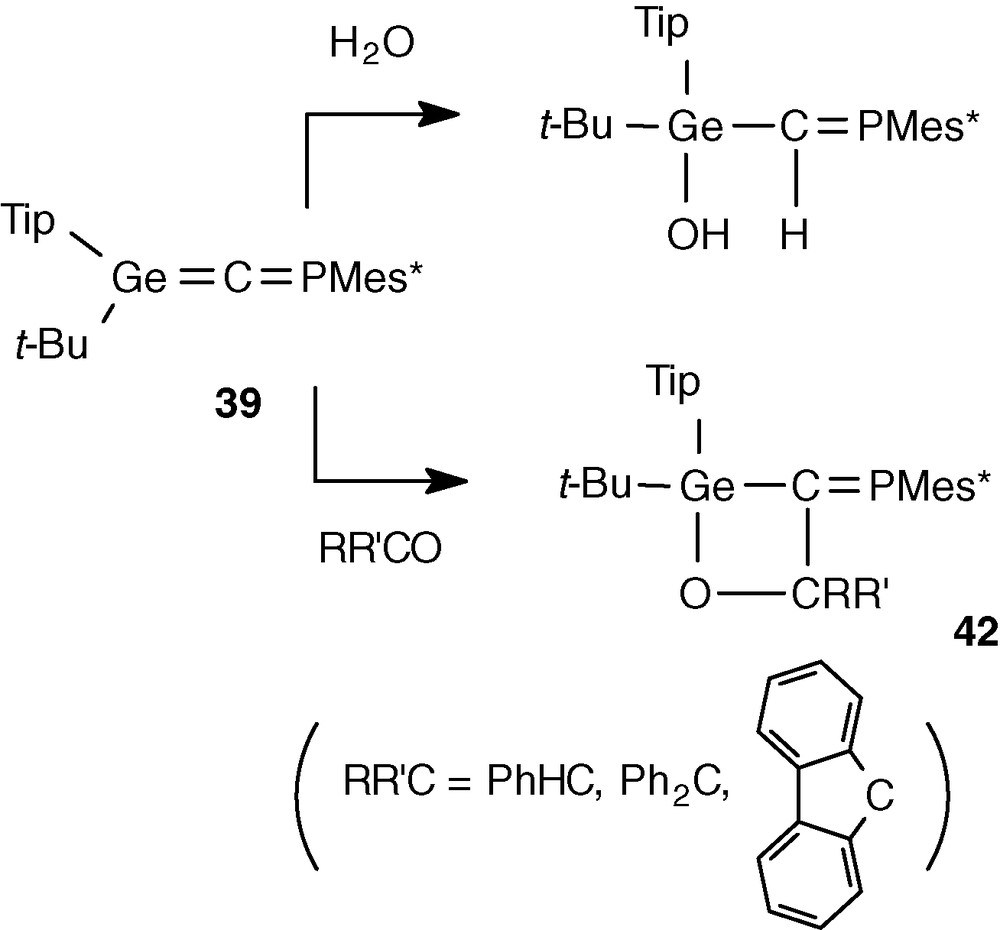
With aldehydes (benzaldehyde) and non-enolysable ketones (benzophenone and fluorenone), [2 + 2] cycloadditions between the Ge = C and C = O unsaturations occurred leading to oxagermetanes 42 with an exocyclic P = C double bond [38] (Scheme 16).
[2 + 1] cycloadditions on the Ge = C double bond occurred with chalcogens: addition of sulphur, selenium or tellurium led to the chalcogenagermiranes 43 [40] (Scheme 17). Such 3-membered ring compounds are very stable, probably owing to the large steric hindance. With one equivalent of chalcogen, only the cycloadduct involving the Ge = C double bond was formed showing the greater reactivity of this unsaturation than that of the lone pair on phosphorus which is generally very reactive towards chalcogens. The presence of the exocyclic P = C double bond is evidenced in 31P NMR by the classic low-field shift (43a: 252.2, 43b: 278.8, 43c: 311.8 ppm). The PSe (107.5 Hz) and PTe (249.6 Hz) coupling constants are larger than those found for selenadiphospholes (50–80 Hz) and telluradiphospholes (130–150 Hz).
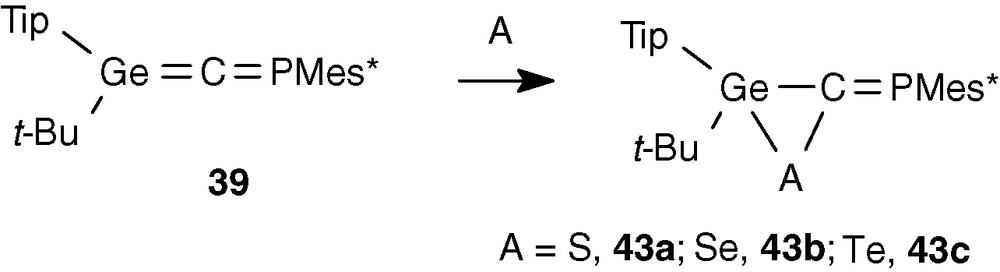
Surprisingly, 43b and 43c present a different configuration around the P = C double bond: Z for 43c (Mes* group cis to Te) and E for 43b (Mes* group trans to Se). The origin for this opposite arrangement is not clear at the moment.
Another striking feature was discovered from the X-ray structure: for 43b and 43c the sum of angles on germanium is close to 360° (43b 358.6°, 43c 353.5° [40] with a short Ge-C bond (1.912.6 Å in 43c) in relation to the standard Ge-C bond length (1.96–1.98 Å) [41]. This partially planar geometry corresponds to a structure intermediate between a π-complex and a normal three-membered ring (Scheme 18).

These results can be interpreted in terms of Dewar-Chatt-Duncanson model of metal olefin bonding. Similar results have been reported in other three-membered ring compounds containing one germanium atom such as GePS [42] or GeCTe [43] or two germanium atoms GeGeX (X = C, N, S, Te) [44–46].
2.5.3 Comparison with SiCN derivatives
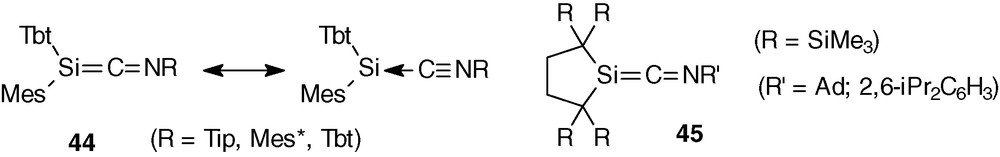
A completely different behaviour is observed between sila- (or germa)heteroallenes of nitrogen and phosphorus: >Si = C = P− and >Ge = C = P− keep their skeleton integrity in all the reactions studied and never behave as silylenes or germylenes >E and isophosphanitriles:C = P−. By a sharp contrast, most of the >Si = C = N− compounds are better described as complexes between silylenes and isonitriles C = N− (for example high field 29Si resonance from −48 to −58 ppm in 44) [47]; however, 45 presents physicochemical data (29Si resonance at lower field: 15.9 and 23.9 ppm, short SiC bond, CN distance longer than that of aryl isocyanide) convenient for an azaphosphaallene structure [48] but also dissociates in solution above −30 °C (Scheme 19).
3 Conclusion and prospects
Transient phosphasilaallene Tip(Ph)Si = C = PMes* and phosphagermaallene Mes2Ge = C = PMes* have been characterized at low temperature and the first stable heteroallene with two heavy elements of groups 14 and 15, the phosphagermaallene Tip(t-Bu)Ge = C = PMes* has been isolated. This study shows the great difficulty to obtain stable doubly-bonded compounds of this type: with a too large steric hindrance on Si and Ge the coupling reaction between the dihalosilane (or -germane) R2EX2 and the phosphacarbenoid Li(X)C = P− does not occur and with a low steric hindrance the final heteroallene dimerizes. Moreover, depending on the substituents at the metal, elimination of lithium halide sometimes does not occur, or gives directly a formal dimer of the phosphametallaallene by an intermolecular condensation with loss of lithium halide without the formation of the expected phosphametallaallene. Thus, the choice of the substituents is extremely crucial and difficult. It seems that at least one aryl group on Si (or Ge) is necessary.
With E14 and E15 belonging to the same row, E14 = C double bond appears to be much more reactive than E15 = C one. For example the phosphagermaallene 39 is much more reactive than its arsenic analogue Mes*As = C = PMes* [29]. Thus, the study of its chemical behaviour appears very promising.
A new challenge is to prepare phosphastannaallenes >Sn = C = P−. A very large steric hindrance on the tin atom will be necessary in order to prevent the dimerization of the Sn = C double bond, much longer than the Ge = C one. Until now, no tin heteroallene has been isolated. Stannaallene Tip2Sn = C = CR2 (CR2 = fluorenylidene) has been postulated as intermediate in a reaction leading to a distannirane [49] and R2Sn = C = Mes is like >Si = C = N− compounds better considered as a complex between stannylene R2Sn and isonitrile: C = NMes than as an azastannaallene [50].
Acknowledgements
We are grateful to the Agence nationale de la recherche (contract ANR-08-BLAN-0105) and to PhoSciNet (CM0802) for financial support.