

1 Introduction
For almost a decade, there has been a great activity devoted to lanthanide-containing coordination polymers [1]. This activity is motivated by their interesting luminescent properties [2] and by their ability to provide potentially porous materials. Several lanthanide-based coordination polymers exhibiting porosity have been reported [3–14]. One of the possible strategies to increase the porosity consists in designing coordination polymers where poly-nuclear complexes would act as metallic centers. This strategy has been successfully applied some years ago and very promising materials involving zinc tetra-nuclear complexes have been obtained [15]. However, up to now, only a few potentially porous coordination polymers involving poly-nuclear lanthanide complexes have been reported [16–23]. In fact, while the cluster chemistry of d-block transition metals is now firmly established [24,25], the analogous chemistry involving the lanthanide ions is rather underdeveloped.
To date, only a few poly-nuclear lanthanide complexes have been reported. Most of them have been obtained by use of ancillary ligands. These ligands pre-occupy part of the coordination sphere of the lanthanide ion. By the way, they prevent the formation of the highly insoluble lanthanide hydroxide and allow the control of the lanthanide ion hydrolysis. Several poly-nuclear lanthanide complexes have so been obtained ranging from di-nuclear to pentadeca-nuclear lanthanide oxo/hydroxo entities [17,26–34]. Unfortunately, these entities are most often very unstable in solution [34] and cannot be used as molecular precursors for further chemistry.
A second synthetic route, first described 15 years ago [35–41], leads to hexa-nuclear lanthanide complexes with general chemical formula [Ln6O(OH)8(NO3)6(H2O)x]2+ with x = 12 when Ln = Sm–Yb, x = 14 when Ln = Pr–Nd and x = 16 for Ln = Ce. This route consists in using lanthanide nitrate as a starting material and in hydrolyzing it by addition of sodium hydroxide. As illustrated by Fig. 1, this synthetic pathway is based on a subtle balance between the various experimental parameters (pH, concentrations, temperature…). The main difficulty encountered during the synthesis of these complexes is to avoid the formation of very stable polymeric species such as Ln(OH)2NO3 [25,36,37], LnONO3 [42] and Ln(OH)3 [43].

Obtaining domain of compounds with general chemical formula [Ln6O(OH)8(NO3)6(H2O)n].(NO3)2.2H2O with Ln = Pr–Tb plus Y at room temperature [44].
Owing to this synthetic method, numerous polymorphic phases of such hexa-nuclear lanthanide complexes have been reported so far [39,40,44–47]. They are listed in Table 1 where it can be noticed that several polymorphic phases can exist for a given lanthanide ion.
Summary of the reported hexa-nuclear lanthanide complexes with general chemical formula [Ln6O(OH)8(NO3)6(H2O)x].(NO3)2.yH2O.
| Ce | Pr | Nd | Sm | Eu | Gd | Tb | Dy | Ho | Y | Er | Tm | Yb | |
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.2H2O | [46] | [45] | [46] | ||||||||||
| [Ln6O(OH)8(NO3)6(H2O)14].(NO3)2.2H2O | [44] | ||||||||||||
| [Ln6O(OH)8(NO3)6(H2O)16].(NO3)2.2H2O | [46] | ||||||||||||
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.3H2O | [47] | [47] | |||||||||||
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.4H2O | [39] | [40] | [39] | ||||||||||
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.5H2O | [39] | [45] | [40] | [45] | |||||||||
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.6H2O | [40] | [45] |
All these compounds, except [Ce6O(OH)8(NO3)6(H2O)16].(NO3)2.2H2O, have been structurally described. All the crystal structures are very similar. They can be described as the juxtaposition of perfect octahedrons with one lanthanide ion in each edge and one μ6–O2− anion at the center. Each face of the octahedrons is shaped by a μ3–OH− anion and each lanthanide ion is bounded in a bidentate manner to a nitrate anion. The main difference between the two families of compounds concerns the coordination water molecules. As shown in Fig. 2, the complexes involving a lanthanide ion comprised between Yb and Sm or Y contain 12 coordination water molecules (two per lanthanide ion) while those involving Nd or Pr include 14 of them. In these two latter complexes, four out of the six lanthanide ion are bounded to two coordination water molecules while the remaining two lanthanide ions are bounded to three coordination water molecules. So, all the lanthanide ions are nine-coordinated in compounds belonging to the former family while for compounds belonging to the latter family two out of the six are 10-coordinated and the remaining four are nine-coordinated.
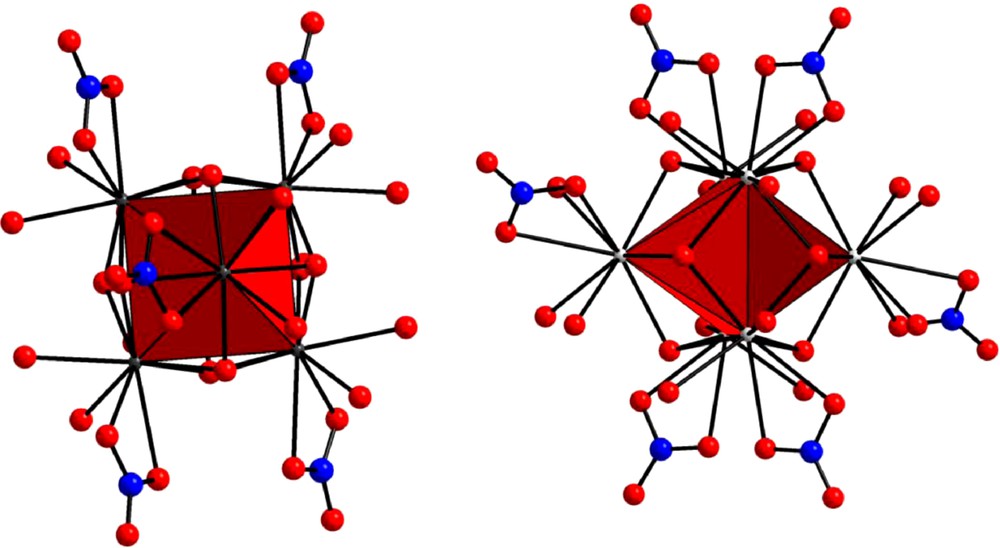
Views of the complexes [Er6O(OH)8(NO3)6(H2O)12]2+ (left) and [Pr6O(OH)8(NO3)6(H2O)14]2+ (right).
These complexes can be used as molecular precursors for further chemistry [48,49]. However, they still present a slight tendency to hydrolyze even in dry organic solvents because of their crystallization and coordination water molecules. In order to increase the chemical stability of the complexes it is possible to dehydrate them. The dehydration of these complexes is easy to realize thermally and leads to stable anhydrous compounds with general chemical formula [Ln6O(OH)8(NO3)8]∞ with Ln = Ce–Yb (except Pm) or Y. It is noticeable that all the dehydrated compounds present similar X-ray powder patterns whatever the initial hydrated complex belongs to one or the other family.
The crystal structure of these compounds has been solved on the basis of the powder X-ray diffraction diagram of [Y6O(OH)8(NO3)8]∞. It can be described as neutral polymeric chains of anhydrous hexa-nuclear complexes. The octahedral complexes are bounded to each other by nitrate groups. These nitrate groups are linked to one yttrium atom in a bidentate manner and to the second in a unidentate manner (Fig. 3). The octahedral complexes are linked to each other by four yttrium atoms in a share-edges manner thereby forming a ribbon-like molecular motif.

Projection view along of a ribbon-like molecular motif.
2 Experimental section
2.1 Synthesis of the micro-crystalline powders of hydrated compounds [Ln6(μ6–O)(μ3–OH)8(NO3)6(H2O)x].2NO3.2H2O with x = 12 for Ln = Sm–Yb plus Y, x = 14 for Ln = Pr, Nd and x = 16 for Ce
Hydrated lanthanide nitrates have been purchased from STREM chemicals and used without any further purification.
Three millimeter of a dilute (0.5 mol·L−1) aqueous solution of sodium hydroxide is carefully added, dropwise, to 10 mL of a concentrated (1 mol·L−1) solution of lanthanide nitrate in a mixture water:ethanol (1:9) vigorously stirred magnetically. This slow addition is crucial in order to avoid any local over-concentration that would lead to the insoluble lanthanide hydroxide. The Ce(III) containing compound has been synthesized under nitrogen atmosphere in order to avoid the cerium oxidation.
During the addition, a polycrystalline powder appears. Once the addition is finished, the mixture is maintained under magnetic stirring for 10 minutes in order to insure the completion of the reaction. The solid phase is then filtrated and dried in air. The yield (relative to lanthanide ions) is 18(1)%. The results of the chemical analysis are summarized in Tables 2–4.
Chemical analysis results for [Ln6(μ6-O)(μ3-OH)8(NO3)6(H2O)12].2NO3.2H2O with Ln = Sm–Yb or Y.
| Anal. Calculated (found) | |||||
| Ln | MW(gmol−1) | Ln (%) | O (%) | N (%) | H (%) |
| Y | 1433.9 | 37.2 (37.0) | 52.5 (52.6) | 7.8 (7.7) | 2.5 (2.7) |
| Sm | 1802.6 | 50.0 (50.1) | 41.8 (41.5) | 6.2 (6.3) | 2.0 (2.8) |
| Eu | 1812.2 | 50.3 (50.0) | 41.5 (41.5) | 6.2 (6.4) | 2.0 (2.1) |
| Gd | 1843.9 | 51.2 (51.1) | 40.7 (40.8) | 6.1 (6.2) | 2.0 (1.9) |
| Tb | 1854.0 | 51.4 (51.4) | 40.6 (40.5) | 6.0 (5.9) | 2.0 (2.2) |
| Dy | 1875.4 | 52.0 (52.1) | 40.1 (40.1) | 6.0 (6.1) | 1.9 (2.1) |
| Ho | 1890.0 | 52.4 (52.2) | 39.8 (40.0) | 5.9 (5.9) | 1.9 (1.9) |
| Er | 1904.0 | 52.7 (52.4) | 39.5 (39.6) | 5.9 (6.0) | 1.9 (2.0) |
| Yb | 1938.6 | 53.6 (53.6) | 38.7 (39.0) | 5.8 (5.6) | 1.9 (1.8) |
Chemical analysis results for [Ln6(μ6-O)(μ3-OH)8(NO3)6(H2O)14].2NO3.2H2O with Ln = Pr or Nd.
| Anal. Calculated (found) | |||||
| Ln | MW(gmol−1) | Ln (%) | O (%) | N (%) | H (%) |
| Pr | 1781.9 | 47.4 (47.5) | 44.0 (44.1) | 6.3 (6.2) | 2.3 (2.2) |
| Nd | 1801.9 | 48.0 (48.2) | 43.6 (43.5) | 6.2 (6.1) | 2.2 (2.2) |
Chemical analysis results for [Ce6(μ6-O)(μ3-OH)8(NO3)6(H2O)16].2NO3.2H2O.
| Anal. Calculated (found) | |||||
| Ln | MW(gmol−1) | Ln (%) | O (%) | N (%) | H (%) |
| Ce | 1813.2 | 46.4 (46.6) | 45.4 (45.4) | 6.2 (6.2) | 2.0 (1.8) |
All compounds present similar IR spectra (cm−1): 3550-3100 (s) ν(OH), 1488 (m) ν(OH), 1461 (m) ν(NO3), 1387 (s) ν(NO3), 1340 (s) ν(NO3), 1040 (w) ν(NO3), 800 (w) ν(NO3), 615 (w) ν(Ln–OH), 421 (w) ν(Ln–OH)[50,51].
2.2 Synthesis of the micro-crystalline powders of anhydrous compounds [Ln6(μ6–O)(μ3–OH)8(NO3)8]∞ with Ln = Ce–Yb plus Y
The anhydrous phases have been thermally obtained by heating the hydrated phases in a furnace at 180 °C for 2 hours. All the anhydrous phases exhibit the same X-ray diffraction powder diagram. The results of the chemical analysis are summarized in Table 5.
Chemical analysis results for [Ln6(μ6-O)(μ3-OH)8(NO3)8]∞.
| Anal. Calculated (found) | |||||
| Ln | MW(gmol−1) | Ln (%) | O (%) | N (%) | H (%) |
| Y | 1181.6 | 45.1 (45.0) | 44.7 (44.8) | 9.5 (9.4) | 0.7 (0.8) |
| Ce | 1488.9 | 56.5 (56.6) | 35.5 (35.4) | 7.5 (7.6) | 0.5 (0.4) |
| Pr | 1493.6 | 56.6 (56.7) | 35.4 (35.5) | 7.5 (7.3) | 0.5 (0.5) |
| Nd | 1513.6 | 57.2 (57.0) | 34.9 (34.8) | 7.4 (7.5) | 0.5 (0.7) |
| Sm | 1550.3 | 58.2 (58.2) | 34.1 (34.3) | 7.2 (7.1) | 0.5 (0.6) |
| Eu | 1559.9 | 58.4 (58.7) | 33.9 (33.8) | 7.2 (7.1) | 0.5 (0.4) |
| Gd | 1591.6 | 59.3 (59.1) | 33.2 (33.2) | 7.0 (7.1) | 0.5 (0.6) |
| Tb | 1601.7 | 59.5 (59.5) | 33.0 (33.2) | 7.0 (6.9) | 0.5(0.4) |
| Dy | 1623.1 | 60.1 (60.2) | 32.5 (32.5) | 6.9 (7.0) | 0.5 (0.5) |
| Ho | 1637.7 | 60.4 (60.5) | 32.3 (32.4) | 6.8 (6.5) | 0.5 (0.6) |
| Er | 1651.7 | 60.8 (60.5) | 31.9 (31.8) | 6.8 (7.0) | 0.5 (0.7) |
| Yb | 1661.7 | 61.0 (61.0) | 31.8 (31.9) | 6.7 (6.6) | 0.5 (0.5) |
All compounds present similar IR spectra (cm−1): 3550-3100 (s) ν(OH), 1488 (m) ν(OH), 1461 (m) ν(NO3), 1387 (s) ν(NO3), 1340 (s) ν(NO3), 1040 (w) ν(NO3), 800 (w) ν(NO3), 615 (w) ν(Ln–OH), 421 (w) ν(Ln–OH)[50,51].
2.3 Synthesis of the micro-crystalline powders of partially dehydrated compounds [Ln6(μ6–O)(μ3–OH)8(NO3)6(H2O)6].2NO3 with Ln = Ce–Yb plus Y
The partially dehydrated phases have been obtained by drying, at room temperature, under a flux of N2 for 2 hours. All these phases exhibit the same X-ray diffraction powder diagram. The results of the chemical analysis are summarized in Table 6.
Chemical analysis results for [Ln6(μ6-O)(μ3-OH)8(NO3)6(H2O)6].2NO3.
| Anal. Calculated (found) | |||||
| Ln | MW(gmol−1) | Ln (%) | O (%) | N (%) | H (%) |
| Y | 1289.6 | 41.4 (41.5) | 48.4 (48.3) | 8.7 (8.6) | 1.5 (1.6) |
| Ce | 1596.9 | 52.7 (52.6) | 39.0 (39.1) | 7.0 (7.1) | 1.3 (1.2) |
| Pr | 1601.6 | 52.8 (52.7) | 38.9 (39.1) | 7.0 (6.8) | 1.3 (1.4) |
| Nd | 1621.6 | 53.4 (53.6) | 38.5 (38.6) | 6.9 (6.7) | 1.2 (1.1) |
| Sm | 1658.3 | 54.4 (54.3) | 37.6 (37.4) | 6.8 (7.0) | 1.2 (1.3) |
| Eu | 1667.9 | 54.7 (54.5) | 37.4 (37.5) | 6.7 (6.8) | 1.2 (1.2) |
| Gd | 1699.6 | 55.5 (55.5) | 36.7 (36.8) | 6.6 (6.5) | 1.2 (1.2) |
| Tb | 1709.7 | 55.8 (55.7) | 36.5 (36.5) | 6.5 (6.5) | 1.2 (1.3) |
| Dy | 1731.1 | 56.3 (56.3) | 36.0 (36.0) | 6.5 (6.5) | 1.2 (1.2) |
| Ho | 1745.7 | 56.7 (56.9) | 35.7 (35.5) | 6.4 (6.4) | 1.2 (1.2) |
| Er | 1759.7 | 57.0 (57.0) | 35.5 (35.4) | 6.4 (6.5) | 1.1 (1.1) |
| Yb | 1769.7 | 57.3 (57.0) | 35.3 (35.5) | 6.3 (6.5) | 1.1 (1.0) |
All compounds present similar IR spectra (cm−1): 3550-3100 (s) ν(OH), 1488 (m) ν(OH), 1461 (m) ν(NO3), 1387 (s) ν(NO3), 1340 (s) ν(NO3), 1040 (w) ν(NO3), 800 (w) ν(NO3), 615 (w) ν(Ln–OH), 421 (w) ν(Ln–OH)[50,51].
2.4 Synthesis of the micro-crystalline powder of [Y6O(OH)8](bdcH)4(NO3)4
Sixty milligram of [Er6(μ6–O)(μ3–OH)8(NO3)8]∞, 30 mg of bdcH2 (where bdcH2 stands for benzene-1,4-dicarboxylic acid) and 3.5 mL of dry acetonitrile have been put in a 24 mL Parr autoclave.
The mixture has been heated at 130 °C for 50 hours and then allowed to slowly cool down at a cooling rate of 1 °C per hour. The resulting solid has been filtered off and dried under N2. Anal. Calcd. (found) for [Y6O(OH)8](bdcH)4(NO3)4: Y 33.4% (31.4%); C 24.1% (21.9%); H 1.8% (2.1%); N 3.6% (3.6%); O 37.1% (41.0%).
The IR spectrum, measured on a KBr pellet clearly shows the characteristic bands of both protonated and deprotonated carboxylate groups that is 1410 cm−1 and 1380 cm−1 for −COOH and –COO−, respectively. The characteristic band of nitrate at 1340 cm−1 is also observed. On the other hand, the spectrum does not show any characteristic band either for water molecule or for nitrile functions.
The TGA measurements confirm that there is no solvent departure. They also confirm that the molecular weight of the compound is 1600 gmol−1 (1594 gmol−1 calculated).
2.5 Solubility and stability of [Er6(μ6–O)(μ3–OH)8(NO3)8]∞
For this study, commercial solvent have been dried on molecular sieves and used without further purification. A suspension of 500 mg of [Er6(μ6–O)(μ3–OH)8(NO3)8]∞ in 20 mL of each tested organic solvent has been left under stirring at room temperature, overnight in a capped vessel. The suspension was then filtered off. The Er3+ concentration of the resulting solution has been was measured by ICP using a Perkin Elmer Optima 3000 DV apparatus. The remaining solid has been analyzed by XRD.
To check the stability of [Er6(μ6–O)(μ3–OH)8(NO3)8]∞, the filtered solution has been evaporated to dryness and the obtained powder has been analyzed by XRD.
2.6 X-ray powder diffraction
The X-ray powder diffraction diagrams have been collected using a Panalytical X’Pert Pro diffractometer with an X’celerator detector. The recording conditions were 40 kV and 40 mA for Cu–Kα (λ = 1.542 Å), the diagrams were recorded in θ/θ mode in 11 hours between 5 and 75° (8378 measurements) with a step size of 0.0084° in 2θ and a scan time of 600 s.
For pattern indexing, the extractions of the peak positions were carried out via the WINPLOTR [52] software. The pattern indexing was performed by the program McMaille [53], and the refinement of the unit-cell parameters by means of the Chekcell program which is a modified version of Cellref from CRYSFIRE suite [54].
The identification of the phases based on their X-ray diffraction diagrams has been done by using the Highscore Software [55] with a JCPDS version PDF2/2000 (sets 1–50 and 70–88) database.
2.7 X-Ray structure determination of compound [Ho6(μ6–O)(μ3–OH)8(NO3)6(H2O)6].2NO3
To solve the structure of the dehydrated compounds, we used the modeling and simulation software Materials Studio Modeling [56]. This software is convenient for molecular materials because it is able to compute a simulated annealing on fixed groups of atoms, here for example the octahedral hexanuclear complex that we expected to be preserved on the basis of the dehydration–rehydration cycle. After thousands of configurations checked the software proposed a chemically suitable molecular structure that we refined using the Rietveld calculation with a global isotropic temperature factor [57]. Crystal and final structure refinement data of the compound are listed in Table 7. The experimental and calculated diffraction diagrams are shown in Fig. 4. Full details of the X-ray structure determination of the anhydrous yttrium containing complex have been deposited by the Fachinformationszentrum Karlsruhe under the depository number CSD-421296 and can be obtained, on request, mentioning the authors and a reference to the present publication.
Experimental data for the X-ray diffraction study of [Ho6O(OH)8(NO3)6(H2O)6].(NO3)2.
| Molecular formula | Ho6O39N8H20 |
| Formula weight | 1745.8 |
| Temperature (K) | 293 |
| Crystal System | monoclinic |
| Space group | P1n1 (no. 7) |
| a (Å) | 13.951(5) |
| b (Å) | 10.227(5) |
| c (Å) | 12.949(5) |
| β (°) | 88.85(1) |
| V (Å3) | 1847 |
| Z | 2 |
| λ (Å) | 1.5418 |
| ρcalc (g·cm−3) | 3.139 |
| Pattern range (2θ deg) | 5–45 |
| Step size (2θ deg) | 0.0084 |
| Step scan time (s) | 600 |
| No. contributing reflns (Kα1 + Kα2) | 274 |
| No. refinement parameters | 18 structural parameters refined (18 DOF) 5 profile parameters refined |
| Rwp | 0.1304 |
| Rp | 0.0850 |

Calculated and experimental diffraction diagrams of compound [Ho6O(OH)8(NO3)6(H2O)6].2NO3.
2.8 Thermal analysis
Thermo-gravimetric and thermal differential analyses were performed in a platinum crucible under a nitrogen atmosphere between room temperature and 1000 °C with a heating rate of 5 °C·min−1 using a Perkin-Elmer Pyris-Diamond thermal analyzer.
The thermal dependence X-ray diffraction experiments have been performed by a Panalytical X’Pert Pro diffractometer with an X’celerator detector using Cu Kα1 radiation in the range 5–75° in 2θ. The heating of the samples (from room temperature to 1000 °C) was performed using an Anton Paar HTK 1200 furnace under nitrogen atmosphere.
2.9 Solid state luminescence measurements
Solid state emission spectra were measured on a Perkin-Elmer LS-55 fluorescence spectrometer with a pulse Xe lamp. The slit width was 5 nm for excitation and 5 nm for emission.
Luminescence spectra were all recorded at room temperature between 200 nm and 800 nm in identical operating conditions without turning the lamp off to ensure a valid comparison between the emission spectra. Reproducibility of the measurements has been carefully checked by reproducing several times. The data were collected, at 100 nm·min−1, in phosphorescence mode with 0.05 ms delay time between the excitation pulse and the emission measurement.
2.10 Optical microscopy and turbidity measurements
Complex particles in suspension have been observed using an optical transmission microscope Zeiss Axiovert 135 equipped with a 63 × objective and a Sony DXC-C33P video camera connected to a computer for image capture.
Turbidity values of the dispersions were obtained using a Perkin Elmer Lambda 35 spectrophotometer at λ = 600 nm, i.e., in a region where neither the complex nor the solvent absorb. Samples were agitated just before acquisition of turbidity data in order to prevent sedimentation of aggregates.
3 Results and discussion
To be able to use hexanuclear complexes as starting materials for further chemistry, we need a synthetic method that leads to well defined entities. For this purpose, we have optimized the synthetic procedure. Therefore, thanks to this optimized synthetic method, it is possible to obtain for each lanthanide ion the hexanuclear complex with two crystallization water molecules (Table 8).
Summary of the hexanuclear complexes obtained by the optimized synthesis.
| Ce | Pr | Nd | Sm | Eu | Gd | Tb | Dy | Ho | Y | Er | Tm | Yb | |
| [Ln6O(OH)8(NO3)6(H2O)12].(NO3)2.2H2O | |||||||||||||
| [Ln6O(OH)8(NO3)6(H2O)14].(NO3)2.2H2O | |||||||||||||
| [Ln6O(OH)8(NO3)6(H2O)16].(NO3)2.2H2O |
The use of these hydrated hexanuclear complexes is possible but because of their high hydration rate (between 2.33 and three water molecule per lanthanide ion) the hydrated complexes hydrolyze during the reaction even in dry organic solvents [48,49]. However, this hydrolysis occurs during the synthesis and leads to original compounds that cannot be obtained from simple isolated lanthanide ions (Fig. 5). This can be understood by considering the different accessibility of lanthanide ions when they are involved in a hexanuclear complex or not.
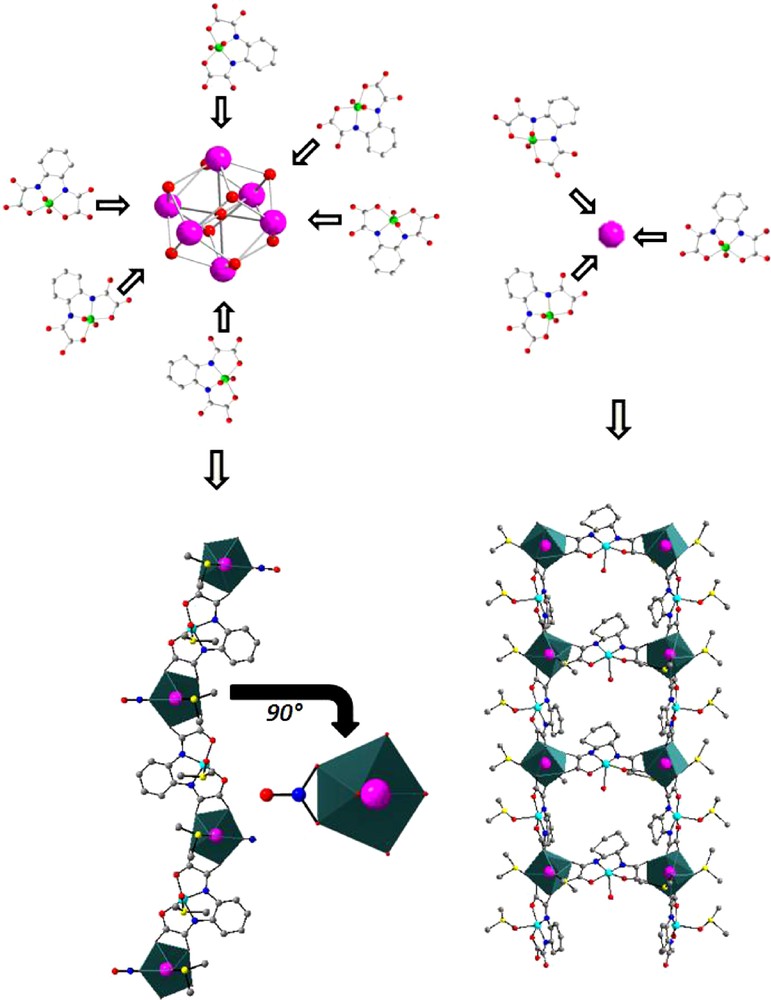
Schematic representation of the “structuring effect” of hexanuclear complexes.
Actually, due to the inner character of their valence orbitals, lanthanide ions can be compared to rigid spheres with no structuring effect. The spatial distribution of the ligands is usually mainly driven by steric effects [58]. On the other hand, when the lanthanide ion belongs to a hexanuclear complex, it can only interact along one direction with the ligand. This structuring effect can be exemplified by comparing the products of the reactions in DMSO of the monomeric Cu(II) complex [Cu(opba)]2− where opba stands for ortho-phenylene-bis-oxamato with respectively an isolated lanthanide ion and the corresponding hexanuclear complex: when reacting with an isolated lanthanide ion, [Cu(opba)]2− leads to an infinite ladder-like molecular motifs with chemical formula {Ln2[Cu(opba)]3(DMSO)6(H2O).H2O}∞ [59] while reacting with a hydrated hexanuclear complex it leads to an infinite bi-metallic chain with chemical formula {Ln(NO3)(DMSO)2[Cu(opba](DMSO)2}∞ [49].
Furthermore, the bidentate nitrates present in the molecular structure of the hexanuclear complexes remain most often bounded to the lanthanide ion even after the hydrolysis (Fig. 5). Overall, one can consider that the hydrolysis of the hexanuclear complex leads to [Ln(NO3)]2+ entities.
Therefore, this hydrolysis is obviously an asset for obtaining linear molecular chains. However, it constitutes a serious drawback for obtaining potentially porous three dimensional coordination polymers. That is why, in order to increase the chemical stability of the complexes, we have decided to study in detail the less hydrated phases.
The thermal behaviors of all these compounds are essentially the same. All the hydrated complexes have been studied by TG-DTA and TDXD under N2 flow (Fig. 6). These studies show that the decompositions occur in five steps and finally lead (above 500 °C) to the rare earth oxides.
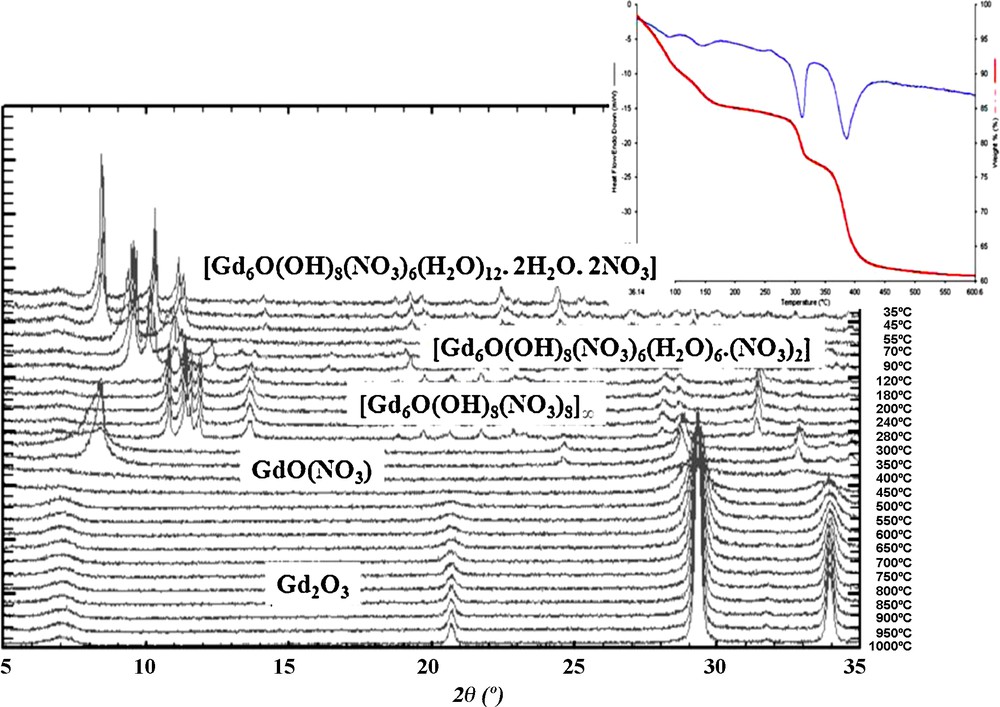
TG-DTA curves and TDXD of compound [Gd6O(OH)8(NO3)6(H2O)12].(NO3)2.2H2O between room temperature and 1000 °C.
The first phenomenon occurs between room temperature and roughly 100 °C. Fig. 7 shows the precise values versus the involved lanthanide ion. It has been attributed to the loss of eight water molecules per molecular unit (the two crystallization water molecules plus one coordination water molecule per Ln(III) ion).

Temperature of appearance of compounds [Ln6O(OH)8(NO3)6(H2O)6].(NO3)2 versus the ionic radius of the Ln3+ ion. Ionic radii are for eight-coordinated ions [60].
These phases with general chemical formula [Ln6O(OH)8(NO3)6(H2O)6].(NO3)2 were not known until this work. They have been structurally characterized in the following.
The six remaining water molecules are lost between 100 and about 170 °C and the resulting phase has been assumed to be the anhydrous hexanuclear compound [Ln6O(OH)8(NO3)8]∞ on the basis of its XRD diagram. This phase is stable over more than 100 °C. Fig. 8 shows the temperatures of appearance and disappearance of the anhydrous phases versus the ionic radius of the involved lanthanide ion. This figure shows an important decreasing of the stability temperature domain of the hydrated phase when the ionic radius of the involved lanthanide ion increases. This behavior has already been observed on other lanthanide-containing compounds [61].
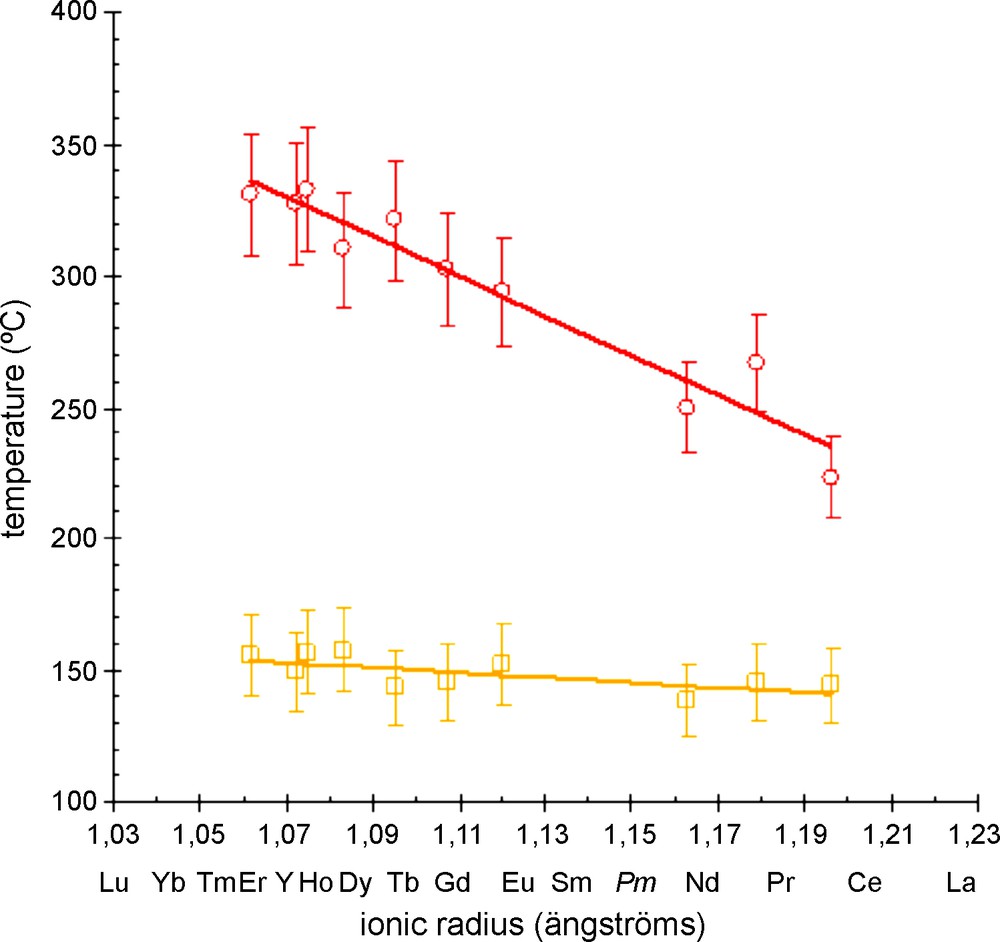
Temperatures of appearance and disappearance of the anhydrous phases [Ln6O(OH)8(NO3)8]∞ versus the ionic radius of the Ln3+ ion. Ionic radii are for eight-coordinated ions [60].
At last, this anhydrous phase decomposes, leading first to LnO(NO3)[42] and finally to the lanthanide oxide.
The XRD diagrams of compounds with general chemical formula [Ln6O(OH)8(NO3)6(H2O)6].(NO3)2 are of poor quality when the compounds are obtained by thermal treatment of the initial hydrated phase [Ln6O(OH)8(NO3)6(H2O)12].(H2O)2.(NO3)2. Fortunately, they can also be obtained by slow dehydration under a N2 flow at room temperature. These synthetic conditions lead to well crystallized micro-crystalline powders exhibiting XRD patterns allowing a structural determination (Fig. 9).

XRD pattern of [Ho6O(OH)8(NO3)6(H2O)6].(NO3)2 obtained by drying under N2 flow at room temperature.
This compound crystallizes in the monoclinic system, space group P1n1 (n°7) with the following cell parameters: a = 13.951 Å, b = 10.227 Å, c = 12.949 Å, β = 88.85° and Z = 2. This crystal structure can be described as the juxtaposition of [Ho6O(OH)8(NO3)6(H2O)6]2+ hexanuclear entities (Fig. 10). The hexanuclear entity can be described as follows: an O2− ion is surrounded by six Ho(III) ions in a slightly distorted octahedral fashion forming a μ6-oxo bridge with an average Ho-O distance equal to 2.375 Å. Each octahedron face is capped by a μ3-hydroxo group with Ho-O distances around 2.25 Å. Each Ho(III) ion is also bounded to a bidentate nitrate and to one coordination water molecule. Actually, all the Ho(III) ions are eight coordinated by four oxygen atoms from μ3-hydroxo groups, the oxygen atom localized at the center of the octahedral, two oxygen atoms from the bidentate nitrate and one oxygen atom from the coordinated water molecule. The coordination polyhedron can be described as a bi-capped trigonal prism (Fig. 10), a rather uncommon geometry already reported [62–64].
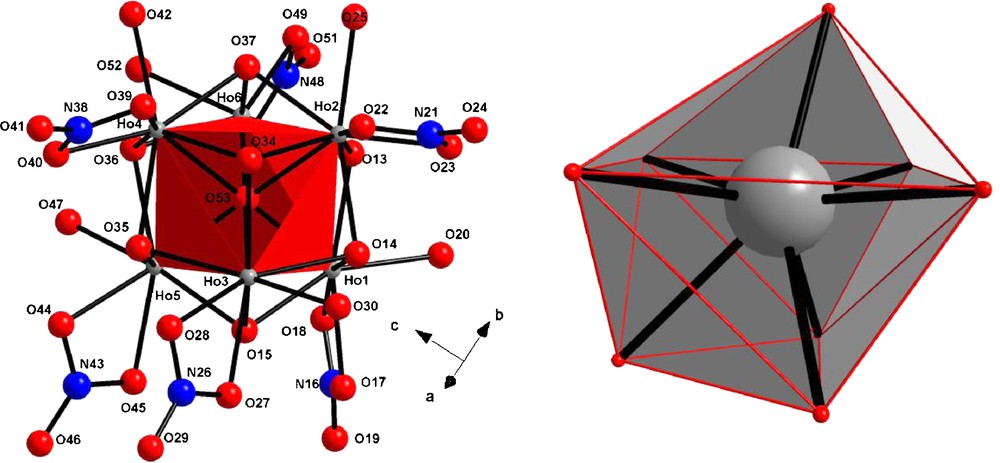
View of the hexanuclear entity [Ho6O(OH)8(NO3)6(H2O)6]2+ along with the labeling scheme (left). View of the bi-capped trigonal prism (right).
The crystal packing of this compound can be described as the juxtaposition of hexanuclear entities (Fig. 11). From the projection view along the a axis, it can be noticed that the complexes present a sort of AB-like arrangement, the A-type complexes and the B-type ones presenting actually a different orientation.

Left: Projection view along the a axis of the crystal packing of [Ho6O(OH)8(NO3)6(H2O)6].(NO3)2. The free nitrates have been omitted for clarity. Right: symbolic representation of the octahedral.
The main difference between these hexanuclear entities and hexanuclear entities encountered in the hydrated phases crystal structures is the relative orientation of the bidentate nitrate groups. In this crystal structure, the nitrate groups are orientated for minimizing the steric hindrance between adjacent hexanuclear complexes, whereas in the crystal structures of the hydrated phases, all the bidentate nitrate groups lay roughly in the same plane (compare Figs. 2 and 10). The neutrality of the crystal network is insured by two nitrate ions.
As shown in Fig. 12, these partially dehydrated compounds reversibly bind water when exposed to wet atmosphere leading to the corresponding hydrated compound [Ln6O(OH)8(NO3)6(H2O)12].(H2O)2(NO3)2.

Dehydration / rehydration cycle for compound [Gd6O(OH)8(NO3)6(H2O)12].(H2O)2.(NO3)2 under respectively dry and wet N2 flow.
The dehydration of the hexanuclear complexes [Ln6O(OH)8(NO3)6(H2O)6] (NO3)2 is accompanied by a condensation: the hexanuclear entities bind together forming infinite chains with chemical formula [Ln6O(OH)8(NO3)8]∞. This condensation occurs in the solid state and implies the free nitrate groups present in the crystal structure of the [Ln6O(OH)8(NO3)6(H2O)6].(NO3)2 complexes. A mechanism based on the analysis of the shortest inter-molecular contacts is proposed in Scheme 1. Our analysis of both crystal structures suggests that, upon dehydration, two out of the six bidentate nitrate groups of each [Ln6O(OH)8(NO3)6]2+ complex bind in a monodentate fashion two Ln(III) ions belonging to another [Ln6O(OH)8(NO3)6]2+ complex. The suggested bounds in Scheme 1 are symbolized by dotted lines and correspond to the shortest Ln-O intermolecular contacts. Simultaneously, the free nitrate groups present in the neighborhood of the hexanuclear complexes establish a monodentate bound with the nearest Ln(III) ions. These Ln(III) ions are the ones on which are involved in the bridging of the hexanuclear complexes. These contacts are symbolized by dashed lines in Scheme 1. It is noticeable than the A-type complexes bind together as well as the B-type complexes. Therefore, the binding of the complexes is accompanied by a rearrangement leading to a centrosymmetric crystal structure in which all the octahedral are equivalent.
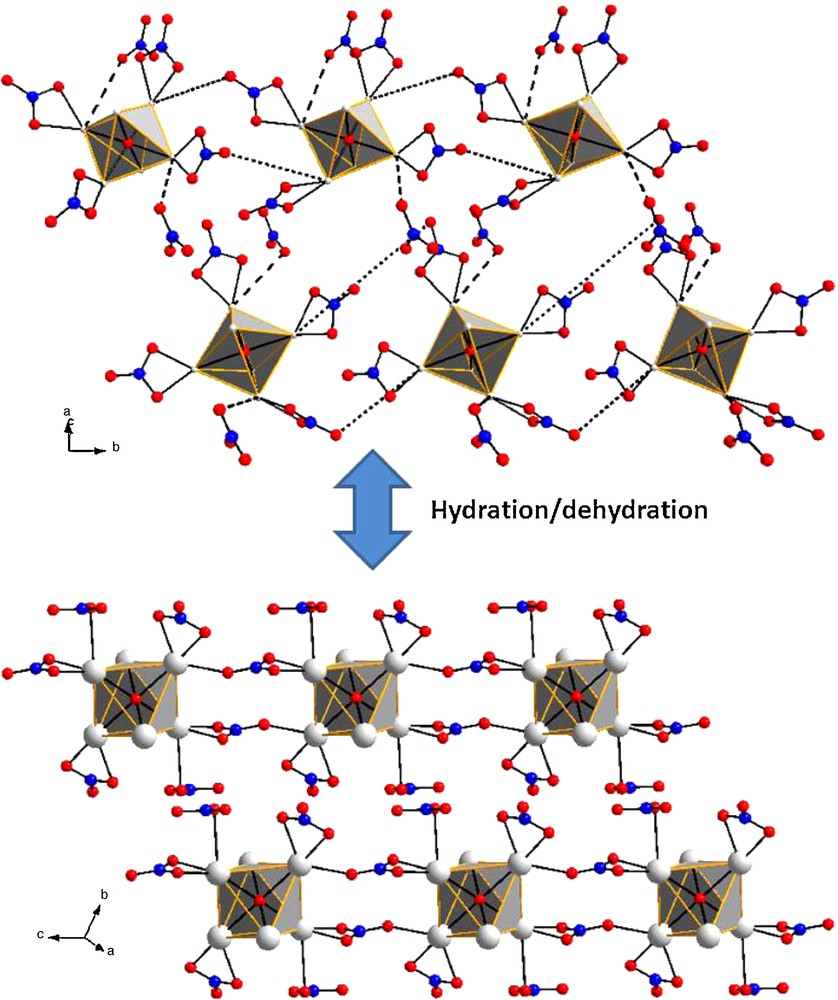
Suggested mechanism of condensation of the hexanuclear complexes. Only the groups involved in the mechanism are represented .
If the rehydration is pursued for several days, the more hydrated phases [Ln6O(OH)8(NO3)6(H2O)12].(H2O)4.(NO3)2, [Ln6O(OH)8(NO3)6(H2O)12].(H2O)5.(NO3)2 and [Ln6O(OH)8(NO3)6(H2O)12].(H2O)6.(NO3)2 are progressively observed. The complete hydration dehydration cycle for the hexanuclear complexes can be outlined as in Scheme 2 where it is apparent that the more hydrated phases are more difficult to obtain when the smaller lanthanide ions are involved.
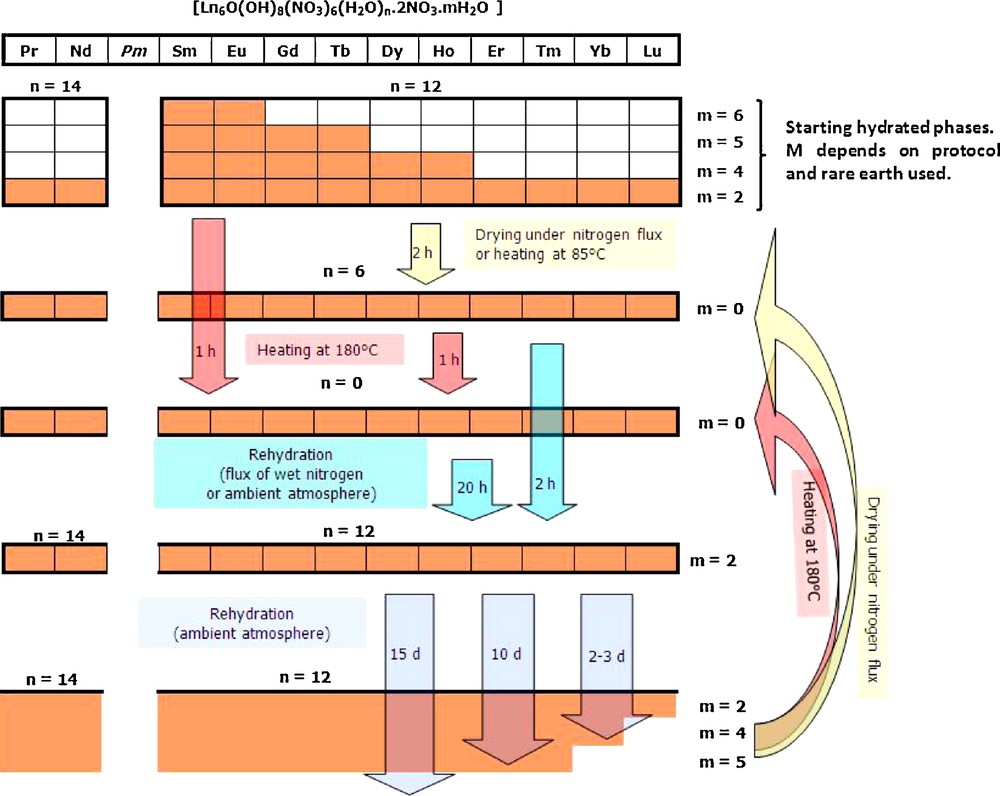
Hydration–dehydration cycle for hexanuclear entities involving one of the rare earth comprises between Pr and Lu (except Pm) or Y.
This hydration–dehydration cycle is particularly interesting as far as hexanuclear complexes are used as molecular precursor for further chemistry. On account of these various processes, it is actually possible, for all lanthanide ions from Pr to Lu (except Pm) and Y, to synthesize well defined monophasic polycrystalline powders of both the hydrated complex [Ln6O(OH)8(NO3)6(H2O)12].(H2O)2.(NO3)2 and the anhydrous phase [Ln6O(OH)8(NO3)6]∞.
As already explained before, the use of hydrated hexanuclear complexes as molecular precursors has always provoked the decomposition of the hexanuclear entities. In order to avoid this decomposition, we have decided to use the anhydrous phase as starting material. For this purpose, we have evaluated the solubility and the stability of this phase in various organic solvents. For practical reasons this study has been realized on the Er(III)-containing compound. Preliminary observations have shown that the compound is soluble in most polar solvents. However, after some hours, it decomposes into ethanol, acetone, DMF, DMSO and THF. Only butan-1-ol, butan-2-ol and acetonitrile do not destroy it, even after several days. Furthermore, the crystallinity of the compound after dissolution in acetonitrile and re-crystallization is of better quality than that of the initial microcrystalline powder (Fig. 13).

Powder XRD diagrams of compound [Er6O(OH)8(NO3)8]∞ before (down) and after (up) re-crystallization in acetonitrile.
This seems to indicate that the coordination polymer [Er6O(OH)8(NO3)8]∞ is really dissolved and not only dispersed in acetonitrile. This point is very important when aiming to use the [Ln6O(OH)8(NO3)8]∞ compounds as molecular precursors. Actually, the use of these compounds as starting materials is hopeless if their dissolution does not liberate free hexanuclear complexes ready to react with organic ligands. Therefore, we have undertaken to verify if the expected equilibrium
Therefore, an oversaturated solution of [Y6O(OH)8(NO3)8]∞ in acetonitrile has been prepared. Complex particles in suspension were large enough to be directly observable using optical transmission microscopy (Fig. 14). Elongated solid particles, with a length up to 10 μm and a diameter of about 2–3 μm, were freely moving in the solvent (Fig. 14 A, arrow). The elongated form of these particles is clearly compatible with the ribbon-like structure. The bigger particles result visibly from partial aggregation of smaller particles (Fig. 14 B, arrow), and are rapidly immobilized on the microscope slide by simple sedimentation. Such particles are evidently large enough to strongly interact with visible light, explaining the turbid aspect of the dispersions.

Observation of [Y6O(OH)8(NO3)8]∞ in acetonitrile as seen by transmission optical microscopy (the bar corresponds to 10 μm). The arrows in images A and B point to an isolated particle and a strongly aggregated particle, respectively.
Turbidity measurements as a function of dilution and agitation gave clear indication of partial solubility of the complexes in acetonitrile. A dispersion containing initially 5 mg/mL of [Y6O(OH)8(NO3)8]∞ in acetonitrile was successively diluted at different volume fractions, from 0.1 to 10−4. Straight after dilution, turbidity decreases about linearly with dilution as expected (Fig. 15). In order to gain some insight about the complexes solubility, the samples were maintained in sealed tubes under a very gentle agitation for three days followed by turbidity measurements. A systematic decrease in the turbidity of the agitated samples was observed compared to those obtained immediately after dilution. A different sample was subjected to rather strong agitation for 90 min. Again, an important decrease in turbidity was detected. Since it is unlikely that the agitation of the dispersions is violent enough to break up the solid particles, the behavior may be attributed to partial dissolution of the particles.

Turbidity measurements of the same suspension of [Y6O(OH)8(NO3)8]∞ in acetonitrile as a function of its dilution, immediately after dilution (●), after 3 days of gentle agitation (○) and following a 90 min strong agitation (×).
In order to confirm these experiments, a spectroscopic study has been realized on the Tb(III)-containing compound in acetonitrile because of the well known luminescent properties of the Tb3+ ions. In order to check the solubilization of [Tb6O(OH)8(NO3)8]∞ in acetonitrile, we have first measured the excitation and the luminescent spectra of the compound in the solid state (Fig. 16).

Solid state excitation (left) an emission (right) spectra of Tb(NO3)(H2O)6 and [Tb6O(OH)8(NO3)8]∞.
Both compounds exhibit similar solid state emission and excitation spectra. The excitation spectra (Fig. 16 left) show seven bands that can be attributed respectively to 7F6→5H7 (316 nm), 7F6→5DJ (J = 1,0) (321–325 nm), 7F6→5LJ (J = 6,7,8) (338–342 nm), 7F6→5L9 (351 nm), 7F6→5G6 (359 nm), 7F6→5L10 (369 nm) and 7F6→5G6 (379 nm) transitions. The excitation wavelength for measuring emission spectra has been chosen at 351 nm (7F6→5L9). The emission spectra are dominated by two multiplets centered around 491 nm and 540 nm attributed respectively to the 5D4→7F6 and 5D4→7F5 transitions.
Then we have prepared an oversaturated solution of [Tb6O(OH)8(NO3)8]∞ in acetonitrile, transferred it in a fluorimetric cell and allowed it to slowly evoluate without stirring. The evolution of the suspension has been followed by measuring excitation and emission spectra from time to time (Fig. 17). The first measurement, realized immediately after the preparation of the oversaturated solution, gave exactly the same spectra than the ones obtained on the solid state: the particles in suspension dominate the optical properties. Then, the characteristic peaks of the solid state excitation spectrum rapidly vanished and another broad excitation band centered on 310 nm appeared: the particles in suspension had sedimentated and the concentration of dissolved compound were still very low. Then the characteristic peaks of the dissolved compound progressively increased and reached a plateau after roughly 150 hours: at this time, the solution was saturated and the equilibrium was reached.

Excitation (Left) and emission (Right) spectra of [Tb6O(OH)8(NO3)8]∞ in acetonitrile versus time. In inset, the integrated intensity of the 5D4→7F5 transition of emission spectra versus time.
From these results, the solubility and the stability in acetonitrile of compounds [Ln6O(OH)8(NO3)8]∞ have been assumed and we have quantitatively estimated the solubility of these compounds in various organic solvents (Table 9).
Solubility of [Er6O(OH)8(NO3)6]∞ in various dry organic solvents.
| Solvent | Solubility (gL−1) |
| DMSO | 21.0 (5) |
| DMF | 17.5 (5) |
| Acetone | 11.0 (5) |
| THF | 3.5 (5) |
| Athanol | 3.5 (5) |
| Acetonitrile | 3.0 (5) |
| Butan-1-ol | 3.0 (5) |
| Butan-2-ol | 3.0 (5) |
| Propan-2-ol | 2.5 (5) |
| Diethyl ether | 1.5 (5) |
| N-hexane | 1.0 (5) |
In order to verify the usability of these anhydrous compounds as molecular precursors of new materials, we have synthesized a new compound. This compound was prepared by a solvothermal method. This synthetic approach has offered us a monophasic microcrystalline powder. The XRD diagram of this powder is very different from that obtained in similar conditions and involving Y(NO3)3.5H2O instead of [Y6O(OH)8(NO3)8]∞. The quality of the XRD diagram was sufficient for indexing it (Fig. 18). This compound crystallizes in the monoclinic system, with the following cell parameters: a = 21.296 Å, b = 9.768 Å, c = 20.762 Å, β = 123.54° V = 3600 Å3. The proposed chemical formula is [Y6O(OH)8](Hbdc)4(NO3)4]. It has been determined on the basis of chemical analysis, thermal analysis and IR spectra. Despite great synthetic efforts, it has not been possible until now to obtain a single crystal suitable for X-ray crystal structure determination. Therefore, we are currently trying to solve the crystal structure on the basis of the powder XRD diagram but the great number of independent parameters makes this determination difficult.
Normalized powder XRD diagram of [Y6O(OH)8](Hbdc)4(NO3)4]. The main diffraction peaks are listed, 2θ (relative intensity) [hkl]): 9,542° (100) [−2 0 2]; 9;957° (69,8) [−2 0 0]; 10,285° (61,3) [−1 1 1]; 14,984° (4,4) [−3 0 0]; 16.621° (3.6) [−4 0 2];16.961° (2.8) [−3 1 3]; 17,366° (9,2) [−3 0 4]; 18,241° (4,7) [−1 0 4]; 19,185° (10,9) [−4 0 4]; 20,074° (4,6) [2 1 2]; 20,703° (6,3) [1 1 3]; 26.771° (3.0) [−6 1 3]; 27.902° (3.0) [0 3 1]; 40.186° (1.2) [−7 0 9]; 40.990° (1.5) [4 0 5].
4 Conclusions and outlook
At this stage, we are able to synthesize hydrated hexanuclear complexes involving almost all the lanthanide ions. These compounds have general chemical formula [Ln6O(OH)8(NO3)6(H2O)x]2+ with x = 12 when Ln = Sm–Yb, x = 14 when Ln = Pr–Nd and x = 16 for Ln = Ce. Despite their hygroscopic character, these complexes can be stocked for a long time because they can be regenerated by a simple hydration–dehydration process.
These complexes can also be dehydrated, leading to a family of polymeric compounds with chemical formula [Ln6O(OH)8(NO3)8]∞ with Ln = Ce–Yb (except Pm) or Y. These anhydrous compounds are of particular interest because they readily dissolved in dry organic solvents such as acetonitrile. Preliminary results show that it is possible to use these anhydrous compounds as starting materials for further chemistry.
However, up to date, we have only demonstrated that the polymeric chains dissolve in acetonitrile but we have no information concerning the chemical form of the dissolved species in solution. We are currently working along this line.