

1 Introduction
During the past decade, ecological requirements have pressed chemists to develop clean catalytic processes and technologies. In this context, the immobilization of homogeneous or heterogeneous catalysts in an aqueous phase appears an environmentally friendly technique to produce organic compounds [1–5]. Indeed, the catalyst can be easily recovered in active form at the end of reaction by decantation of the aqueous and organic phases, and the production costs are significantly lower (Fig. 1).
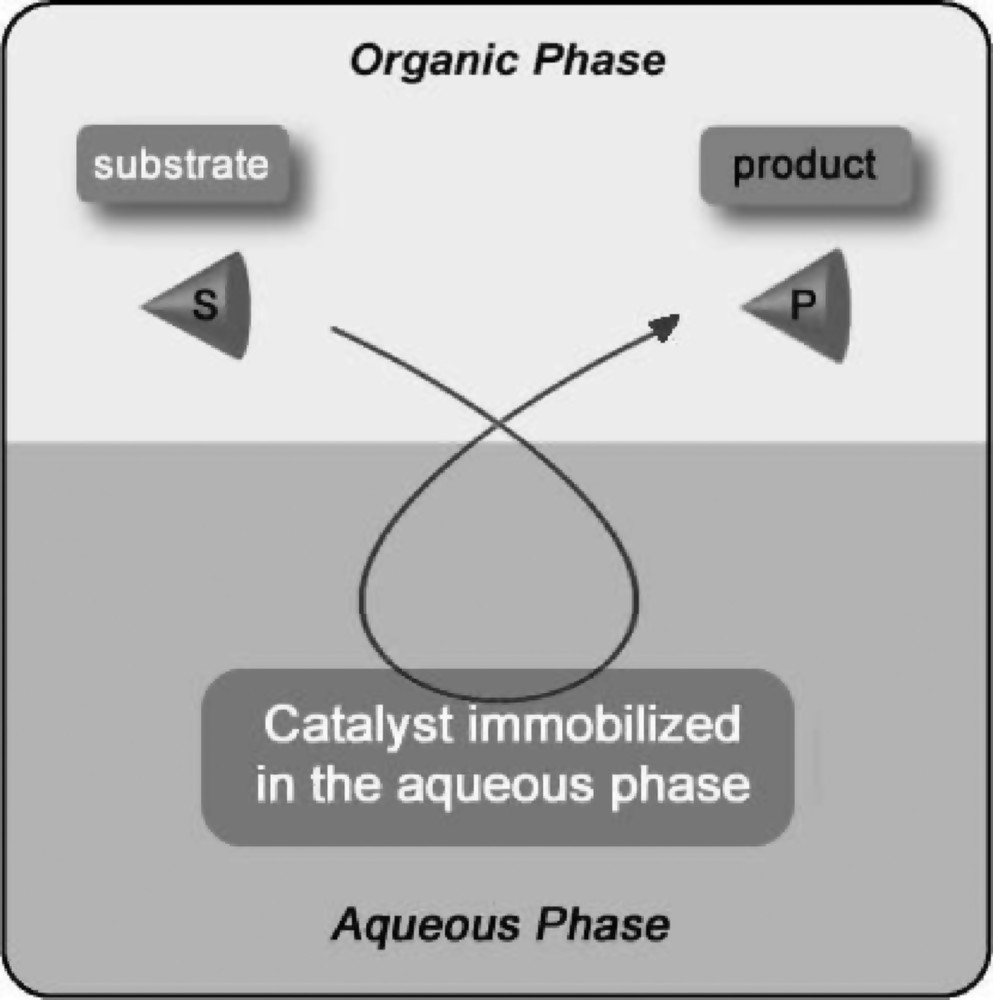
Principle of aqueous catalysis.
This methodology has proven its viability with the industrial Ruhrchemie/Rhône-Poulenc process (now OXEA company), in which the rhodium-catalyzed hydroformylation of propene or butene has been conducted since 1984 producing 800,000 t y−1 of C4 and C5 aldehydes. In this process, the rhodium catalyst was efficiently anchored in the aqueous phase via a water-soluble phosphine ligand P(C6H4SO3Na)3 (TPPTS), leading to very low metal leaching, i.e., on a ppb scale [6]. However, the scope of aqueous catalysis is greatly reduced by the low solubility of most organic substrates in water and by the need to synthesize water-soluble ligands or stabilizing agents to immobilize the catalyst in water. Indeed, poorly water-soluble substrates react far too slowly under aqueous biphasic conditions for economically viable industrial applications, and the synthesis of water-soluble ligands or stabilizing agents usually requires expensive or non-commercially available reagents, tedious work-up or long multistep reaction sequences [7].
Among the different approaches proposed to circumvent these problems, the use of cyclodextrins (CDs) proves to be very promising [8]. Very concisely, CDs are a class of naturally occurring receptors which are cyclic oligosaccharides constituted of six (α-CD), seven (β-CD), or eight (γ-CD) D-glucopyranose units [9]. Their shape is a conical cylinder whose inner surface is hydrophobic and outer surface hydrophilic. The wider opening is surrounded by the whole of the secondary hydroxyl groups, while the narrower opening contains all the primary ones (Fig. 2).
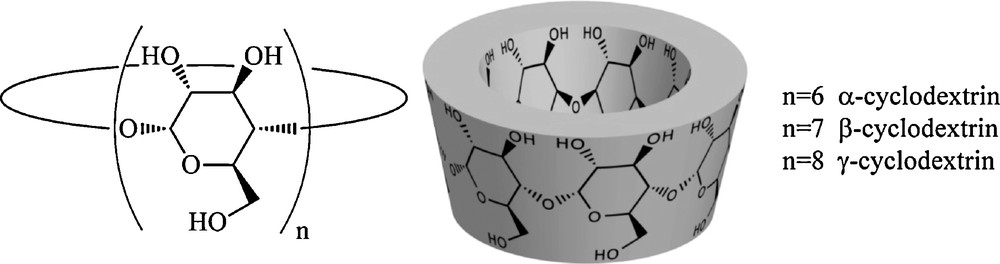
Chemical structure of native cyclodextrins.
The reactivity difference between the primary and secondary hydroxyl groups allows selective functionalization on the primary and secondary rims of CDs. Well-established synthesis protocols for selective modification of CDs have been extensively studied by many researchers and provide the opportunity to fine tune their physicochemical properties (solubility, surface activity, etc.) [10]. Their ability to encapsulate a wide range of guest molecules into their cavity has been widely demonstrated and makes them suitable for applications in analytical chemistry, agriculture, pharmaceutical industry, foodstuffs, toilet articles, textile processing and catalysis [11].
In this review, we will demonstrate that chemically modified CDs allow one to:
- • significantly increase the rate and selectivity of reactions catalyzed by water-soluble organometallic catalysts;
- • design new water-soluble catalysts;
- • stabilize catalytically active noble metal nanoparticles in water;
- • facilitate reactions catalyzed by supported metals or metallic powder in water.
- • These outstanding results will be attributed to the complexing and surface-active properties of the chemically modified CDs.
2 Results and discussion
2.1 Cyclodextrins as mass transfer additives in aqueous organometallic catalysis
The possibility of using CDs as mass transfer additives in reactions catalyzed by metal transition complexes was first reported by Alper in 1986. This author successively published two papers on the use of native CDs in palladium-catalyzed olefin oxidation and rhodium-catalyzed conversion of carbonyl compounds to hydrocarbons (Table 1) [12,13].
Aqueous organometallic catalysis mediated by native CDs.
| Catalytic reaction | Scheme | References |
| Olefins oxidation (Wacker reaction) | [12,14] | |
| Reduction of carbonyl compounds | [13] | |
| Deoxygenation of allylic alcohols | [15] | |
| Hydrogenation of α,β-unsaturated acids and derivatives | [16] | |
| Asymmetric hydrogenation of α-keto ester | [17] | |
| Hydrogenation of conjugated dienes | [18] | |
| Alkylation of aldehydes | [19,20] | |
| Hydrogenation of aldehydes | [21] | |
| Hydroformylation of higher olefins | [22] |
In these papers, two main features of the CD-based catalytic system already appeared. First, the activity of the system strongly depended on the nature of the CD. Thus, in the oxidation reaction of 1-decene by means of oxygen, palladium chloride and copper chloride in water, the native β-CD proved to be more efficient than the native α-CD, itself better than the native γ-CD. Second, CDs modified the chemoselectivity of the reactions. Thus, when styrene underwent carbon–carbon bond cleavage to benzaldehyde when oxidation was attempted using a quaternary ammonium halides or PEG-400 as the phase transfer catalyst, styrene was mainly converted to acetophenone when the reaction was performed using native CDs. These pioneer works were swiftly followed by Takahashi et al. who confirmed the efficiency of native CDs as additives in palladium-catalyzed olefin oxidation [14]. In 1990, the use of native CDs was extended to deoxygenation of allylic alcohols [15], and the reduction of α,β-unsaturated acids [16], α-keto esters [17] and conjugated dienes [18]. Recently, Yorimitsu and Oshima reported that the reaction of benzaldehyde with a trialkylborane having a benzyl ether moiety could be greatly improved by adding native β-CD into the reaction medium [19]. The authors assumed that β-CD suppresses an unfavorable interaction between the nickel catalyst and the substrate by including the substrate aromatic ring into its cavity [20]. However, it should be emphasized that only β-CD promoted the reaction. Indeed, the yields in the presence of α-CD were lower than those observed without CD. Adverse effects of native CDs on transition metal catalyzed reactions have also been reported in two other reactions. The reduction of aldehydes using the ruthenium complex [RuCl2(P(C6H5)2(C6H4SO3Na))2] [21] and the hydroformylation of 1-hexene using the rhodium catalyst [HRh(CO)(P(C6H5)2(C6H4SO3Na))3] [22] were inhibited by addition of β-CD and α-CD to the reaction medium, respectively. Although no spectroscopic evidence has been obtained, the detrimental effect of the CD was attributed to interactions between the CD and the catalyst.
A major breakthrough was achieved in 1994 by using chemically modified CDs [23]. Chemically modified CDs are obtained by substitution of the alcohol functions of CD by one or several chemical groups. The modified CDs usually used in aqueous organometallic catalysis are gathered in Table 2.
Structure of the cyclodextrin derivatives usually used in aqueous organometallic catalysis.
| Abbreviation | n | Substituent (R) | Carbon bearing the OR group | Average number of OR group by CD | Source |
| HP-α-CD | 6 | 2, 3, 6 | 3.6 | Commercially availablea | |
| DIME-α-CD | 6 | 2, 6 | 12 | Commercially availablea | |
| RAME-α-CD | 6 | 2, 3, 6 | 10.8 | Ref. [132] | |
| HTMAP-α-CD | 6 | 2 | 1 | Ref. [72] | |
| DIME-β-CD | 7 | 2,6 | 14 | Commercially availablea | |
| RAME-β-CD | 7 | 2, 3, 6 | 11.8–12.6b | Commercially availablea | |
| HP-β-CD | 7 | 2, 3, 6 | 4.2 | Commercially availablea | |
| HP-β-CD | 7 | 2, 3, 6 | 5.6 | Commercially availablea | |
| SBE-β-CD | 7 | 2 | 1 | Commercially availablea | |
| HTMAP-β-CD | 7 | 2 | 1 | Ref. [72] |
a This cyclodextrin is commercially available from chemical reagent suppliers or companies such as Sigma/Aldrich, Across, Cyclolab, CyDex, Wacker–Chemie or Roquette Frères.
b The average number of methyl group depends on the chemical reagent suppliers or companies.
Some of them are commercially available. Others have been synthesized and fully characterized. The reader is urged to consult the cited references for more details. Among these modified CDs, the randomly methylated β-CD (RAME-β-CD) which is highly water soluble, cheap and industrially available in large quantities appeared as the most efficient mass transfer promoter. Indeed, this methylated CD exhibited a better catalytic activity than native CDs in numerous reactions such as the oxidation of olefins [24–28] or aromatic compounds [27,29,30], hydroxylation of benzene or phenol [27,28,31], hydrogenation of aldehydes [32,33], Suzuki cross-coupling reaction [34], cleavage of allylic substrates [35], hydroformylation [36–42] or hydrocarboxylation [43,44] of olefins (Table 3).
Aqueous organometallic catalysis mediated by chemically modified CDs.
| Catalytic reaction | Scheme | References |
| Olefins Oxidation (Wacker reaction) | [23–28] | |
| Oxidation of alkyl aromatic compounds | [27,29] | |
| Hydroxylation of benzene or phenol | [27,28,31] | |
| Hydroformylation of higher olefins | [36–42,51–52,64–66,68–72] | |
| Aldehydes hydrogenation | [32,33] | |
| Hydrocarboxylation of higher olefins | [43,44] | |
| C-C coupling reaction | [34] | |
| Cleavage of alkyl allyl carbonates | [35,47–51,62,63,67,76] | |
| Telomerization of butadiene with glycerol | [45] | |
| Polymerization of substituted acetylene monomers | [46] |
Furthermore, it was also demonstrated that the catalytic system consisting of metal, water-soluble ligand and RAME-β-CD could be quantitatively recovered. Indeed, the phase separation between the organic and aqueous phases was fast with this modified CD and no increase in catalyst leaching into the organic phase was observed. Additionally, gravimetric analyses demonstrated the reusability of the system as no trace of CD was found in the organic phase at the end of the reaction. In the case of butadiene telomerization with glycerol, Behr et al. even reported that RAME-β-CD reduced the catalyst leaching. Thus, the amount of palladium catalyst in the organic phase was found to be 70 ppm without mass transfer additive and only 42 ppm in the presence of RAME-β-CD [45]. Finally, it is worthwhile to point out that a catalytic amount of modified CDs is sufficient to observe a positive effect on the reaction rate and selectivity. Indeed, a substrate or product inhibition is a phenomenon rarely observed with modified CDs. However, a stoichiometric amount of CD with respect to the substrate can also be used. For instance, Deng and Yang reported that hydroxypropyl β-CD/acetylene derivatives inclusion complexes could be successfully polymerized in aqueous solution using a water-soluble Rh-based catalyst, [Rh(cod)2BF4] or [Rh(nbd)(H2O)OTs]. The polymers could be obtained in very high yields and exhibited little difference to their counterparts obtained via copolymerization in organic solvents [46].
Interestingly, chemically modified CDs can also be used successfully to perform substrate-selective reactions that are difficult to achieve with conventional transition metal catalysts [47]. In fact, when the organic phase contains an isomers mixture, the water-soluble catalyst reacts with the isomer that preferentially interacts with the CD cavity, inducing substrate selectivity (Fig. 3).
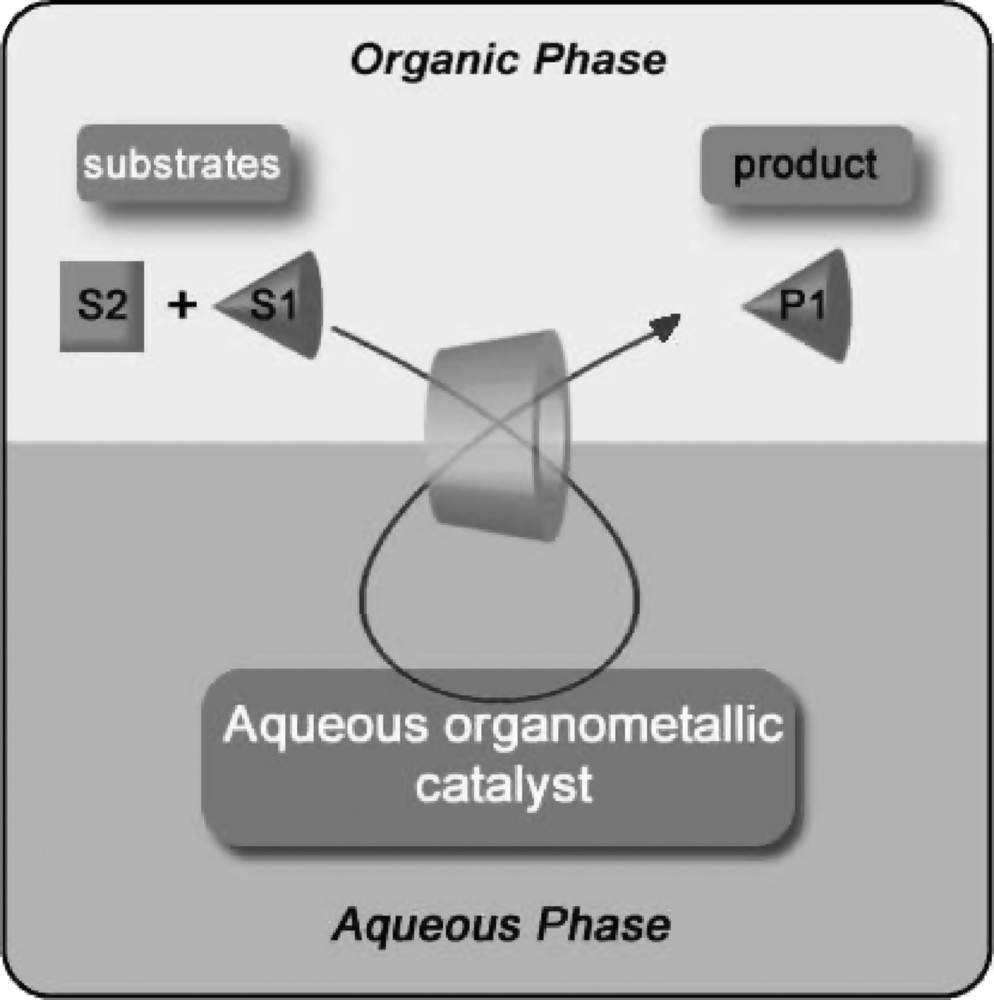
Substrate selective catalysis mediated by cyclodextrins
This type of catalytic system clearly exhibits enzyme-like properties. The possibility to use CDs as discriminating agents was demonstrated by performing competitive experiments. For instance, the deprotection of a 50/50 mixture of ortho- and para-biphenylmethylallyl carbonate was carried out in the presence of acetonitrile or methylated CDs. The ratio of the products (o-biphenylmethanol and p-biphenylmethanol) was used as a measure of substrate selectivity. No substrate selectivity was observed in a control experiment in which acetonitrile was used as a mass transfer promoter. By contrast, the use of RAME-α-CD or RAME-β-CD led to substantial substrate selectivity. Indeed, substrate selectivities of 7.2 and 3.6 were observed in the initial stages of the reaction with RAME-α-CD and RAME-β-CD, respectively [48].
By using different pairs of structural isomers, it was found that the substrate selectivity strongly depended on the water solubility and the isomers, structure. Thus, high substrate selectivity could only be observed with highly water-insoluble isomers that presented pronounced structural differences. The size-fit concept that postulated the highest reactivity for the best size-matched host–guest pair provided a very useful tool for predicting the values of substrate selectivity [48,49]. However, it was noteworthy that the presence of small organic water soluble molecules, such as amine or alcohol derivatives, appeared to be crucial in the discriminating process. Indeed, the presence of such additives greatly affected the substrate selectivity. The existence of ternary CD/substrate/additive complexes appears the most probable explanation of this unexpected result. In fact, the presence of small polar organic compounds near or inside the CD cavity reduce the size of the cavity, which is less disfavored for linear substrates than for bent ones, inducing a higher substrate selectivity. For instance, during the palladium catalyzed decarboxylation of two alkylallylurethane isomers into alkylallylamines, it has been observed that addition of triethylamine to the reaction medium improved the discriminating power of RAME-β-CD by a factor of 7 [50].
The precise role of chemically modified CDs was established by a careful analysis of experimental data (surface tension and association constant measurements) [51,52] and molecular dynamics simulations [53,54]. It is now well established that chemically modified CDs favour the contact between the organometallic catalyst and the substrate at the interface or into the bulk aqueous phase according to the nature of the substrate. In fact, when a mechanism of inverse phase catalysis is proposed for partially water-soluble substrates such as polar styrene derivatives (Fig. 4a), the reaction of highly water-insoluble substrates such as higher α-olefins is believed to occur by an interfacial catalysis mechanism (Fig. 4b).
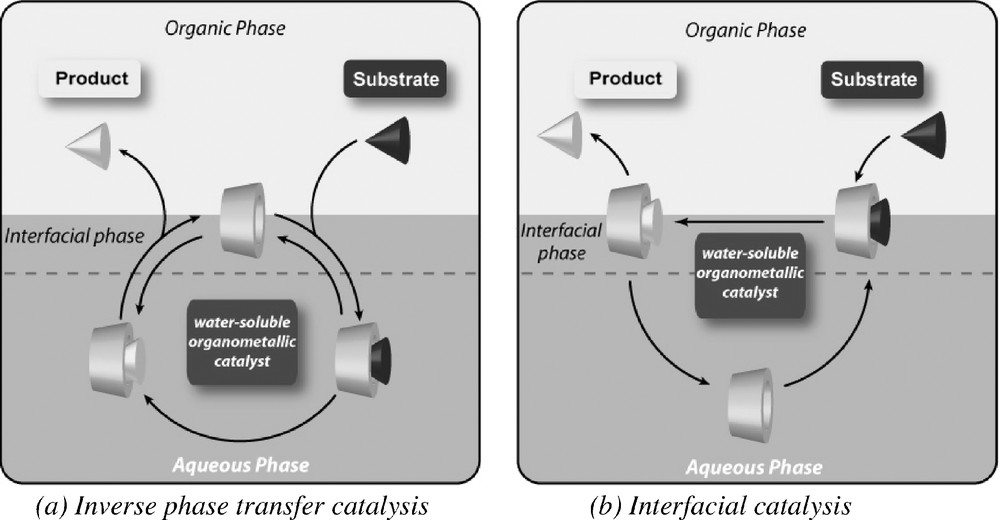
Principle of aqueous biphasic organometallic catalysis mediated by modified CDs.
Unexpectedly, recent spectroscopic studies have demonstrated that β-CD and RAME-β-CD could interact with the monosulfonated triphenylphosphine (TPPMS) and the trisulfonated triphenylphosphine (TPPTS), two water-soluble ligands widely used in aqueous organometallic catalysis [55–60]. For instance, titration and continuous variation plots obtained from 31P- and 1H-NMR data proved the formation of a 1:1 inclusion complex between β-CD and TPPTS. T-ROESY NMR experiments showed that a sulfonated aromatic ring was included into the β-CD hydrophobic cavity from the secondary hydroxyl groups side (Fig. 5).
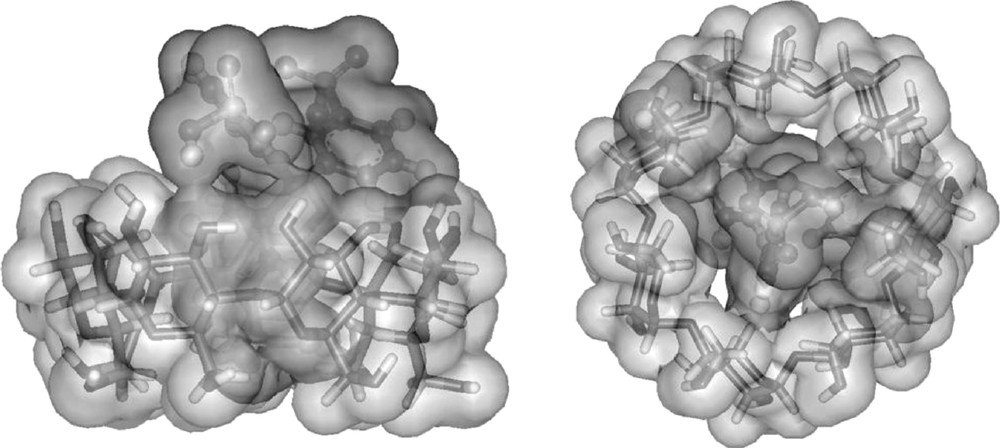
Side view and top view of β-CD/TPPTS inclusion complex.
Formation of inclusion complexes between the β-CD and the TPPTS ligand was strongly suspected to affect the equilibria between the different catalytic species in the rhodium catalyzed hydroformylation reaction [40]. The experimental proof of this phenomenon was established by High Pressure NMR experiments [61]. In the presence of RAME-β-CD under CO/H2 pressure at 80 °C, the 31P NMR spectra of the HRh(CO)(TPPTS)3 rhodium precursor showed chemical shift variations indicative of the formation of phosphine low-coordinated rhodium species (Fig. 6).
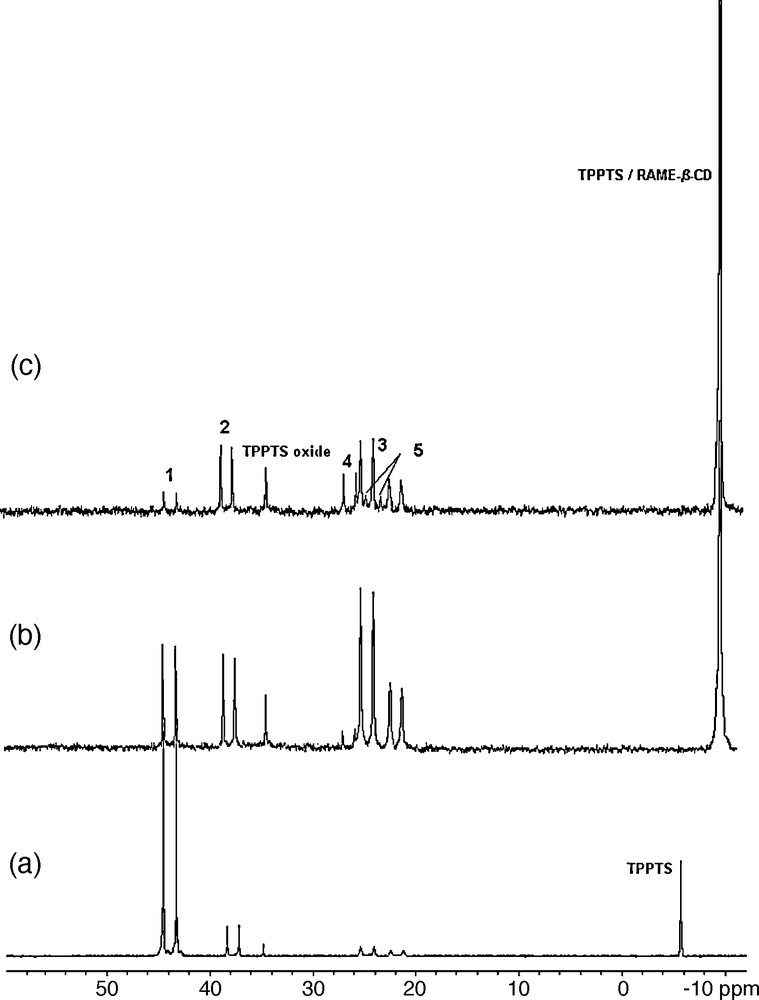
31P{1H} NMR spectra at room temperature in D2O of a) [RhH(CO)(TPPTS)3] (1) under 50 atm (CO/H2) after 1 h at 80 °C; b) 1 + 3 equiv RAME-β-CD under 50 atm (CO/H2) after 1 h at 80 °C; c) 1 + 9 equiv RAME-β-CD under 50 atm (CO/H2) after 1 h at 80 °C. [RhH(CO)2(TPPTS)2] (2); [Rh2(CO)5(TPPTS)3] (3); [Rh2(CO)6(TPPTS)2] (4); [Rh2(CO)7(TPPTS)] (5) (Reproduced with permission from ref [61]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA).
Knowing that HRh(CO)(TPPTS)2 and HRh(CO)2(TPPTS) constitute the active species responsible for the formation of the linear and branched aldehydes respectively, the inclusion of TPPTS in the RAME-β-CD cavity led to a decrease in the concentration of the former and an increase in the concentration of the latter (Fig. 7).
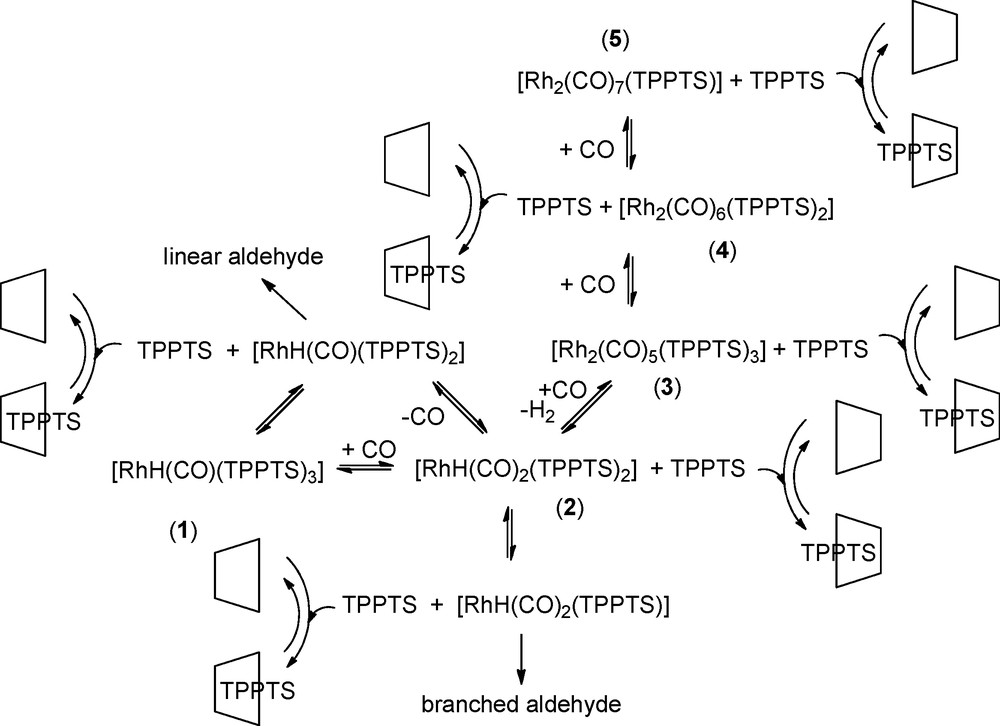
Shift of the equilibra towards phosphine low-coordinated rhodium species in the presence of RAME-β-CD
As a consequence, the linear/branched aldehyde ratio significantly dropped from 2.7 without CD to 1.8 when RAME-β-CD was used as additive. Detrimental effects of the CD/phosphine interaction have also been demonstrated in the palladium-catalyzed cleavage of water-insoluble alkyl allyl carbonates (Tsuji–Trost reaction) [62]. Indeed, when the TPPTS/RAME-β-CD ratio was too high, the CD was poisoned by the sulfonated ligand and the substrate transfer significantly decreased.
The interaction between β-CD and TPPTS could be avoided by using properly modified β-CDs. Thus, introduction of sulfobutyl ether groups on the secondary face of β-CD impeded the formation of inclusion complex by electronic repulsion between the anionic groups of the CD and the sulfonate groups of the water-soluble phosphine [63]. A full methylation of the β-CD secondary face was also an efficient solution to impede inclusion by greatly reducing the accessibility to the cavity [64,65].
The behavior of native β-CD or RAME-β-CD towards other water-soluble phosphines has also been explored [66–70]. Studies have also been carried out with the carboxylated analogue of TPPTS, namely the meta-tricarboxylated triphenylphosphine (TPPTC). As TPPTS, TPPTC also proved to interact with CDs. NMR measurements (Job's plots) were consistent with a mixture of 1:1 and 1:2 β-CD/TPPTC complexes. The existence of 1:2 complexes resulted from the natural tendency of carboxylates to form dimeric species [66]. Interestingly, bulky water-soluble monodentate phosphines such as TPPTS bearing a methyl group in ortho position on each aromatic ring [67] or 1,3,5-triaza-7-phosphaadamantane derivatives [68] have been considered as non-interacting phosphines, as no alteration of the catalytic performance was detected with these phosphines in the presence of RAME-β-CD. The affinity of bidentate phosphines for β-CD or RAME-β-CD was found to be dependent on the nature of the linker between the phosphorous atoms. Thus, 1,ω-bis(diarylphosphino)alkanes such as tetrasulfonated 1,4-bis(diphenylphosphino)butane [69] interacted more strongly with β-CDs than sulfoxantphos [70]. The difference of affinity was attributed to the great flexibility of the alkyl chain compared to the xanthene skeleton. Interestingly, in the case of the sulfoxantphos ligand, the catalytic results showed that, concurrently to the constraint generated by the bulky sulfonated xantphos ligand, the additional steric stress of the CD cavity on the substrate compelled the latter to react preferentially by its terminal carbon, leading to very high regioselectivity towards the linear aldehyde (Fig. 8) [70].
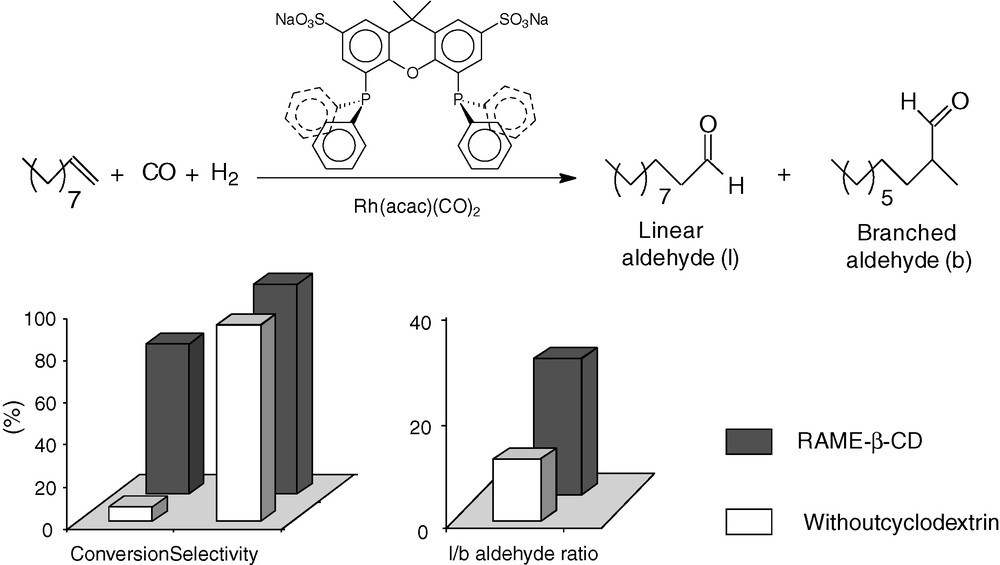
Hydroformylation of 1-decene catalyzed by rhodium/Sulfoxantphos system in the presence of RAME-β-CD.
Contrary to β-CDs, it has been reported that α-CDs cannot interact with TPPTS [61,62,71]. Indeed, these α-CDs have smaller cavities, which prohibit the inclusion processes of the phosphine aromatic cycles inside the CD cavities. Moreover, grafting an alkylammonium chain on a α-CD resulted in new catalytic supramolecular species, where CD behaved both as a transient ligand and as a supramolecular shuttle that can bring the substrate to the water-soluble organometallic catalyst (Fig. 9) [72].
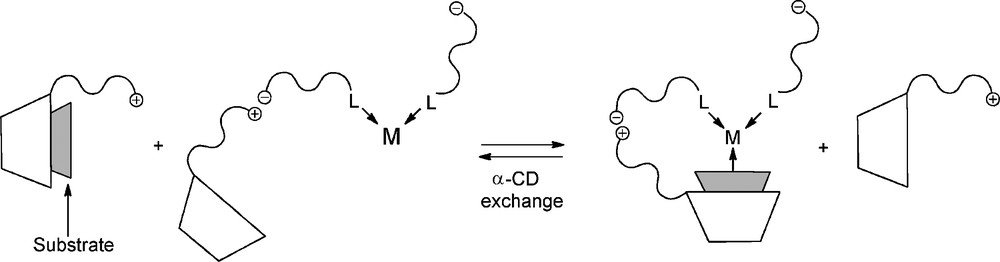
Transient formation of organometallic complexes in the presence of a α-CD modified by an alkylammonium chain. S represents a substrate included into the CD cavity.
2.2 New water-soluble catalysts based on cyclodextrins
The ability of chemically modified CDs to interact with phosphines can be used to generate water-soluble phosphine low-coordinated organometallic complexes that are generally accepted as the catalytically active species in numerous transition-metal-catalysed reactions [73,74]. The strategy relies on the ability of RAME-β-CD to interact strongly with tert-butylphenyl group of phosphine 1 (Fig. 10).
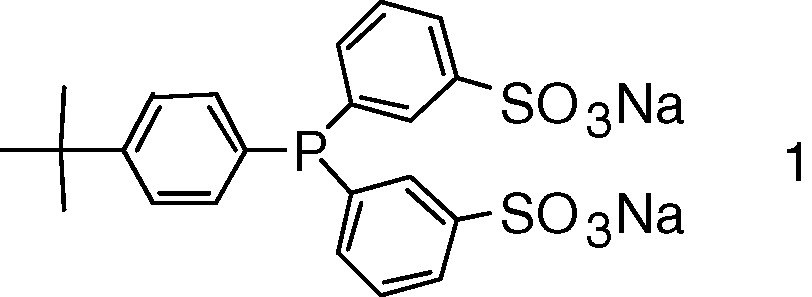
Phosphine used to generate phosphine low-coordinated organometallic complexes.
As a consequence of this strong interaction (KRAME-β-CD/1: 250,000 M–1), RAME-β-CD could induce a shift of the equilibrium towards the phosphine low-coordinated complexes M(1)n–1 by trapping the free phosphine 1 (pathway A in Fig. 11) and/or binding to an organometallic complex M(1)n (pathway B in Fig. 11). In this last case, the local bulkiness around the metallic centre of the obtained supramolecular adducts become too high and a steric decongestion by dissociation of the RAME-β-CD/phosphine inclusion complex from the metal occurs, leading to phosphine low-coordinated complexes as schematically represented in Fig. 11.
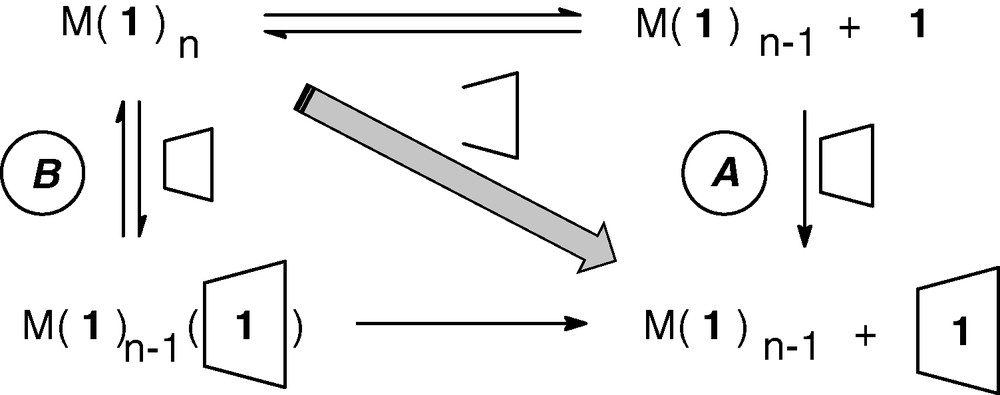
Mechanisms postulated for the generation of phosphine low-coordinated organometallic complexes.
For instance, addition of the native β-CD or RAME-β-CD to an aqueous solution of Cl[Pt(1)3Cl] or [Pd(1)3] complex allowed to generate stable phosphine coordinatively unsaturated [Pt(1)2Cl2] or [Pd(1)2] complexes, respectively. Furthermore, the amounts of [Pd(1)2] complex were found to be dependent on the temperature. Indeed, when a solution containing 12 equiv. of RAME-β-CD with respect to [Pd(1)3] was heated at 85 °C, the signals corresponding to the [Pd(1)2] complex notably increased. Thus, the percentage of phosphine-low coordinated palladium species could reach 40% at 85 °C (vs. 14% at 25 °C) regarding the total amount of palladium.
The ability of phosphine 1 to form very stable inclusion complexes with chemically modified CDs has also been used to design water-soluble supramolecular bidentate ligand for aqueous organometallic catalysis [75]. Thus, the inclusion of phosphine 1 into the cavity of 6I-amino-6I-deoxycyclo-maltoheptaose by the NH2-containing face resulted in the formation of a rigid chelating bidentate ligand, with the nitrogen and the phosphorus atoms on the same side of the supramolecular edifice. This self-assembled bidentate ligand could coordinate platinum complexes to efficiently catalyze the hydrogenation of a water-soluble allylic alcohol (Fig. 12).
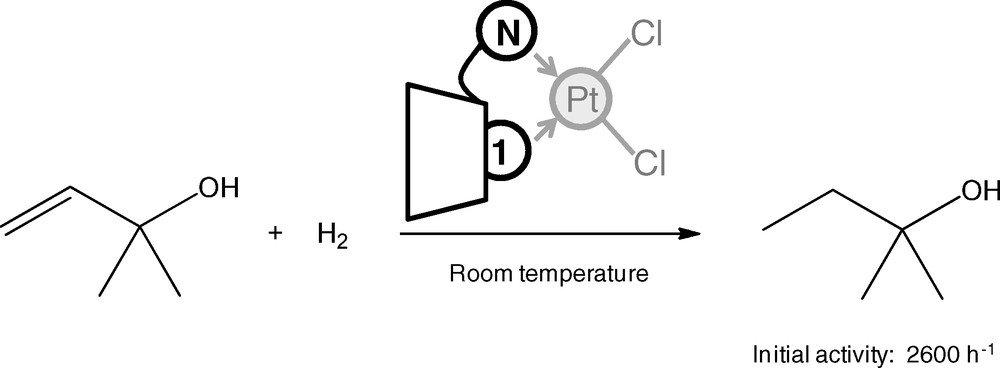
Platinum-catalyzed hydrogenation of 2-methyl-3-buten-2-ol in water.
In contrast to phosphine 1, phosphine 2 allowed the formation of stable second-sphere coordination adducts (Fig. 13). In this case, RAME-β-CD could bind to the metal-coordinated phosphine without inducing dissociation of the phosphine because CD remained far from the coordinated phosphorus atom. Interestingly, these second sphere coordination adducts derived from 2 were active in a Trost–Tsuji reaction [76].
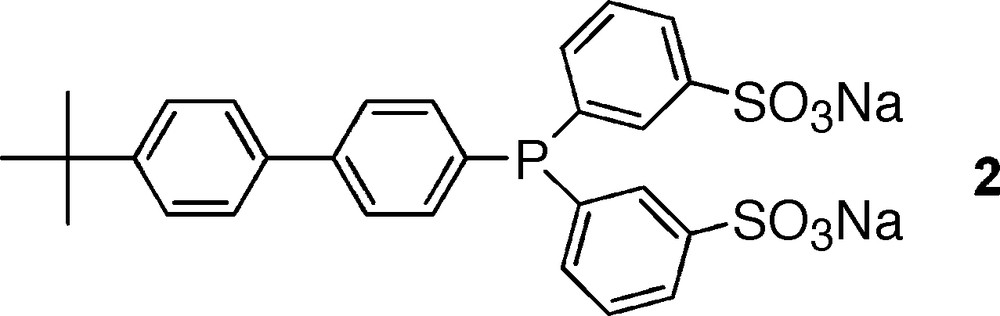
Phosphine used to generate stable second-sphere coordination adducts.
Interestingly, more sophisticated approaches involving the covalent attachment of catalyst to CD through a spacer have also been performed to combine molecular recognition, phase transfer property and metal catalysis. In this case, modified CDs may be regarded as enzyme mimics. Indeed, these catalysts generally show substrate specificity for molecules that can bind into the CD cavity and are highly selective, including stereospecificity. Breslow is one of the pioneers in this field [77]. Indeed, he reported as far back as 1970 that the hydrolysis of p-nitrophenylacetate was four times faster in the presence of the compound displayed in Fig. 14 than in the presence of nickel complexes without CD unit (Fig. 14) [78]. Since this seminal work, many research groups have attached catalytically active centers to β-CD and have used them to perform oxidation–reduction reactions or hydrolysis of esters and phosphates [79–83]. Surprisingly, the possibility to use modified CDs as ligands in reactions catalyzed by traditional transition metals such as Rh, Pd or Ru has hardly been investigated [84–93]. This is all the more surprising since these transition metals are involved in important aqueous biphasic reactions such as hydrogenation, hydroformylation or Wacker oxidation. The first significant work in this field was reported by Reetz et al. in 1997. The author achieved the synthesis of various β-CD-based diphosphines and successfully used them as ligands in hydrogenation and hydroformylation reactions [84–87] (Fig. 15). Unfortunately, the author reported that the catalytic systems cannot be recovered quantitatively due to partial transfer of the catalyst into the organic phase [84,86].
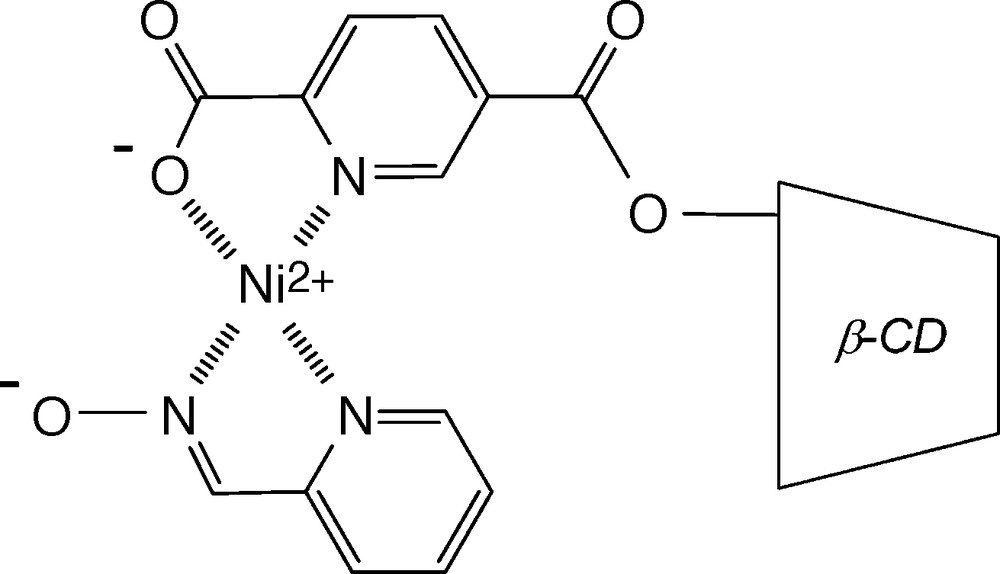
First CD modified by a metal-binding group [78].
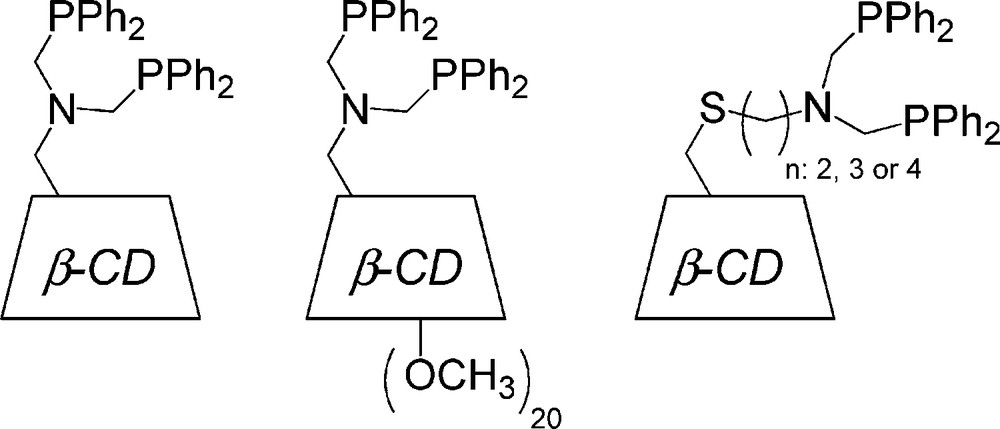
CD-based diphosphines for aqueous organometallic catalysis.
Water-soluble palladium complexes based on nitrile-modified β-CD were also reported to be very active and can serve as selective catalysts for the Wacker oxidation of 1-alkenes in a two-phase system [90]. The high catalytic activities of these catalysts were explained by a simultaneous interaction of alkene with both the CD cavity and the palladium ion attached to the CD [91]. The catalyst efficiency was also affected by the position of the palladium ion with respect to the cavity of the macrocyclic receptor. For example, the activity of the palladium complex in which the nitrile group was attached to the CD secondary face significantly differed from the activity of the complex in which the palladium ion was bound to the complexing group attached to CD primary face [26–28]. Woggon et al. reported a similar phenomenon during the enantioselective hydrogenation of ketones catalyzed by ruthenium complexes linked to β-CD [92,93]. Thus, ruthenium-η-arene complexes attached to the secondary face of β-CD catalyzed more efficiently the enantioselective reduction of ketones in the presence of sodium formate than ruthenium-η-arene complexes attached to the primary face. Enantioselectivities from 70% to 98% were observed for a series of aliphatic and aromatic ketones, which represent the highest ee values for these challenging substrates reported to date. Note that this unprecedentedly high enantioselectivity also resulted from a catalyst in which β-CD was the only chiral auxiliary.
2.3 Water-soluble catalytically active metallic nanoparticles stabilized by cyclodextrins
The first example of metallic nanoparticles stabilized by native CDs was reported by Komiyana et al. in 1983 [94]. The authors reported that refluxing an aqueous solution of rhodium (III) and α-CD or β-CD, followed by further refluxing in the presence of ethanol gave a colloidal dispersion of rhodium particles of 28 Å in diameter. Interestingly, this colloidal dispersion effectively catalyzed the hydrogenation of various olefins such as acrylonitrile, 3-buten-2-one and 3,4-dimethyl-3-penten-2-one at 30 °C under atmospheric pressure (Table 4).
Catalytically active metallic nanoparticles stabilized by cyclodextrins.
| Catalytic reaction | Scheme | References |
| Hydrogenation of α,β-unsaturated ketones | [94,102] | |
| Hydrogenation of α,β-unsaturated nitrile | [94] | |
| Reduction of bicarbonate | [95,96] | |
| Hydrogenation of α,β-unsaturated acids | [97] | |
| Reduction of 4-nitrophenol | [98,99] | |
| Hydrogenation of allylic alcohol or amine | [99,100] | |
| Hydrogenation of olefins | [101,106] | |
| Hydrogenation of imines | [102] | |
| Suzuki reaction | [103,108] | |
| Sonogashira reaction | [104] | |
| Hydrogenation of styrene derivatives | [105–107] | |
| Heck reaction | [108] |
The ability of native β-CD to stabilize metallic nanoparticles was also observed by Willner et al. in 1987 [95]. The authors showed that heterogeneous Pd colloids stabilized by β-CD catalyzed the photoreduction of bicarbonate to formate. Furthermore, the Pd/β-CD colloids acted as artificial enzymes. For example, the catalytic system exhibited high specificity and effectiveness towards CO2/HCO3- reduction and was competitively inhibited towards the substrate activation and reduction [96]. The influence of β-CD on the hydrogenation of α,β-unsatured carboxylic acids in water catalyzed by a RhCl3/TPPTS colloidal system has been investigated by H. Arzoumanian et al. [97]. A positive effect of β-CD was obtained with α,β-unsaturated carboxylic acids which could form inclusion complexes via the olefinic double bond. Experiments conducted with NaHSO3 instead of TPPTS suggested that the presence of sulfonate ions is required to obtain high selectivities. Unfortunately, the real nature of the RhCl3/TPPTS/β-CD colloidal system was not determined in this work.
Recently, Qi et al. reported the controlled synthesis of Au nanoparticles with adjustable sizes and narrow size distributions by directly reducing hydrochloroauric acid with α-CD in an alkaline aqueous solution. The obtained α-CD capped Au nanoparticles exhibited good catalytic activities for the reduction of 4-nitrophenol in the presence of NaBH4 [98]. The reaction rate constant was inversely proportional to the nanoparticles diameter, suggesting that the catalysis takes place on the nanoparticles surface.
A multifunctional FePt/γ-CD nanoparticle exhibiting catalytic activity, magnetic properties and water compatibility has recently been developed by H. Yamashita et al. [99]. Magnetic FePt nanoparticles stabilized by oleic acid and oleylamine were dispersed in water by addition of γ-CD. The FePt nanoparticles capped with γ-CD had a mean diameter of 2.5 nm and exhibited superparamagnetic behavior at 300 K with zero remanence and coercivity. The FePt/γ-CD nanoparticles were successfully employed as an efficient nanocatalyst for the hydrogenation of allyl alcohol and the reduction of p-nitrophenol. Surprisingly, the FePt/γ-CD nanoparticles showed a higher catalytic activity in water than in organic solvents.
The synthesis of metallic nanoparticles stabilized by chemically modified CDs has also been investigated. Kaifer et al. reported that per-6-thio-β-CD effectively stabilized Pd or Pt nanoparticles without passivating their surface [100]. FTIR spectra indicated that per-6-thio-β-CD was chemisorbed on the surface of the metal nanoparticles as the weak S–H stretching peak, which was clearly visible in the spectrum of free CD, disappeared in the spectrum of the nanoparticles. Interestingly, it was found that these modified CDs were effective catalysts for the hydrogenation of CC or CN double bonds [101,102] and for CC coupling reactions such as the Suzuki [103] and Sonogashira [104] reactions. In these works, it was also demonstrated that the catalytic activity of the nanoparticles could be modulated by host-guest interactions between the surface immobilized CDs and properly chosen hosts. Finally, the authors mentioned that the solubility mismatch between the highly hydrophilic per-6-thio-β-CD capped metallic nanoparticles and the hydrophobic reactants tended to limit the applications of this type of CD capped metallic nanoparticles.
The low affinity of CD capped nanoparticles for hydrophobic substrates could be solved by using randomly methylated CDs (RAMECDs) [105]. Contrary to native or perthiolated CDs, methylated CDs could not only stabilize metallic nanoparticles, but also facilitate the mass transfer between the organic phase and the catalyst-containing phase (Fig. 16).
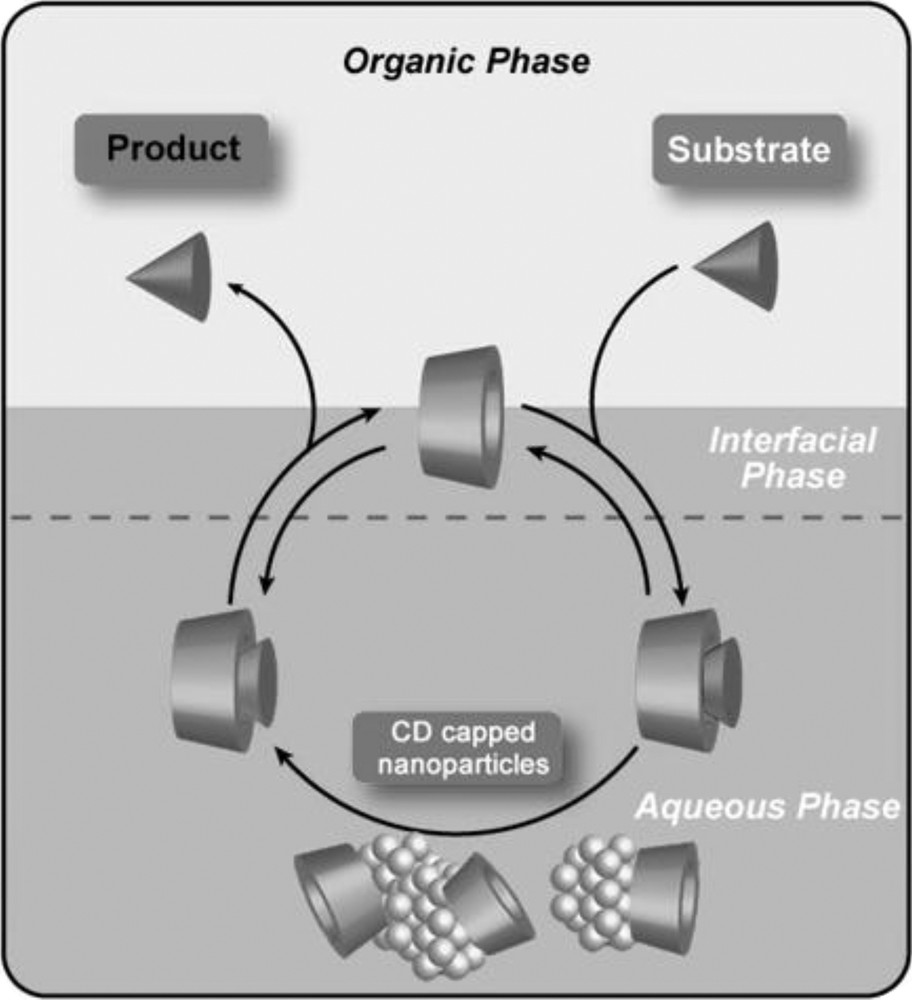
Methylated CDs as protective agents and mass transfer additives in metallic nanoparticles catalyzed reactions.
Indeed, RAMECDs are weakly adsorbed on a metal surface and their mass transfer ability remains high. The RAMECDs/Ru molar ratio was found to be a key parameter to obtain stable and catalytically active colloidal suspension. An agglomeration of metal nanoparticles in aqueous suspension was noticed after the catalytic reaction for ratio lower than 5. For a ratio greater than 10, a notable decrease in the catalytic activity was observed, probably due to a steric hindrance of the nanoparticles surface, avoiding the substrate to come close to the metal. Consequently, the adequate ratios for an efficient steric stabilization to maintain catalytic species within the aqueous phase and good activities were found to be in the range between 5 and 10. In this range of ratios, the RAMECDs-capped Ru(0) nanoparticles proved to be very active in hydrogenation of olefins such as α- or β-pinene, and more particularly of aromatic compounds under biphasic conditions at room temperature and atmospheric hydrogen pressure [106]. Moreover, interesting chemoselectivity has been observed in the hydrogenation of monofunctionalized arene derivatives thanks to a relevant choice of cavity and methylation degree of the CD. Interestingly, these nanocatalysts could be easily reused without significant loss of catalytic activity.
Hydrogenation of arene derivatives has also been successfully performed in water by using Ru(0) nanoparticles stabilized by 1:1 inclusion complexes formed between RAME-β-CD and an ammonium salt bearing a long alkyl chain [107]. These nanoparticles stabilized by CD inclusion complexes appeared more active than the nanoparticles stabilized by classical surfactants. The origin of this better efficiency was attributed to a dynamic organization of the protective agents around the nanoparticles. Indeed, it was assumed that the polar head of the surfactant was directed towards the metallic surface and that RAME-β-CD (which interacted with the alkyl chain of the surfactant) could be partially released into the bulk aqueous phase as schematically shown in Fig. 17.
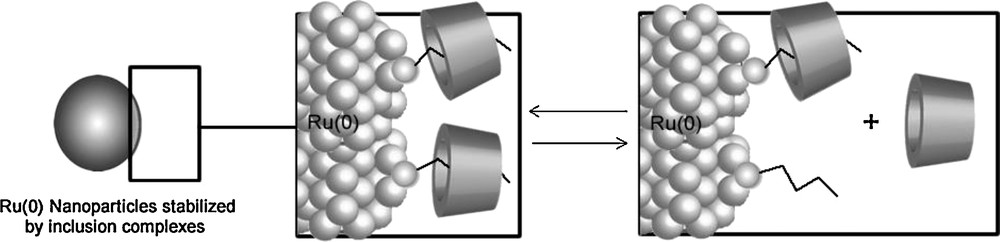
Dynamic organisation of the stabilizers around the Ru(0) nanoparticles.
Moreover, it was also postulated that RAME-β-CD could exert some control on the surfactant adsorption process, acting as a suitable spacer between the alkyl chains of the surfactants, reducing their intermolecular interactions and consequently allowing a better diffusion of substrates towards the metallic surface.
A new approach for the preparation of metallic nanoparticles in water has been recently reported by L. Malta et al. [108]. In this approach, 2-hydroxypropyl-α-CD (HP-α-CD) acted as a reducing and capping agent during the synthesis of palladium nanoparticles. From FT-IR, 1H NMR and MALDI-TOF experiments, it was demonstrated that the reduction of Pd(II) occurred through the oxidation of alcohol groups of the CD substituent and that HP-α-CD is physically adsorbed on the nanoparticles surface limiting the mutual coalescence of nanoclusters. Importantly, these nanoparticles promoted Heck, Suzuki and Sonogashira couplings in aqueous medium in good to excellent yields and selectivity. Furthermore, the nanocatalyst showed significant stability and remained active for four subsequent runs.
2.4 Reactions catalyzed by supported metals or metallic powder in aqueous medium
Fornasier et al. first reported the use of CD and palladium on charcoal in 1987 [109]. They examined the hydrogenation of acetyl and benzoyl-substituted pyridines in the presence of native β-CD in aqueous solution. Results indicated that native β-CD had a rather modest effect on the reaction (Table 5). In fact, the experimental data suggested that the effect exerted by native β-CD in this reaction could be merely a type of solvent effect. More positive results were reported by Shimizu et al. in 1990 [110]. Indeed, these authors showed that native CDs functioned as inverse phase transfer catalysts for the reduction of aryl bromides with sodium formate in the presence of a charcoal-supported palladium catalyst. The CD efficiency was found to be dependent on the CD size and the best results were obtained with β-CD. It was also concluded that the inclusion complex formed between the CD and substrate did not interact with the palladium catalyst, and that most of the reaction was probably carried out by the free species.
Reactions catalyzed by supported metals in the presence of cyclodextrins.
| Catalytic reaction | Scheme | References |
| Hydrogenation of acyl substituted pyridines | [109] | |
| Reduction of bromoasinoles | [110] | |
| Hydrogenation of aromatic ketones | [111] | |
| Hydrodechloration of carbon tetrachloride | [112] | |
| Suzuki reaction | [113] | |
| Heck reaction | [114,116,117] |
Hydrogenation of β-CD/aromatic ketone inclusion complexes catalyzed by various supported metals (Ir/Al2O3, Rh/Al2O3, Pt/C) was investigated by M. Graziani et al. in 1994 [111]. The results showed that the reaction rate and selectivity could be affected by inclusion of the aromatic ketone into β-CD cavity. However, the results were found to be very dependent on the nature of the aromatic ketone, the CD and the heterogeneous catalyst. For instance, when an inclusion complex between the dimethyl-β-CD (DIME-β-CD) and acetophenone was used as substrate, the reaction rate significantly decreased and no hydrogenation of the aromatic ring of acetophenone was observed with Ir/Al2O3 catalyst. With the Pt/C catalyst, inclusion of acetophenone into DIME-β-CD cavity did not affect the reaction rate, but the ring reduction was always observed. Surprisingly, with the Rh/Al2O3 catalyst, the selectivity was lower than that observed without CD as reduction of the carbonyl group was also observed (Table 6).
| Catalyst | Substrate | Conv.(%) | Products | Yields (%) |
| Ir/Al2O3 | PhCOCH3 | 100 | PhCH(OH)CH3 | 68 |
| C6H11CH(OH)CH3 | 32 | |||
| PhCOCH3/DIME-β-CDb | 42 | PhCH(OH)CH3 | 42 | |
| Rh/Al2O3 | PhCOCH3 | 100 | C6H11CH(OH)CH3 | 100 |
| PhCOCH3/DIME-β-CDb | 100 | C6H11CH(OH)CH3 | 72 | |
| C6H11COCH3 | 20 | |||
| C6H11CH2CH3 | 8 | |||
| Pt/C | PhCOCH3 | 100 | PhCH(OH)CH3 | 59 |
| C6H11CH(OH)CH3 | 41 | |||
| PhCOCH3/DIME-β-CDb | 100 | PhCH(OH)CH3 | 85 | |
| C6H11CH(OH)CH3 | 15 |
a Reaction conditions: reaction time 24 h, Solvent: H2O, Substrate/catalyst: 50, P(H2): 1 atm, temperature: 25 °C.
b Inclusion complex prepared by adding 1 equivalent of acetophenone to an aqueous solution of DIME-β-CD.
Effect of catalytic amount of chemically modified CDs in Pd/C catalyzed reactions such as the hydrodechlorination of CCl4 [112], the Suzuki–Miyaura reaction [113] and the Heck reaction [114] has recently been reported. The modified CDs, particularly RAMECDs, appeared much more efficient than native CDs to increase reaction rates and selectivities. The high efficiency of RAMECDs was attributed to a combination of several effects: a well-known mass transfer effect but also a dispersing and activation effect (Fig. 18).
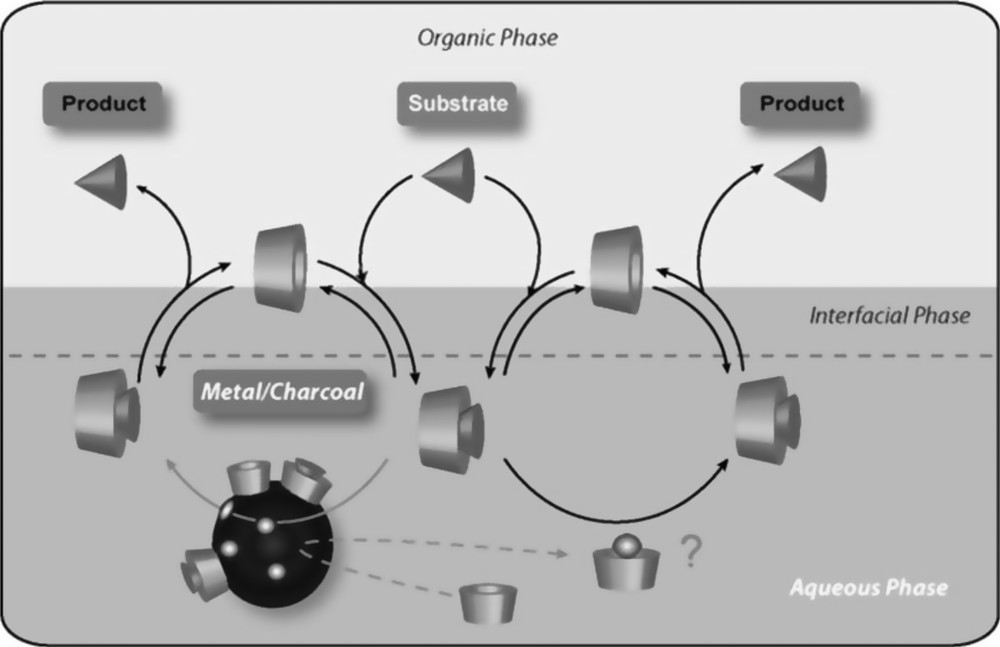
Multiple role of methylated CDs in aqueous biphasic Pd/C catalyzed reactions. The question mark indicates that the nature of the solubilized palladium species has not yet been elucidated. In fact, it was assumed that RAME-β-CD could stabilize Pd clusters leached from the support and that these Pd clusters could also be catalytically active.
The beneficial effect of methylated CDs on the dispersion of the Pd/C catalyst in water was easily demonstrated by observing the distribution of Pd/C in a two-phase system (Fig. 19). Indeed, a much more homogeneous distribution of the Pd/C catalyst in the aqueous phase was obtained with RAME-β-CD, compared to native β-CD. The dispersion of Pd/C in water in the presence of CD was attributed to the CD adsorption on the Pd/C catalyst surface. Indeed, CD adsorption increased the hydrophilic character of the support, making its dispersion in the aqueous phase easier.

Pd/C distribution in a mixture of water/heptane (10 mg/10 mL/10 mL): (a) without CD; (b) with 57 mg of native β-CD (0.05 mmol); (c) with 66 mg of RAME-β-CD (0.05 mmol).
The activation effect of methylated CDs was confirmed by elemental analyses of aqueous and organic phases [115]. Elemental analyses of the organic phases showed that RAMECDs did not notably increase palladium leaching in solution. Indeed, the amount of palladium in the organic phase in the presence of RAMECDs was identical to that obtained without CD, confirming the reusability of the catalytic system. However, it was found that the amount of Pd in the aqueous phase was higher in the presence of RAMECDs. The nature and the role of this solubilized palladium species have not yet been elucidated. It must be pointed out that a similar phenomenon has also been described by Antunes et al. in Heck reactions catalyzed by Pd/CaCO3 and hydroxylpropylated CD. It was assumed that hydroxylpropylated CD increased the Pd/CaCO3 dispersion and stabilized Pd clusters leached from the support. In fact, the heterogeneous Pd/CaCO3 solid could act as a reservoir of catalytically active Pd species [116,117].
The selective carboxylation of aromatic acids with carbon tetrachloride in the presence of metallic copper powder and β-CD in aqueous alkali medium has been investigated by H. Hirai et al. [118–120]. Dicarboxylic derivatives were obtained in good yield (from 67 to 74%) and very good to excellent selectivity (from 84 to 100%). This was a consequence of two concomitant inclusion processes. First, hydrophobic carbon tetrachloride was solubilized in water and could then react with copper powder dispersed in water to give copper(I) chloride and the trichoromethyl radical species which was included into β-CD cavity. Second, radical species selectively attacked the aromatic carboxylates included into another CD cavity to give p-trichloromethylcarboxylates which were rapidly hydrolyzed, resulting in p-dicarboxylate derivatives (Fig. 20).
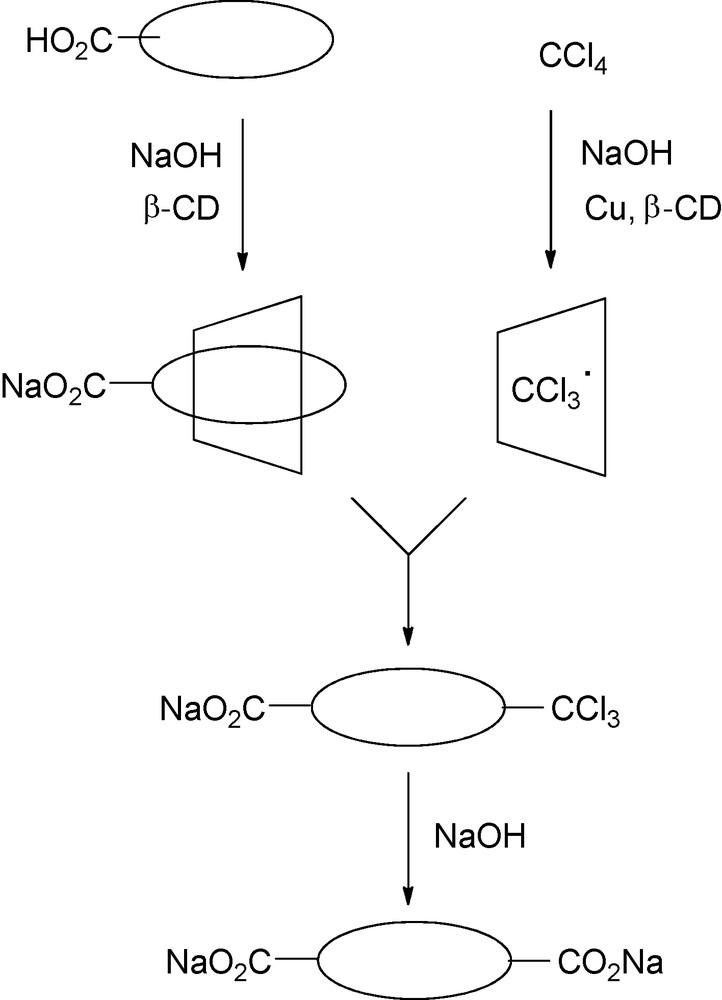
Proposed mechanism for carboxylation of aromatic acids in the presence of copper powder and β-CD.
The dechlorination of trichloroethylene by iron powder in the presence of various substituted β-CDs has been reported by G. vanLoon et al. [121]. It was found that, in the presence of 100-mesh (0.145 mm particle size) zero-valent electrolytic iron powder, reaction rate decreased as the CD concentration increased. A satisfactory interpretation of the kinetic data could be given by the formation of an inclusion complex between β-CDs and trichloroethylene and also by a new mechanism involving CD binding to the iron-surface. More precisely, the overall rate was governed by the extent of CD binding with the iron surface (which depended on the CD unsubstituted –OH groups), the binding constant between CD and the substrate, and the degradation of the CD-included trichloroethylene, the CD being associated with the ion surface. These observations were made both in batch (static) and column (dynamic) systems.
2.5 What else with chemically modified CDs?
In addition to the CD multi-functional character described above (mass transfer additive, stabilizing and/or dispersing agent, building block for the synthesis of new water soluble ligands), CDs could also be used in the following applications. The first application concerns molecular imprinting, defined as design of macro-receptors by means of template synthesis. Using CDs as building blocks, a pre-organisation of the macro-receptors took place by partial inclusion of an organic template in the CD cavities. The CD network was then made rigid by reticulation with an organic linker (epichlorhydrine for example) to give the template-included macro-receptors. The template molecule is then removed to give macro-receptors able to recognize substrates having structures similar to that of the template [90,122,123] (Fig. 21).
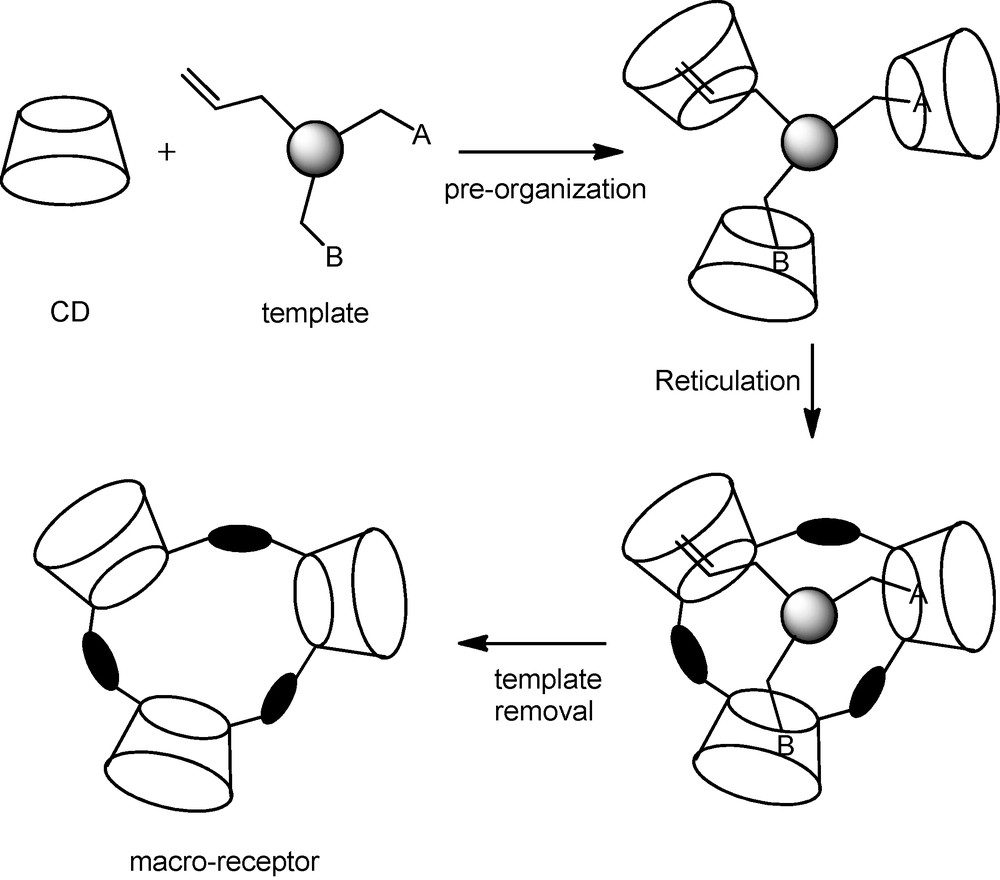
Molecular imprinting process involving cyclodextrins.
The proof of concept was given by activity enhancements in several catalytic reactions. Actually, using CD-based macro-receptors with 1-dodecene and 1-hexadecene as template molecules, the reaction rates obtained in oxidation of 1-dodecene (PdSO4/CuCl2/heteropolyacid catalyst) were 4- and 5.5-fold higher than those obtained without molecular imprinting, respectively [90]. Similarly, using p-tert-butylstyrene as template molecule, 1.7- and 2.4-fold reaction rate increases were observed for styrene and p-methylstyrene oxidation, respectively. This approach also appears successful in a Pd-catalyzed oxidative coupling of styrene and benzene leading to stilbene with a 2-fold reaction rate increase when the CD-receptor was built by molecular imprinting using stilbene as template [122]. In a biphasic chloroform/water system, using 1,1’-binaphtol as template and epichlorhydrine as reticulating agent, FeCl3-catalyzed dimerization of 2-naphtol into 1,1’-binaphtol also showed a 2-fold increase in the reaction rate [123]. It has been shown that an increase in the reaction rates were obtained when an analogue of the reaction transition state is taken as a template. The best results were obtained with rigid template molecules. In fact, thanks to their template-induced geometry, the CD-based macro-receptors fitted the substrate structure more accurately, leading to a better recognition process and a subsequent improvement of the catalytic performances.
Another application of CDs concerns the solubilization of hydrophobic organometallic catalysts in aqueous media. Two examples are given in the literature. The first one is related to Mo-catalyzed epoxidation of cyclooctene [124]. Using CpMo(CO)3CH2CONH2 as catalyst and n-hexane and water as solvents, it was demonstrated that 2,3,6-trimethyl-β-CD (TRIMEB) was more efficient than native β-CD in stabilizing the tricarbonyl molybdenum complex in the presence of water, allowing an easier access to the active catalyst by oxidative decarbonylation. Indeed, when the activity dropped after recycling the β-CD/Mo-catalyst system, the catalytic performances remained constant with the TRIMEB/ Mo-catalyst system. The second example highlighted the influence of native CDs onto the activity and stereoselectivity of Ru-catalyzed ring-closing and cross metathesis of non-polar substrates in pure aqueous medium. Comparing α-, β- and γ-CDs, β-CD appeared the best additive in terms on yield and E/Z ratio [125].
Finally, the last application demonstrated that the simultaneous use of α-CD and ionic liquid surfactant allowed the hydroformylation of higher alkenes in a micellar system without phase separation problems [126]. In fact, α-CD helped breaking emulsion at room temperature by including the hydrophobic moiety of the ionic liquid surfactant in its cavity, resulting in a faster decantation process. In contrast, the formation of emulsion at reaction temperature (80 °C) was always possible due to weak interactions between the ionic liquid surfactant and α-CD.
3 Conclusions
This review highlights that chemically modified CDs are very attractive compounds to develop catalytic processes in aqueous media. Indeed, a wide range of catalytic reactions including hydrogenation, hydroformylation, oxidation, reduction and carbon–carbon coupling reactions have been successfully achieved in this environmentally friendly medium thanks to these cheap and bulk commercially-available compounds. The possibility of performing substrate-selective reactions as enzymes offers a further dimension to CD-based catalysis. In the future, chemically modified CDs may also give the opportunity to promote reactions in other green solvents such as ionic liquids [26,127] or supercritical carbon dioxide [128–131].
Acknowledgements
This work was supported by the ministère de l’Enseignement Supérieur et de la Recherche, the Centre national de la recherche scientifique (CNRS), the institut de recherche en environnement industriel (IRENI) and the Agence nationale de la recherche (ANR). Roquette Frères (Lestrem, France) is gratefully acknowledged for generous gifts of cyclodextrins. The authors are grateful to G. Crowyn for his technical contribution.