

1 Introduction
Macrocycles are promising building blocks for the creation of nanosized molecular devices and nanomaterials on the “bottom-up” principle. Their unique properties are based on the relative rigidity of their spatial characteristics in comparison with acyclic compounds of similar size and on the presence of an empty intramolecular space, the so-called intramolecular cavity. The fragments of organic and organoelement guest molecules or even whole molecules are able to penetrate into these cavities and to be binded inside. The macrocyclic structure provides the effective binding of the guests (so-called macrocyclic effect) due to multiple host–guest interactions. It should be mentioned that these binding and orientational interactions may vary from very weak (for example, hydrophobic, Van-der-Waals, π–π, H–π interactions) to relatively strong ones (hydrogen and coordination bonds) [1]. The penetration of the guest molecules inside the macrocyclic cavities may result in their isolation from the environment (molecular containers and reactors), may change their characteristics (molecular sensors, solubilisers and molecular transport) or may even cause the appearance of the unique properties (molecular devices and machines) [1].
The availability of the macrocyclic compounds is one of the key points for their use in the field of supramolecular chemistry and nanomaterials. If some types of macrocycles with a unique shape, distinct architecture, and set of functional groups become easily available from natural or synthetic sources, they stimulate the chemists to the wide and effective search for novel molecular materials and devices.
At present three main strategies of the macrocyclic synthesis may be separated:
- 1. the macrocyclization under high-dilution conditions;
- 2. the template synthesis of macrocycles;
- 3. the covalent self-assembly of macrocycles.
The first two approaches have been used for a long time and have a number of well-known advantages and disadvantages. The high-dilution macrocyclization, as a rule, requires fast irreversible reactions, and the participation of more than two molecules is undesirable. Another disadvantage is a large amount of a solvent and/or the use of special dosing devices for the slow simultaneous addition of both reagents. It increases the reaction time and complicates the technique. In addition, the regio- and stereoselectivity of the irreversible macrocyclization reactions are usually low [1].
The template macrocyclization requires that one of the reagents contains the donor center (or centers) which is able to form the coordination compound of the desirable structure. Another problem is the final demetallation step for the release of the obtained macrocyclic ligand because the metal complexes are usually very stable due to the macrocyclic effect [1].
The last approach, namely the covalent self-assembly of macrocycles, has been formed recently. The self-assembly conception came from supramolecular chemistry. J.-M. Lehn used the biochemical conceptions of templating, self-assembly and self-organization for the explanation of the drawing forces of the supramolecular structures formation [1e]. Later in the 1990s, the self-assembly phenomenon was successfully used for the directed design of the macrocyclic systems formed by the coordination bonds [2]. It has been shown that the main necessary conditions for the self-assembly of metallamacrocycles are the reversibility of all steps of the reaction, the mutual spatial correspondence of the building block sizes and their geometry for the formation of the definite type of the macrocycle with higher thermodynamic stability or lower solubility in comparison with other possible products for the desirable equilibrium shift. The distinctive feature of the self-assembly processes is their ability for the self-correction, then the “incorrect” intermediate or product is able to decompose into initial compounds due to the reversibility of the reaction. These compounds react further to give the more thermodynamically stable “correct” combination. The self-correction is the reason of the high selectivity of the self-assembly processes [2c]. So similar principles are expected to be applicable to macrocyclization processes via the reversible formation of covalent bonds.
Thus a series of the self-assembly processes [2b] of organic and organoelement macrocyles synthesis via covalent bonds formation was found during the last two decades. Most of them are based on several types of reversible reactions:
- 1. esterification of organic acids (and reesterification of esters) [3] or organoelement compounds (boronic [4], iodine [5] and organophosphorus acids [6]);
- 2. the amidation of organic [7] and organophosphorus [8] acid derivatives;
- 3. the olefine metathesis in the presence of catalysts [9];
- 4. the formation of imines (Shiff bases) and hydrazones [10] including organophosphorus ones [11];
- 5. the formation of Mannich bases [12];
- 6. the formation of organic disulfides or diselenides under reversible oxidation conditions [13].
In addition, the formation of calixarenes and, especially, of calixresorcines [14] may be probably considered as covalent self-assembly processes. There is also a number of reactions whose high yields and selectivity have been explained by the templating effects of some particles which were weakly bound by the substrates and (or) final macrocycles. It is very probable that the templating plays only an auxiliary role in such cases. It may accelerate the covalent self-assembly process or shift its equilibrium position to the macrocyclic product by the increase of the thermodynamic preference of the last one [10f,13c] or by its removal from the reaction sphere.
The indication of self-assembly phenomenon is the high yield of macrocycles and the regio- and stereoselectivity of their formation in the course of the reversible reaction (often at elevated temperature) between two or more initial molecules (at least one of them must possess spatially divided functional groups) under conditions of relatively high concentration and without any templating reagents.
This synthetic strategy has been applied for the synthesis of P,N-containing macrocycles by our research group in A.E. Arbuzov Institute of Organic and Physical Chemistry (Kazan) for the last 10 years and appeared to be very effective [15].
1.1 P,N-Containing cyclophanes with large hydrophobic molecular cavities
The Mannich-type condensation of primary and secondary phosphines as proton-donating reagents (Scheme 1) has been chosen as the basic reaction for the use of covalent self-assembly approach to the synthesis of P,N-containing macrocycles. These condensations are an effective tool for the targeted synthesis of aminomethylphosphine ligands including chiral, water-soluble and polymer or dendrimer-supported ones. At the same time, the direct experiments, interconversions of different types of aminomethylphosphines or stereoisomers of cyclic aminomethylphosphines showed the reversibility of these reactions, the equilibrium being especially fast in the presence of donors of protons [16].

The reversible addition of phosphines to imines.
The condensation of bis(hydroxymethyl)phenylphosphine and bis(4-aminophenyl)methane as the bifunctional reagent with spatially divided amine groups in hot DMF (110 °C) at the phosphine concentration of 0.1–0.2 M indeed led to a cage P,N-containing macrocycle 1 in a very good yield (Scheme 2) [17].
Synthesis of P,N-containing cyclophanes on the basis of bis(4-aminophenyl)methane and its heteroanalogues.
The formation of 1 was the first example of the covalent self-assembly of the macrocycle in the reactions of this type. But if some types of macrocycles can form spontaneously from the relatively large number of smaller molecules, their analogues seem to be also available on the basis of the starting reagents with the changed periphery, but with the same (or similar) main framework. The suggested reaction appeared to be applicable to a wide series of primary phosphines including sterically hindered aryl-, benzyl- and l-menthylphosphines and to heteroanalogues of bis(4-aminophenyl)methane (Scheme 2) [18].
The 31P NMR spectra of the resulting reaction mixtures showed that the cyclophanes were the predominant products in comparison with other phosphorus-containing products and their relative content in the reaction mixtures was about 80% [17,18]. So nine individual representatives of this new class of cyclophanes were isolated and characterized (Table 1). It should be mentioned that the chiral P-l-menthyl substituted macrocycle 9 appeared to be one of the first representatives of aminomethylphosphines on the basis of l-menthylphosphine in spite of this phosphine being known since 1997 [19].
The yields of cyclophanes 1-9.
| Compound | R/X | Yield (%) |
| 1 | Ph/CH2 | 51 |
| 2 | Ph/S | 41 |
| 3 | Ph/SO2 | 21 |
| 4 | CH2Ph/CH2 | 21 |
| 5 | CH2Ph/S | 21 |
| 6 | Mes/S | 60 |
| 7 | Mes/SO2 | 76 |
| 8 | C6H2iPr3/CH2 | 71 |
| 9 | Ment/CH2 | 59 |
As expected, all cyclophanes obtained have similar molecular structures. Their molecules can be described as truncated rhombohedral prisms with the side faces formed by four phenylene rings and 1,5-diaza-3,7-diphosphacyclooctane fragments in the truncated acute angles (Fig. 1) [17,18].

The molecular structure of the macrocycle 1 (solvate molecules and hydrogen atoms are omitted for clarity).
The intramolecular cavities are cylindrical and their free volumes are 110–130 Å3 [18]. It is sufficient for the penetration of small organic molecules or their fragments. It should be noted that the formation of macrocyclic tetraphosphines is stereoselective and leads to one isomer with phosphorus lone electron pairs directed into the cavity. The diameter of the internal section of the cavity is higher than the external rim diameter because the substituents on the phosphorus atoms restrict the cavity and control the access into it from the sides of lower and upper rims of the macrocycle.
It is typical that in crystals the intramolecular cavities are filled with N-methyl groups of the solvent (DMF or N,N-dimethylacetamide (DMA)) (Fig. 2). The location of these groups indicates the existence of the binding H–π-interactions between their protons and electron-excessive phenylene fragments of the macrocycles [17,18].
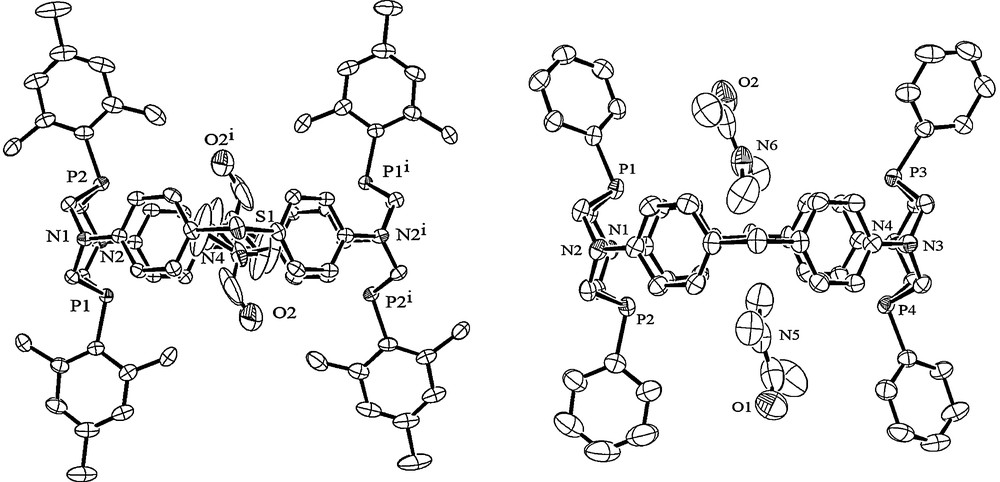
Side view of macrocycle 6 with two molecules of DMF (left) and macrocycle 1 with two molecules of DMA (right) inside the cavity.
The existence of the similar interaction was confirmed by the study of the structure and solution behaviour of the tetrasulfide 10, which was obtained by the addition of sulfur to the macrocyclic tetraphosphine 2. The oxidation of 2 by hydrogen peroxide gave the corresponding tetraoxide 11 (Scheme 3) [20].
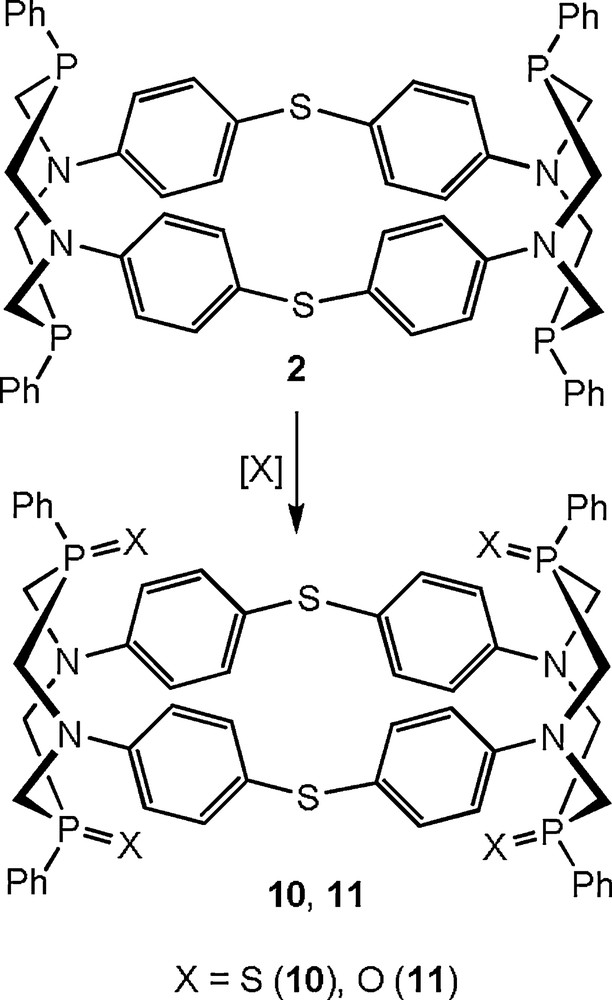
Synthesis of the derivatives of macrocyclic tetraphosphines.
In the crystal, the macrocycle 10 has a distorted conical conformation unlike the cylindrical conformations which were typical for cyclophanes 1–9. The conformational changes were caused by the DMF molecule penetrating the cavity (Fig. 3) [20].

The side view of macrocycle 10 with DMF molecule penetrating the cavity. Other solvate molecules and hydrogen atoms are omitted for clarity.
The access to the cavity of 10 is additionally shielded by four sulfur atoms. So the penetration of the guest molecule requires the change of the calculated optimal symmetrical cylindrical conformation to the conical one. The energy cost of this transformation is compensated by the energy of the inclusion complex formation due to the above-mentioned C–H–π interactions. The study of the behaviour of the 10-DMF complex in solutions showed that the processes of the formation and the dissociation of the complex were reversible and fast in the NMR time scale. These results demonstrate the ability of this class of macrocycles to increase noticeably the square of their rims if it is necessary for the penetration of the relatively large molecules [20].
The size of the intramolecular cavities of these cyclophanes is not very large and the form of the cavities may vary from cylindrical to conical. Since the sizes and the form of the cavities are the key characteristics of macrocycles as building blocks for the design of molecular devices and nanomaterials, it was important to search for the synthetic paths to similar cyclophanes with enlarged free intramolecular volumes.
The first attempts to expand the set of starting diamines by the introduction of thionine acetate, 3,6-diaminoacridine, 2,7-diaminofluorene and 4,4′-ethylenedianiline (Scheme 4) led only to the formation of the mixtures of oligomeric aminomethylphosphines.
Diamines of unfavourable structures for the spontaneous macrocyclization.
The approach elaborated for the synthesis of the coordination macrocycles [2] and based on the spatial complementarity of the building blocks was used for the more well founded choice of starting diamines. The key peculiarity of the macrocycles 1–9 is the presence of two cup-like fragments formed by the diazadiphosphacyclooctane ring in chair-chair conformation and two phenylene fragments on nitrogen atoms of this ring. The structure-forming 1,5-diaza-3,7-diphosphacyclooctane fragments determine the turning angle of 60–70° and require the diamine turning angle value of about 110–120° according to the complementarity principle (Fig. 4). In addition, the phenylene fragments of diamine must provide the formation of planar conjugated systems with the participation of nitrogen atoms included into the eight-membered heterocyclic fragment of the target cyclophane. Thionine acetate, 3,6-diaminoacridine, 2,7-diaminefluorene did not satisfy the last requirement, whereas 4,4′-ethylenedianiline showed too high conformational lability.
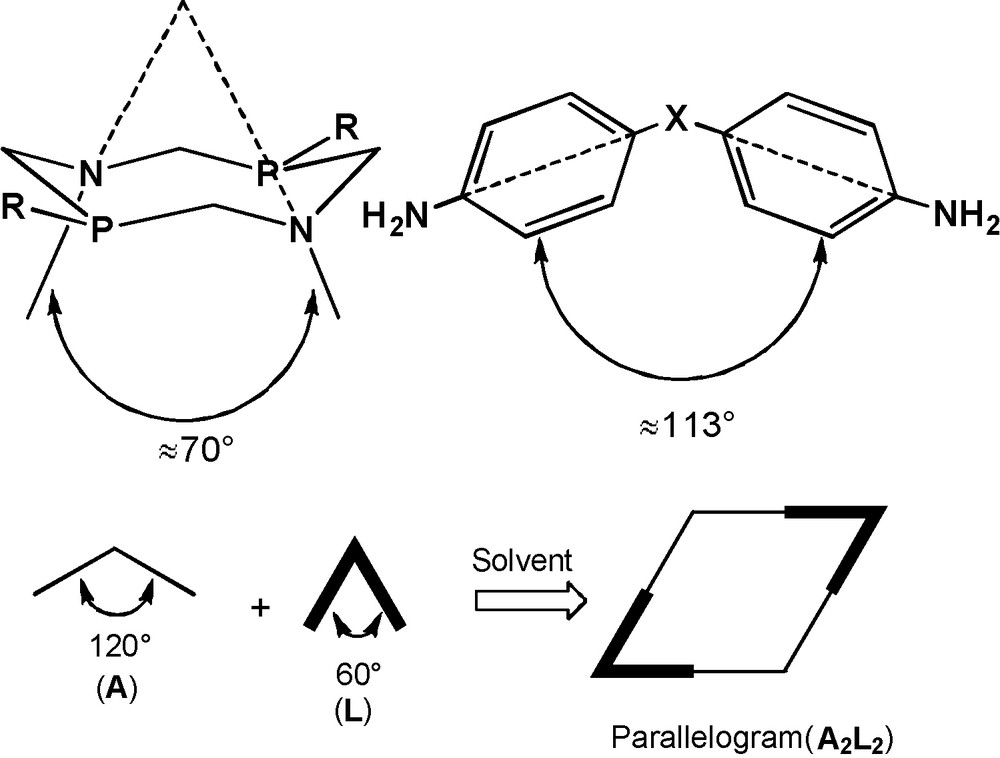
The complementarity of the building blocks for the macrocycles 1–9.
The first type of diamines chosen on the basis of the complementarity principle was 2,6-diamino-9,10-dihydro-9,10-ethanoanthracenes 12–14 [21] (Scheme 5) which had the total chirality.

Synthesis of 2,6-diamino-9,10-dihydro-9,10-ethanoanthracenes.
The racemic diamines 12–14 reacted with bis(hydroxymethyl)phenylphosphine under the covalent self-assembly conditions (DMF, the elevated temperature and phosphine concentrations of 0.1–0.3 M) to give the corresponding cyclophanes 15–17 as the mixtures of two regioisomers (Scheme 6) [22].

Synthesis of P,N-containing cyclophanes on the basis of 2,6-diamino-9,10-dihydro-9,10-ethanoanthracenes.
The less soluble isomers of all cyclophanes 15–17 were isolated as individual compounds. The structure of methyl-substituted macrocycle 15a was established by X-ray analysis which showed that it was a racemic isomer formed by identical enantiomers of the starting diamine. Its conformation appeared to be chiral and may be described as slightly twisted cylindrical one (Fig. 5). The similarity of NMR spectra of isolated isomers 15a, 16a and 17a indicates that 16a and 17a are also the racemic isomers of the close structure. More soluble isomers of 15b–17b are probably meso-isomers formed by the opposite enantiomers of the diamines.

The molecular structure of the macrocycle 15a. The solvate molecules and hydrogen atoms are omitted for clarity.
In the crystal the chiral cavity of the cyclophane 15a is filled with the methyl group of solvate DMSO molecule like in the case of cyclophanes 1–9. The isolation of the racemic isomers of 15–17 allows one to expect that the use of enantiomerically pure diamines 12–14 may lead to the synthesis of optically active macrocycles with chiral cavities, which are of interest as sensors and containers for chiral substrates or enantioselective molecular reactors.
However, the increase of the intramolecular cavity's size remained an unsolved important problem. The diamines with longer spacers have higher conformational lability so the mutual location of the amine groups favouring the self-assembly can be realized only in certain conformations. However, if these conformations of diamines are present in the reaction mixtures, they can give “correct” intermediates which later on can lead to the most thermodynamically stable desired macrocycles [3a]. Recently several diamines with spacers of the desirable length formed by three phenylene fragments became commercially available.
The proposed strategy appeared to be successful. The interactions of two diamines, namely 1,3-bis(4′-phenoxy)benzene (18) and 1,4-bis[α-(4′-aminophenyl)isopropyl]benzene (19), with primary phosphines and formaldehyde under the conditions of the covalent self-assembly led to the formation of two novel types of cyclophanes with large intramolecular cavities.
The Mannich-type condensations with the participation of 18 gave the macrocycles 20–23. Their cyclophane structures are formed by two diazadiphosphacyclooctane, four p-phenylene and two m-phenylene fragments (Scheme 7) [23].
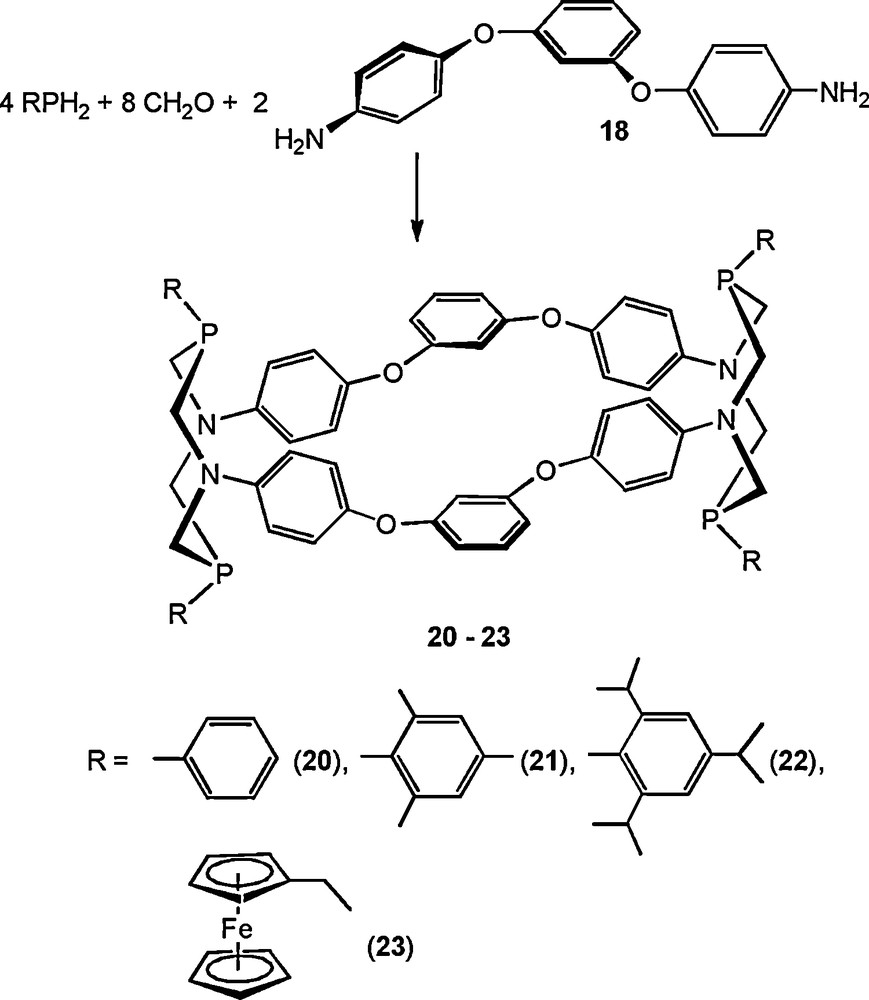
Synthesis of P,N-containing cyclophanes on the basis of 1,3-bis(4′-phenoxy)benzene.
According to X-ray analysis, the total structure of the cyclophane 20 may be described as two key cup-like fragments which are moved apart and mutually shifted with the central resorcinol fragments (Fig. 6) [23]. These fragments are almost coplanar to the reference plane formed by four trigonal-planar nitrogen atoms. As a result, the cavity of 20 is elongated and its width is determined by the P,N-containing heterocycle, whereas its length is determined by the diamine. The distance P(1)–P(5i) and the free volume of the intramolecular cavity are increased in comparison with the macrocycles 1–9 (12.3 Å [23] vs. 8.6–9.8 Å [17,18] and 165 Å3 [23] vs. 100–120 Å3 [17,18], respectively).

An ORTEP view of 20 (left) and the side view of 20 with the two closest DMF molecules (right). Hydrogen atoms and other solvate molecules are omitted for clarity.
The methyl groups of two DMF molecules penetrate into the macrocyclic cavity from the opposite sides of the macrocycle. Each of these methyl groups is located between two p-phenylene groups bound with a diazadiphosphacyclooctane fragment and their positions indicate H–π interactions between methyl protons and p-phenylene fragments (Fig. 6) [23].
The macrocycles 24 and 25 were obtained in satisfactory yields on the basis of diamine 19 under the covalent self-assembly conditions (Scheme 8). The yield of the cyclophane 24 was increased by the use of toluene as the solvent [24]. The successful replacement of the usually used DMF with toluene showed the absence of the templating effect of the first solvent.

Synthesis of P,N-containing cyclophanes on the basis of 1,4-bis[α-(4′-aminophenyl)isopropyl]benzene.
The spatial structure of cyclophane 24 differs more essentially from the structures of the prismatic macrocycles 1–9 than the structure of cyclophane 20. Crystals of 24 suitable for X-ray analysis, were obtained on recrystallization from benzene and contained eight benzene molecules per one molecule of 24 [24].
As expected, the intramolecular cavity of cyclophane 24 is formed by six phenylene units and two heterocyclic fragments. The P,N-containing heterocyclic fragments have a crown conformation with equatorial substituents on the phosphorus atoms. Due to this geometry, the electron lone pairs of the phosphorus atoms point towards the center of the cavity. Two 1,5-diaza-3,7-diphosphacyclooctane moieties are linked by two 2,2,4,4-tetramethyl-1,3,5(1,4)tribenzenapentaphane fragments in a chiral, helically twisted manner (Fig. 7a). The observed “figure-of-eight” structure differs from the cylindrical geometries found for the cyclophanes described above. The single crystal of 24 is a true racemic mixture of the right- and left-handed helicates [24].
(a) ORTEP view of 24 including one benzene molecule inside the cavity. H atoms and other benzene molecules are omitted for clarity; (b) Space-filling model (view along the a axis) showing encapsulation of a benzene molecule in the macrocyclic cavity of 24.
The encapsulation of one of the benzene molecules inside the macrocyclic cavity demonstrates the ability of new P,N-containing cyclophanes to bind aromatic hydrocarbon guests. The binding of the guest is provided by the H–π interactions between the protons of the central phenylene rings of the cyclophane and the π-system of the guest. The benzene molecule is isolated from the environment due to the helical conformation of the macromolecule and the presence of bulky substituents on the phosphorus atoms. A view on the least shielded side of the macrocycle (along the a axis, Fig. 7b) shows that conformational changes of the macrocycle are necessary for the guest to leave the cavity [24].
The chemical shifts of the central phenylene protons of the macrocycles 24 and 25 appeared to be highly diagnostic for the presence of the aromatic guests in the macrocyclic cavities in solutions. Their signals undergo dramatical shifts to high field (Δδ are about −0.5 ppm) in aromatic solvents (benzene or toluene) due to the anisotropic effect of the aromatic guest molecules. Therefore, NMR investigations of 24 and 25 indicate that the supramolecular host–guest organization with the aromatic compound located in the macrocyclic cavity is retained in aromatic solvents [24].
The results obtained show that the condensations of the three-component system primary phosphine/formaldehyde/primary diamine with preorganized spatially divided amine groups are a convenient tool for the design of various heterocyclophanes with diazadiphosphacyclooctane fragments.
1.2 The cyclophanes with linear P,N-containing fragments
The study of the total scheme of the cyclophane formation was necessary for the further development of the methodology of their design. The slow proceeding of the condensation allowed us to perform the 31P NMR-monitoring of the reaction mixture during the synthesis of macrocycle 4 (Scheme 9) [18].
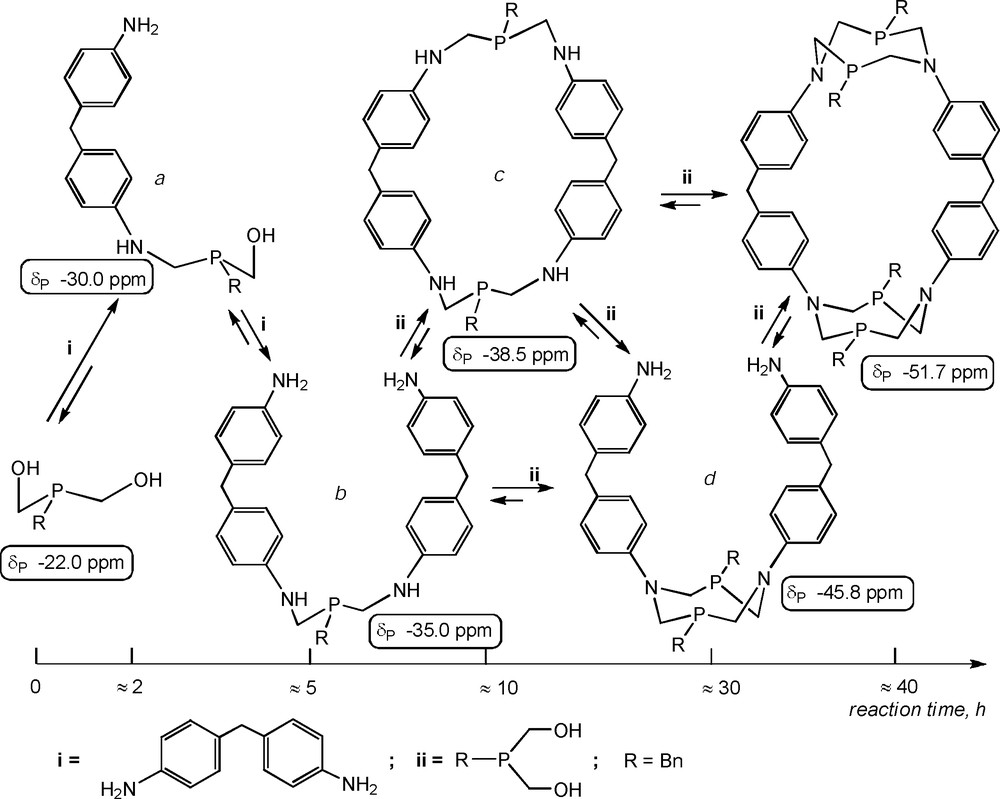
The supposed key intermediates of the macrocyclization process.
At the very beginning of the reaction, a peak of the corresponding (hydroxymethyl)-(aminomethyl)benzylphosphine (a) at δP −30 ppm appeared besides the signal of the starting bis(hydroxymethyl)benzylphosphine (Scheme 9). Later, the signal of corresponding bis(aminomethyl)benzylphoshine (b) and the peak at δP −38.5 ppm appeared. The last peak probably corresponded to the macrocyclic intermediate with linear aminomethylphosphine fragments (c) and became prevalent in the middle stage of the reaction. Only later signals in the 1,5-diaza-3,7-diphosphacyclooctane region (−46 to −52 ppm) appeared, including the increasing signal of the cage macrocycle 4. It should be mentioned that a clear separation of the reaction steps was not observed [18].
The macrocyclic intermediate (c) with phenylene fragments linked by flexible linear aminomethylphosphine bridges drew special attention, because there was the reaction stage when this intermediate prevailed. It indicates that the formation of the intermediate (c) may also be a result of the covalent self-assembly, so the synthesis and the isolation of similar macrocycles are possible. The secondary diamines with di(p-phenylene)methane spacers, namely N,N’-dimethyl- (26), N,N’-di(3-pyridylmethyl)- (27) and N,N’-di(4-pyridylmethyl)-4,4′-diaminodiphenylmethane (28), were used for the synthesis of this new type of macrocycles. Bis(4-amino-3-carboxyphenyl)methane (29) was also chosen as the starting diamine, because it was known that o-aminobenzoic acid reacted with hydroxymethylphosphines as a secondary amine due to the strong H-bonding between amino and carboxy groups [16b].
The condensations of phenyl- and mesitylbis(hydroxyphenyl)phosphines with secondary diamines 26–28 were performed in DMF at the reagent concentrations of 0.1–0.3 M. In the case of diamine 26, the reaction temperature was 100–110 °C, in the cases of N-pyridylmethyl substituted compounds 27 and 28, the ambient temperature appeared to be optimal in spite of the long reaction times: the especially slow condensation of 27 required 22 days for completion. 31P NMR control of the reaction mixtures showed the similar course of the condensations which was practically identical to the first stages of the formation of cage macrocycles 1–9 (Scheme 10). In the spectra of the resulting reaction mixtures, there were one or two very close prevailing peaks in the range of δP −37 ÷ −42 ppm. The relative content of the corresponding products was about 75–80%, but only macrocycles 30 and 31 were isolated in the yields of 21 and 67%, respectively. The similar product 32 was obtained as a very viscous oil of 85% purity which contained also the unidentified oligomeric aminomethylphosphine byproducts [25].
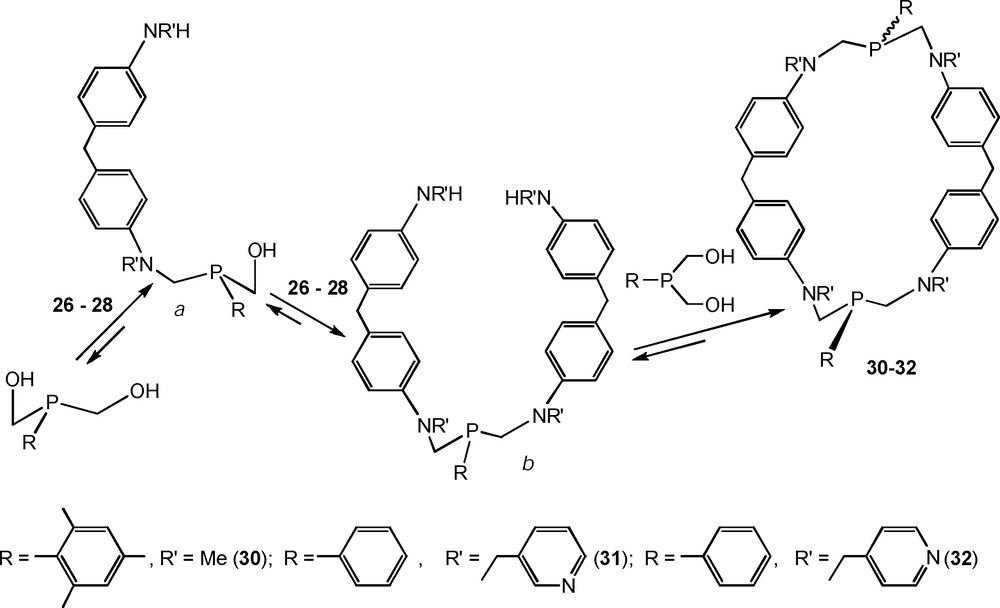
Synthesis of P,N-containing cyclophanes on the basis of secondary diamines.
The macrocycles 30–32 were formed as the mixtures of cis- and trans-stereoisomers. The trans-isomers prevailed in the reaction mixtures only slightly, but the fractional crystallization of 30 from DMF led to the individual trans-stereoisomer [25].
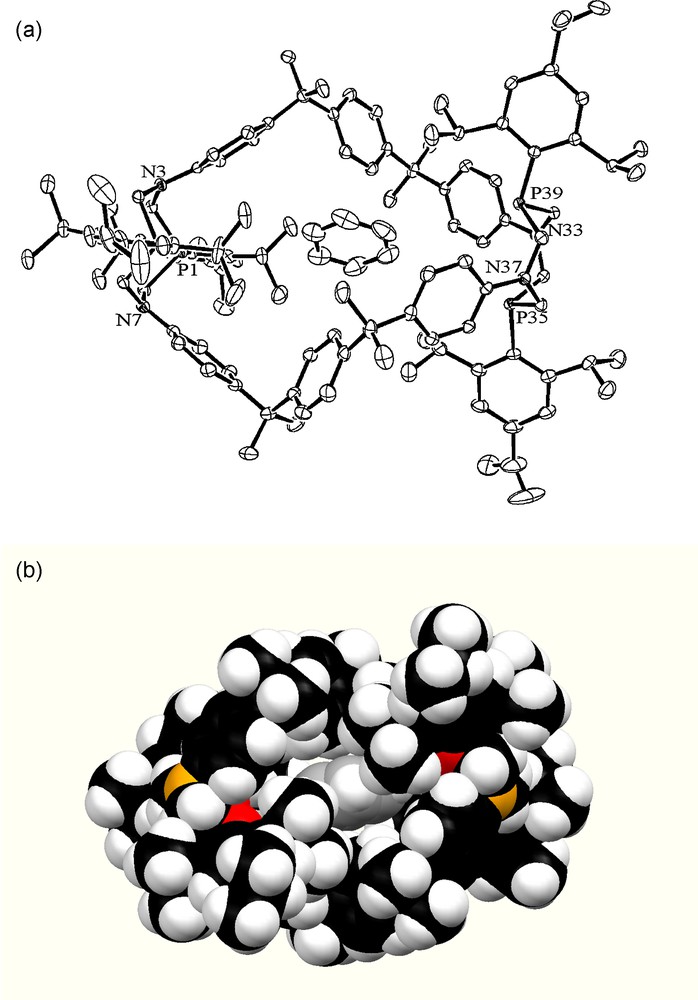
Its structure was confirmed by X-ray analysis. The structure of the centrosymmetric molecule of trans-30 (Fig. 8) differs strongly from the structures of cage cyclophanes with similar phane fragments, namely 1,5(1,5)-di(1,5-diaza-3,7-diphosphacyclooctana)-2,4,6,8(1,4)-tetrabenzenacyclooctaphanes 1–9. Their phenylene fragments form the side faces of the prismatic cavities and are almost orthogonal to the reference planes of macrocycles determined by four nitrogen atoms [17,18]. All nitrogen atoms of trans-30 are also located in the same plane, but only two of them are coordinated in trigonal-planar fashion and their lone electron pairs are conjugated with the π-systems of the phenylene rings. Two other ones are coordinated in near trigonal-pyramidal fashion (the sums of their bond angles are 346.2°) which indicates the low extent of the conjugation. The neighbouring phenylene rings are strongly twisted and the angle between their planes is 69.7°. The opposite rings are coplanar, and different pairs of rings are inclined to the reference plane of the macrocycle at the angles of 73.1 and 138.6° respectively, so the macrocyclic cavity is practically collapsed and elongated: the P–P-distance is 11.80 Å, whereas the distance between the opposite aromatic rings is only 5.85 Å [25].
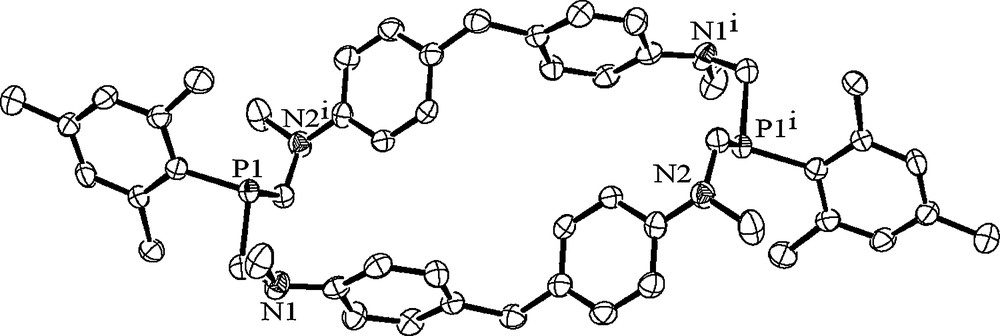
The molecular structure of cyclophane trans-30. The hydrogen atoms are omitted for clarity.
It should be mentioned that the equivalence of all phenylene rings and methyl groups on nitrogen atoms in 1Н NMR spectra of 30 gives the evidence of the fast conformational exchange in solutions of this macrocycle [25].
As expected the condensation of bis(hydroxymethyl)mesitylphosphine with the primary diamine 29 also gave the corresponding 1,5,19,23-tetraaza-3,21-diphospha[5.1.5.1]paracyclophane 33 as a major product (Scheme 11). The cyclophane 33 was isolated in the yield of 71% as the mixture of trans- and cis-diastereomers with their ratio 1.4: 1 according to 1H NMR spectra. It was stable to oxidation both in the solid phase and in solutions and soluble in aqueous sodium hydroxide solution due to the presence of four carboxy groups. The formation of macrocycle 33 instead of the cage macrocycle is probably caused by the deactivation of the amine groups by the strong intramolecular hydrogen bonds with carboxy groups [25].

Synthesis of P,N-containing cyclophane on the basis of bis(4-amino-3-carboxyphenyl)methane.
As has been already mentioned, the most important peculiarity of the structure of the cyclophanes with the linear N-C-P-C-N fragments is the absence of the conjugation between two of the four nitrogen atoms and the phenylene fragments [25]. It means that the structural restrictions for the secondary diamines become less rigid in comparison with primary ones. It was indeed found that N,N’-diphenylbenzidine with linear biphenylene spacer was able to participate in the covalent self-assembly of macrocycles in spite of its apparent difference with the previously used angular diamines. Its condensation with bis(hydroxymethyl)arylphosphines led to the new type of cyclophanes. The macrocycles 34 and 35 were also formed as the mixtures of cis- and trans-diastereoisomers. The data from the mass spectrometry and the cryoscopy (for the highly soluble compound 35) gave the unambiguous evidences that these cyclophanes were the products of [2 + 2]-condensation (Scheme 12).

Synthesis of P,N-contaning cyclophanes with linear biphenylene fragments.
So it has been shown that the covalent self-assembly methodology is also effective for the synthesis of more flexible macrocyclic systems, the requirements to the building blocks geometry being less rigid in these cases.
1.3 P,N-containing corands and cryptands
All macrocycles described above were synthesized with the use of the covalent self-assembly approach on the basis of diamines with spatially divided amine groups as the structure-forming building blocks. However, in the course of the systematic studies in the field of the synthesis of heterocyclic diphosphines by the condensation of secondary diphosphines with formaldehyde and primary amines, we unexpectedly met another example of the covalent self-assembly of flexible macrocyclic tetraphosphines.
The interaction of bis(arylphosphino)methanes and -ethanes with the wide set of primary amines in the presence of formaldehyde led to expected six- [26] and seven-membered heterocycles which were obtained in good yields [27]. The reactions of 1,3-bis(arylphosphino)propanes 36 and 37 with formaldehyde and 5-aminoisophtalic acid proceeded similarly to give corresponding water-soluble 1-aza-3,7-diphosphacyclooctanes 38 and 39 (Scheme 13) [28].

The interaction of 1,3-bis(arylphosphino)propanes with 5-aminoisophtalic acid.
The replacement of the arylamine by more basic and active in the condensation reactions benzylamine unexpectedly led to the formation of a new class of macrocyclic tetraphosphines. Its first representatives 40 and 41 (Scheme 14) were formed in good yields and with excellent stereoselectivity [29]. The starting diphosphines exist as the mixtures of rac- and meso-stereoisomers, and the macrocycles 40 and 41 contain four phosphorus asymmetric centers. It must lead to the formation of five possible diastereoisomers, whereas both macrocycles are formed as single RSSR-isomers. Therefore, in the course of the reaction, not only the mutual recognition of the intermediate stereoisomers takes place but also the inversion of the starting diphosphine configuration proceeds [29].

Synthesis of P,N-containing corands on the basis of benzylamine.
The reactions were performed in the concentration range 0.1–0.5 M at elevated temperatures (80–110 °C) and in the absence of the templating reagents. The NMR monitoring of the reaction mixtures showed that the reactions proceeded with the formation of plenty of intermediates and the reaction mixtures were enriched by the isolated products only at the final stage, as in the case of macrocyclizations described above [29]. So, we again met the phenomenon of the covalent self-assembly. The amine activity provided the sufficient reversibility of the condensation for the self-assembly process and the 16-membered macrocycle appeared to be the most thermodynamically stable product even in comparison with eight-membered 1-aza-3,7-diphosphacyclooctane.
In the solid state 40 forms a centrosymmetrical 16-membered ring. One half of the macrocycle,–CH2P(CH2)3PCH2N–, has a distorted crown conformation with a nearly axial orientation of the lone pairs of the heteroatoms, and the other half is its inverted analogue in which the lone pairs point to the opposite side of the macrocycle. The two centrosymmetric –CH2P(CH2)3PCH2N– fragments are linked by N1–C1 and N1*–C1* bonds. Two phosphorus atoms of each halves have different configurations (Fig. 9) [29]. The compounds 40 and 41 may be considered as a new type of P,N-containing corands.
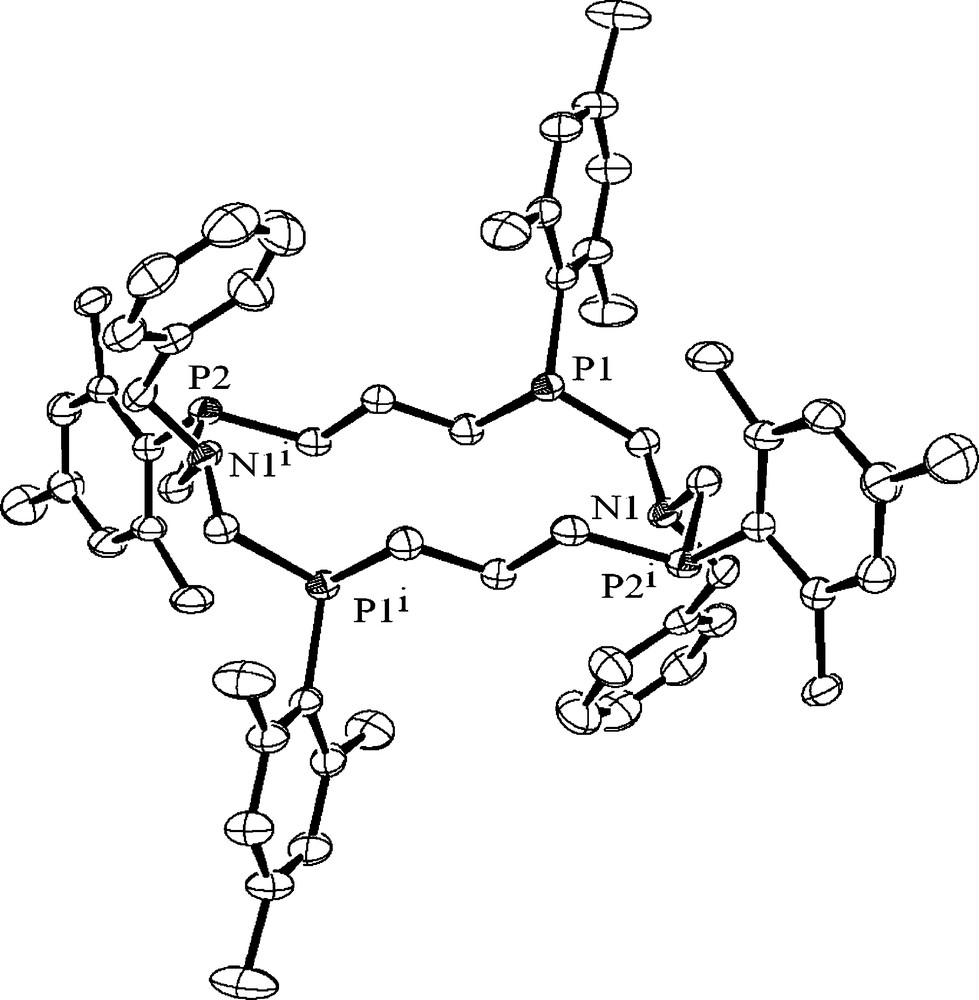
The molecular structure of the corand 41. Hydrogen atoms are omitted for clarity.
The covalent self-assembly process found in the course of the condensation in the systems secondary diphosphine/formaldehyde/primary amine allowed us to design the ligands of the desirable structure and properties on the basis of our experience in this field. The macrocyclic ligands 42–46 (Scheme 15) with additional donor centers of various nature and in different positions were obtained in very good yields [30]. The synthesis of the compound 46 is especially important because this macrocycle is water-soluble in the presence of the bases [30].

Synthesis of functionalized P,N-containing corands.
The study of the structure of the macrocycles 42–46 showed that all of them were RSSR-stereoisomers like compounds 40 and 41 and their conformations in the solid state were similar to ones of these N-benzyl substituted macrocycles [29].
One of the most interesting features of aminomethylphosphine chemistry is the possibility to obtain a wide set of optically active phosphine ligands on the basis of the various available chiral amines. The use of chiral amines must theoretically increase the number of possible stereoisomers to 16. However, in these cases, the reactions proceeded with the excellent stereoselectivity and led to the formation of the single stereoisomer for each of the corands 47–50 (Scheme 16) [31].
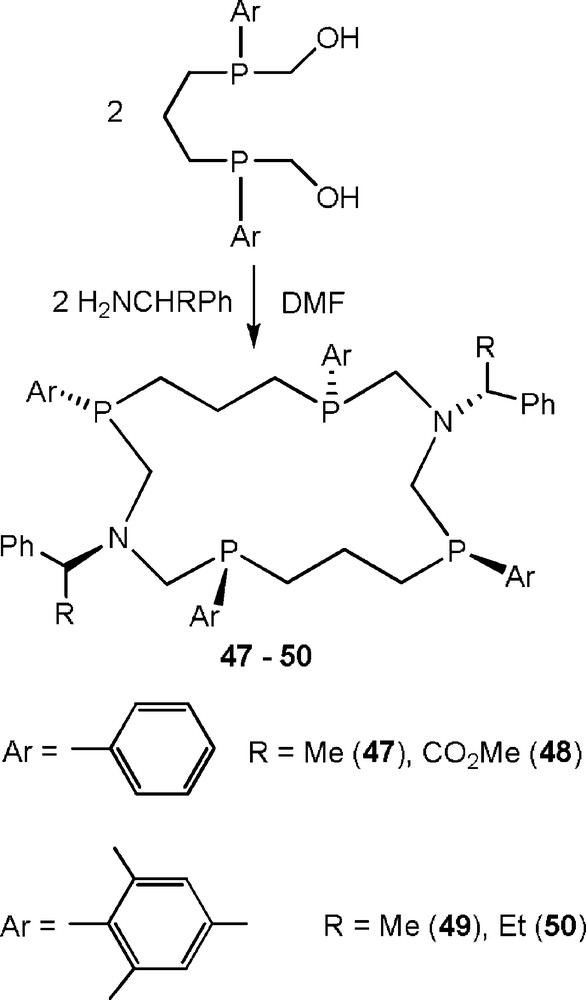
Synthesis of optically active P,N-containing corands.
The configurations of the phosphorus atoms of these corands (RSSR) were not changed in comparison with optically inactive analogues 42–46, so the total structure of the optically active macrocycles 47–50 remained also similar to the structures of corands 40–46 (Fig. 10) [31].

The molecular structure of the corand 49. Hydrogen atoms are omitted for clarity.
However, NMR spectra of the macrocycles 47–50 appeared to be more complicated. The presence of chiral exocyclic groups on nitrogen atoms led to the non-equivalence of the phosphorus atoms in each half of the molecule and to doubling most of the signals in 1H and 13C NMR spectra, so the full assignment of the signals was performed by a variety of 1D and 2D NMR experiments supported by quantum-chemical calculations [31].
Cryptands are one of the most effective “hosts” in coordination and supramolecular chemistry [1]. The condensation of the secondary diphosphines with formaldehyde and primary diamines could become a convenient method of the synthesis of unique P,N-containing cryptands, but only if it proceeded as the covalent self-assembly. We indeed found the first example of the cryptand formation as a result of the reaction between 1,3-bis(mesitylphosphino)propane, formaldehyde and m-xylylenediamine (Scheme 17) [32].

Synthesis of P,N-containing cryptand on the basis of two bifunctional reagents.
The cryptand 51 is also formed stereoselectively as a single diastereoisomer. According to NMR and X-ray analysis data (Fig. 11), it is a racemate of RRRR/SSSS-isomer. The lone electron pairs of the phosphorus atoms have axial orientation relative to the 16-membered ring and their directions alternate. Nitrogen atoms are coordinated in trigonal-pyramidal fashion and their lone electron pairs are directed into the cavity [32].
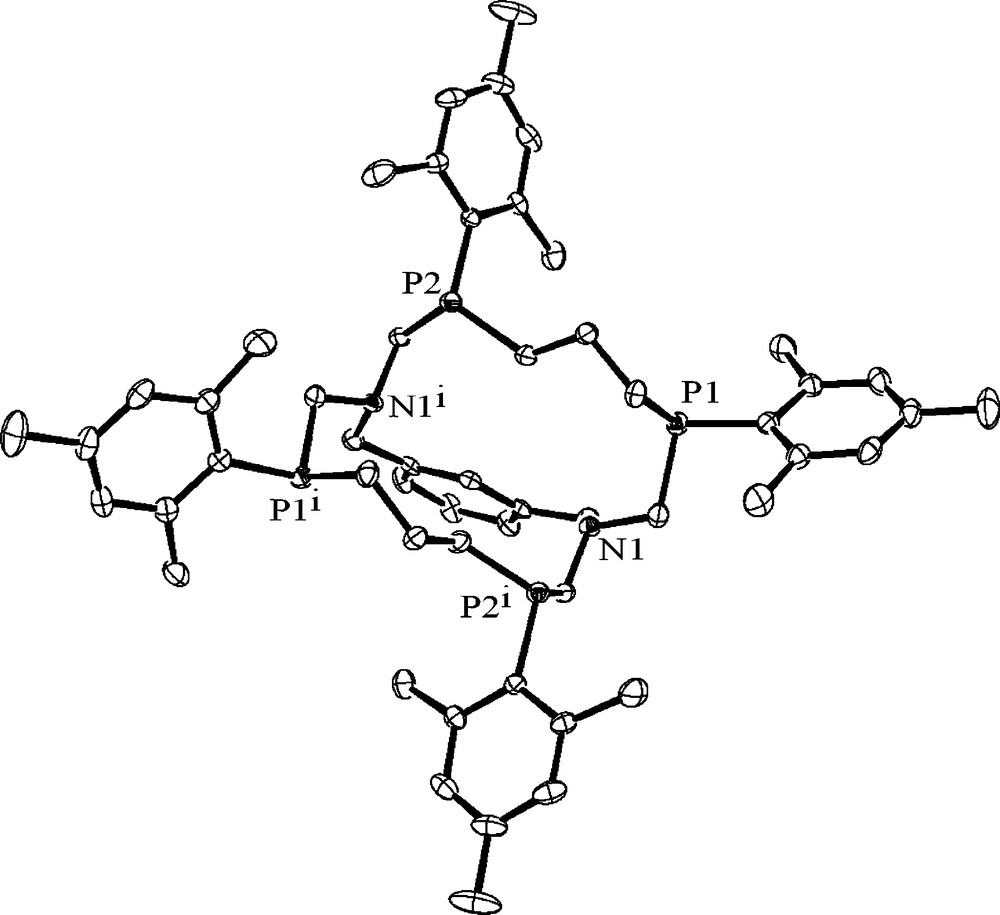
The molecular structure of the cryptand 51. Hydrogen atoms are omitted for clarity.
These results show the effectiveness of the covalent self-assembly approach to the synthesis of P,N-containing corands and cryptands.
2 Conclusions
In the course of the present study, an effective methodology for the synthesis of original P,N-containing macrocycles has been developed. It is based on the covalent self-assembly phenomenon and provides the possibility of the synthesis of the wide set of these unique compounds which become available for the specialists in the fields of coordination and supramolecular chemistry. The study of the properties of these macrocycles, their behaviour in solutions and especially their complexing properties towards organic, organoelement and metal-containing substrates has now just started but will probably become a matter of active investigation in the nearest future. It seems to be probable that these macrocycles may be used as the building blocks for the creation of new unusual catalysts of organic reactions due to the specific interactions of the substrates with the intramolecular cavities of the catalysts, for the design of nanosized molecular containers or reactors, selective sensors for organic and organometallic compounds and molecular devices.
Acknowledgements
This work was supported by the RFBR and DFG (No.09-03-99011_r_ofi, 10-03-00380-а, RFBR-DFG 09-03-91338-DFG-a), a President of Russia grant for the support of leading scientific schools (No.3831.2010.3), and Federal Agency of Science and Innovations (state contract 02.740.11.0633).