

1 Introduction
Cyclodextrins (CDs) are water-soluble cyclic oligosaccharides with six, seven or eight α-1,4-linked d-glucopyranose units, and named α, β and γ-cyclodextrin, respectively. The geometry of CDs gives a hydrophobic inner cavity having a depth of 7.8 Å, and an internal diameter of 5.7, 7.8 and 9.5 Å for α, β and γ-CD, respectively (Fig. 1) [1]. In aqueous solution, the slightly apolar CD cavity is occupied by water molecules which are energetically unfavored, and therefore can be readily replaced by appropriate “guest molecules” which are less polar than water. Such guest molecules thus lead to the formation of an inclusion complex with CD, acting as the host molecule. Such inclusion complexes are characterized by an association constant Ka and a stoichiometry (host:guest ratio) which is most frequently 1:1. Due to their unique property to include hydrophobic guest molecules in their internal cavity, natural cyclodextrins have been widely used as carriers for drugs to enhance their solubilization, stabilization and bioavailability [2–5]. β-CD is by far the most widely used host compound due to the optimal size of its internal cavity [5]. However, because of its low aqueous solubility, chemical derivatives of β-CD were prepared in order to extend its physicochemical properties as well as inclusion ability [3,5].
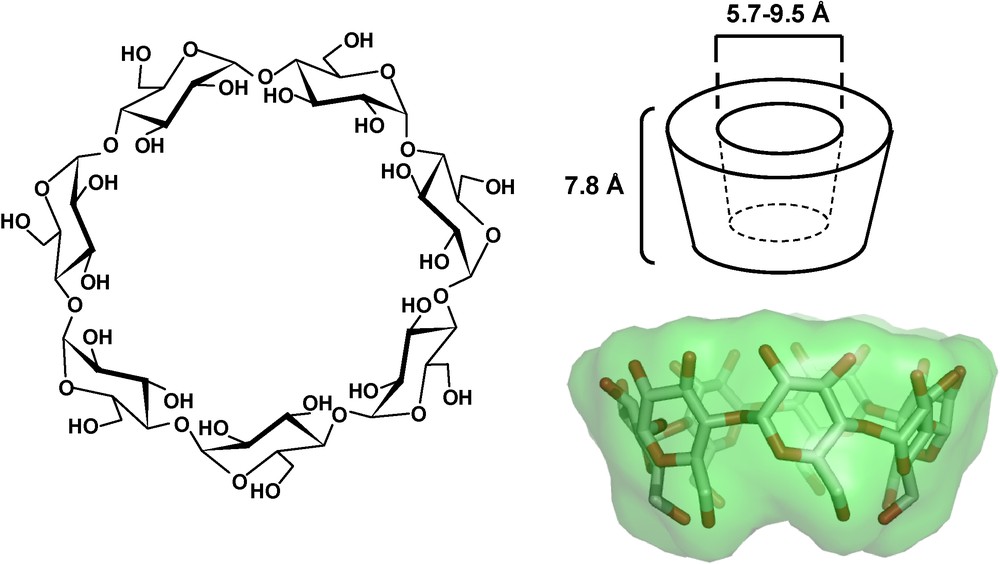
Structure of β-CD and schematic representation of CD molecules exhibiting the shape of a truncated cone or torus.
Besides their application as drug carriers, CDs and their derivatives have been used as building blocks for the development of a wide variety of polymeric networks and assemblies. Over the last 10 years, several works have thus been dedicated to the development of hydrogels, nano/microparticles, nano/microcapsules and thin films. Such polymeric materials have gained significant interest for application in diagnostics, drug delivery, and tissue regeneration, as they can be designed with unique mechanical properties, stimuli-responsiveness and tunable drug release characteristics [6–12].
Recent reviews have summarized research progress in the synthesis of cyclodextrin-based polymeric materials [13], the construction of cyclodextrin-based supramolecular assemblies and their interactions with biologically important substrates [14], the synthesis and applications for drug and gene delivery of cyclodextrin-based supramolecular architectures [15], biomedical applications of cyclodextrin based polyrotaxanes [16]. This review will describe the recent developments of self-assembling polysaccharide systems based on cyclodextrin complexation, with special emphasis on advances in understanding the structure–property relationship in these host–guest multivalent systems.
Compared to the commonly self-assembled synthetic polymers, polysaccharides exhibit interesting and original properties which make them a special class among polymer materials. They are generally biocompatible and biodegradable and may also exhibit a biological activity which can be advantageously exploited for applications in the biomaterials field [17]. This is highlighted in the case of chitosan (CHI) and hyaluronic acid (HA) which are two charged polysaccharides with various biomedical and cosmetic applications. Chitosan is one of the most abundant natural biopolymers and the only natural polycationic polysaccharide, which is widely used in the biomedical field [18–21]. Chitosan increases the healing rate of open wounds by stimulating the immune response and tissue reconstruction. It is also a suitable substrate for cell culture and additionally stimulates cell growth. Hyaluronic acid is an anionic natural polysaccharide (at physiological pH) which finds many uses in the biomedical field (ophtalmological surgery, treatment of the osteoarthritis of the knee, prevention of post-surgical adhesion, tissue engineering…) [22–25]. HA belongs to the family of glycosaminoglycans and is a constituent of the extracellular matrix of biological tissues where it plays major structural and biological roles. Alginates, which are extracted from brown algae, are also largely exploited for the design of biomaterials due to their ability to form biocompatible gels by a cooperative ionic interaction with bivalent cations [26]. All these polysaccharides are half-rigid polymers, which is also the case for most neutral polysaccharides owing to their polyglucidic structure [27,28]. Dextran, which is a biocompatible neutral polysaccharide of bacterial origin, is thus distinguished from other polysaccharides, by the higher flexibility of its chain [29]. Fig. 2 shows the chemical structure of these polysaccharides which have been used for the synthesis of networks and assemblies based on cyclodextrin complexation.

Chemical structure of biocompatible polysaccharides used for the synthesis of networks and assemblies based on cyclodextrin complexation.
2 Supramolecular hydrogels based on host–guest cyclodextrin inclusion complex
Hydrogels are appealing for biological applications because of their high water content and biocompatibility. In the last couple of decades, these three-dimensional polymeric networks have attracted a great deal of attention, and significant progress has been made in designing, synthesizing, and using these materials for many biological and biomedical applications. Recent developments include the design and synthesis of novel hydrogels and their use in tissue engineering, drug delivery, and bionanotechnology [10]. A particularly interesting and important polymeric system is hydrogel forming solutions by a simple sol–gel transition in water without any chemical reaction or external stimulation [30,31]. These systems provide simplicity and safety in in vivo situations. Indeed, an injectable matrix can be implanted in the human body with minimal surgical wounds, and bioactive molecules or cells can be incorporated simply by mixing before injection. In the case of biocompatible and biodegradable polysaccharides, specific interactions can be advantageously used to produce such reversible networks with promising applications as injectable scaffolds for tissue engineering, therapeutic implants, and smart vehicles. Polysaccharides additionally offer the advantage of reducing the critical gelation concentration and the dissolution rate of the network in vivo due to the high solution viscosity provided by their semi-rigid character [30]. It is also worth noting that owing to their unique biological and/or physicochemical properties, polysaccharides may also promote interactions between the gel and native tissues, which represents a challenging topic in drug delivery and tissue engineering. As far as a drug vehicle is concerned, the penetrability of various drugs, especially hydrophobic ones, and precise control of their release profiles are also persistent challenges [30]. In this context, several groups have investigated the formation of physical networks based on CD inclusion complexation between polysaccharide chains.
2.1 Specific interaction in modified charged polysaccharide systems
Several supramolecular assemblies based on charged natural polysaccharides have been developed over the last 10 years. The properties of such systems strongly depend on the balance between attractive interactions due to CD inclusion complexation and repulsive electrostatic interactions, which may be modulated by external parameters (ionic strength, pH) as well as intrinsic factors (degree of substitution of polymers, association constant of the host–guest complex). As developed below, interesting thickening properties or transparent gel formation due to complex formation between polysaccharide chains were observed under optimal conditions.
Our group synthesized associating polysaccharides based on inclusion complex formation between β-cyclodextrin and adamantane (AD) molecules, each grafted along the polysaccharide chains [32–35]. AD was selected as a guest molecule as AD derivatives form deep and snug-fitting complexes with the host β-CD molecule, leading to very high association constants (Ka ∼ 104–105 M−1) [36]. The natural polysaccharides chitosan and hyaluronic acid were thus selectively modified by β-CD and AD (Fig. 3), leading to “guest” (CHI- and HA-AD) and “host” (CHI- and HA-CD) polymers with degrees of substitution (DS) ranging from 0.03 to 0.08.

Chemical structure of host and guest derivatives of hyaluronic acid and chitosan leading to physical networks through CD/AD complexation.
The covalent grafting of the β-CD and AD molecules on these semi-rigid polyelectrolytes was based on a reductive amination-type reaction performed in homogeneous aqueous solution. Such conditions allow not only to achieve a random distribution of the substituents along the chain, but also avoid polymer chain depolymerization. These are essential to control the properties of the resulting networks. When a solution of CHI- or HA-CD was added to a solution of CHI- or HA-AD, respectively, at a total polymer concentration ∼ 1.5-fold higher than the critical overlap concentration C* of the initial polysaccharide, the rapid formation of transparent “gels” could be observed macroscopically. It should be noted that the salt concentration used for the formation of the assemblies in aqueous medium was lower than 0.05 M as above this critical concentration a phase separation phenomenon was observed [33,35]. Dynamic rheological measurements demonstrated a viscoelastic behavior for the assemblies in which inclusion complexes play the role of sticky point between the polysaccharide chains, contrary to the solutions of the native CHI and HA samples which exhibit only a viscous character. Indeed, from Fig. 4, it can be seen that the storage (elastic) modulus, G′, is larger than the loss (viscous) modulus, G″, within the whole range of frequencies covered for CHI-AD/CHI-CD and within a large range of frequencies for the HA-AD/HA-CD mixture. Moreover, the values of the storage and loss moduli for the mixtures of modified CHI and HA samples are much higher than those obtained for the solution of initial CHI and HA, although the concentration of the latter is, respectively, eight- and three-fold higher than that of modified polymers in the mixtures.
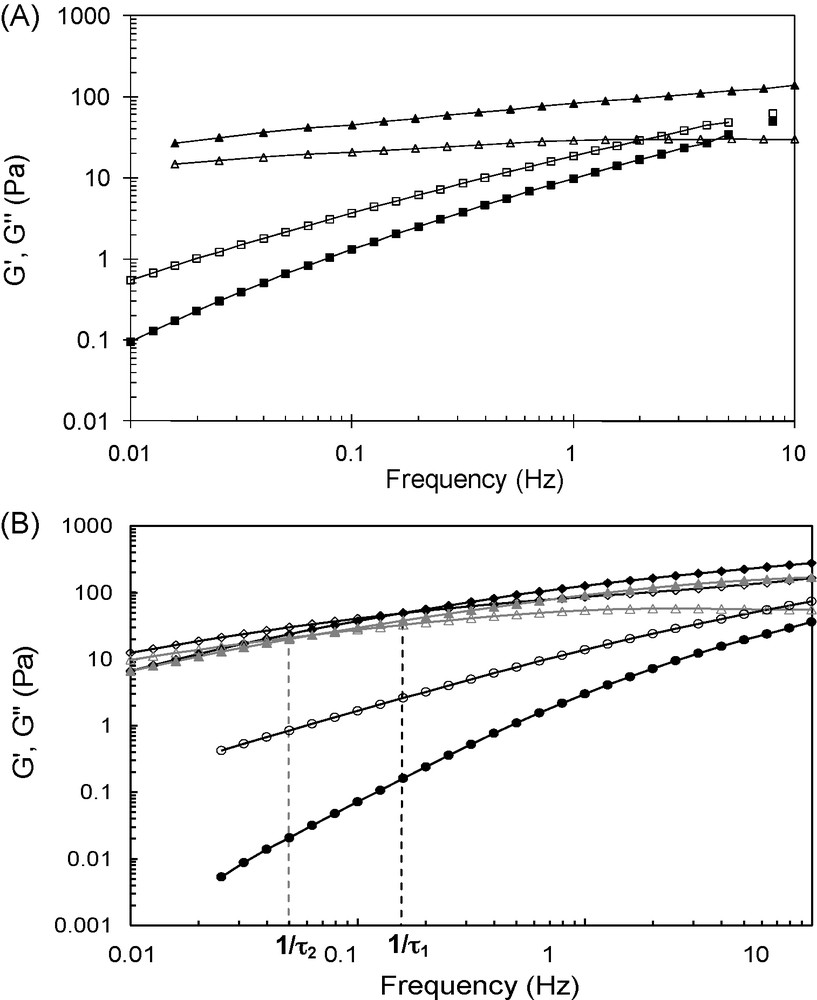
Comparison of the storage (filled symbols) and loss moduli (opened symbols) as a function of frequency for (A) a chitosan solution (■, □, 30 g/L) and a CHI-AD (2.46 g/L, DS = 0.07)/CHI-CD (1.97 g/L, DS = 0.1) mixture (▴, ▵) in 0.3 M CH3COOH/0.03 M CH3COONa at 25 °C and, (B) a hyaluronic acid solution (, , Cp = 30 g/L), a HA-AD (DS = 0.06)/HA-CD (DS = 0.05) mixture (, , Cp = 10 g/L) and a HA-AD2 (DS = 0.03)/HA-CD2 (DS = 0.025) mixture (▴, ▵, Cp = 10 g/L) in 0.025 M NaCl at 25 °C (adapted from [33,35]).
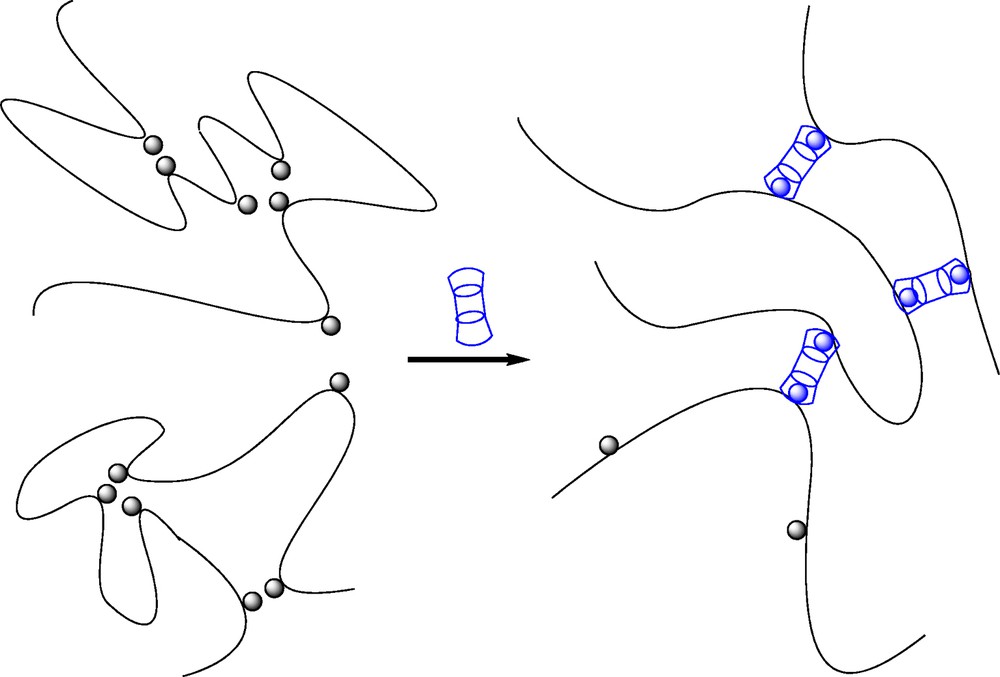
The effects of different external parameters such as polymer chain concentration, temperature and addition of free competitive host molecules on the viscoelastic properties were intriguing and similar for both CHI-based and HA-based systems. They were related to a peculiar mechanism of association resulting in a network of double-chain strands connected by fourfold junction points (Fig. 5) [34,37]. Indeed, in semi-dilute solution and in dilute solution as well, the mixtures of CD-grafted and AD-grafted HA/CHI may result in formation of double chain complexes (also called railway or ladder complexes), which benefit from the high energy of the CD/AD complex [37]. Above a given concentration, which is around the HA polymer coil overlap concentration C*, the ladder complexes form a reversible network in which the cross-links are junction points where the crossed double-chains interchange their partners. The theory developed to elucidate the most essential features of the rheological behavior of these assemblies thus considered two types of cross-links in the reversible network: (1) the “strong” junctions where two double chains interchange their partners; the lifetime of a strong junction point is at least τstr; (2) the “weak” junctions formed when a CD molecule in one double-chain fragment makes a bond through complexation with an AD molecule in another double chain. The lifetime of such interchain complex is ∼τd. As the strong junction points quench drastically the motions of the chain, the theory was focused on the network relaxation at timescales t ≥ τstr. The weak junction points dissociate much faster (τd ≪ τstr), and were therefore neglected. It was additionally demonstrated that the irregularity of chemical positions of grafted stickers plays a major role on the dynamical properties. It has a slowing effect, which is controlled by the parameter β = B × (DS × N)1/2, where N is the total number of units per chain. The parameter B is related to the distribution of the stickers along the chain. B increases with the polydispersity of the main-chain spacers between the stickers. This implies that matched double-chain fragments coexist with mismatched ones. The former are very stable, and consequently, contribute to significantly slow down the structural relaxation of the network. From these considerations, chain concentration may have a negligible effect on the dynamical properties, which was observed experimentally.

Schematic representation of (A) double-chain strands connected by fourfold junction points constituting the reversible network made of host and guest polysaccharides and, (B) a physical network in which bivalent β-CD/AD complexes play the role of junction points (adapted from [33,34]).
The theoretical parameter B was thus considered as a fitting parameter to calculate the theoretical curves representing the frequency dependencies of G′ and G″ of the host–guest systems at different concentrations. Indeed, B could not be calculated, as the degree of polydispersity of inter-sticker length distribution is not known. A quite reasonable agreement could be observed between theory and the experiments, both for G′ and G″, with a single essential fitting parameter B for the whole set of concentrations [37].
Interestingly, adding competitive host molecules remarkably diminished the characteristic time τx = 1/τx corresponding to G′ = G″. These molecules compete for pairing with grafted bond-forming stickers, thus increasing the probability of the dissociated state of a pair of grafted stickers. Actually, they were assumed to decrease the high elastic energy penalty resulting from the double chain structure with all stickers forming bonds. This effect thus crucially depends on the flexibility of the backbone spacer between the neighboring CD or AD (called stickers), which can be derived from its length (L) and the backbone persistence length (Lp) of the polysaccharide. As the CHI derivatives were characterized by stiff spacers (L ≪ Lp with Lp ≈ 9 nm [27]) contrary to the HA derivatives (L ≫ Lp with Lp ≈ 7.5 nm [27]), the decrease of the characteristic time τx due to addition of free stickers was particularly dramatic in the former case [34,35,37]. The relaxation time indeed decreased by many orders of magnitude even when the number of added free stickers is less than the amount of grafted stickers.
Moreover, HA was grafted with bivalent β-CD or AD moieties, which were able to form dimeric β-CD/AD complexes with a higher association constant as compared to monovalent β-CD/AD inclusion complexes (Fig. 5B, Ka(CD/AD) = 75 × 103 M−1 and Ka(CD2/AD2) = 1.14 × 106 M−1) [33,34]. The dynamics for the “bisticker” system was shown to be slower than that for the corresponding “monosticker” system. Indeed, as can be observed on Fig. 4B, the crossover point of the G′ and G″ curves is lower for the mixture stabilized by pairs of CD/AD complexes with respect to the other one made of single complexes (1/τ2 < 1/τ1), as a result of the higher energy and lifetime of the junction points. However, given the higher association constant of the dimeric β-CD/AD complex compared to that of the monomeric CD/AD one, a much slower dynamics for the system with bistickers was expected. This was related to significant elastic energy penalty for the formation of the (CD)2/(AD)2 complex. Moreover, the storage and loss moduli for the mixture stabilized by pairs of CD/AD complexes were found to be lower than those for the network stabilized by single complexes. Since the mesh size of networks is related to the number of junction points, this result appeared to be consistent with expected ones as the content in AD and CD is similar for both mixtures, but they are distributed by pairs along the polymer chain in the case of the bisticker system.
As highlighted by Soltès et al., such host–guest associating HA derivatives can be classified as a novel potential in the viscosupplementation of (osteo)arthritic joints [38]. This therapeutical concept consists in intra-articular injection of HA in order to compensate the loss of viscoelastic properties of endogeneous HA. Thus, such host–guest networks may be advantageously used for such a therapy owing to the cumulative effect of HA, the viscoelastic properties resulting from host–guest complexation and possible inclusion of anti-inflammatory drugs in the free CD cavities.
Besides adamantane moieties, other molecules have also been used as guest compounds for the formation of CD inclusion complexes. It was found that the probability to stabilize a three-dimensional network was lower for the CD-dodecyl (C12) alkyl chain complex [39]. The effect observed when CHI-CD was added to CHI-C12 was thus completely different to that observed with CHI-AD. While transparent “gels” could be observed macroscopically in the latter case for total polymer concentrations 1.5-fold higher than the critical overlap concentration C* of the initial polysaccharide, no strengthening of the associative network formed by CHI-C12 due to its amphiphilic character was found in the former case. This different behavior may be related to the strength of the β-CD/AD and β-CD/C12 chain inclusion complexes which is significantly lower in the latter case (Ka (β-CD/sodium dodecyl sulfate) = 8150 M−1) [40].
It should be noted, however, that when a neutral polymer of β-CD (poly(β-CD-EPH)), obtained by polycondensation of β-CD and epichlorohydrin (EPH), was used as a cross-linking agent of alkylated derivatives of alginate showing associative properties, a strengthening of the associative network was demonstrated [41]. The differences observed between the two latter systems may be related to the presence of additional electrostatic repulsions for the CHI-CD/CHI-C12 which tend to destabilize the physical network.
As developed below, this β-CD polymer was used in combination with neutral hydrophobically modified polysaccharides (dextrans), giving rise to self-assembling systems whose properties are mainly controlled by the strength of the interaction between the alkyl group and β-CD. However, the randomly branched structure of poly(β-CD-EPH) did not allow one to precisely assess effects of the molecular architecture on the final properties of the macromolecular assemblies. Remarkable thickening effects were also demonstrated when neutral dimeric species of β-CD were combined with a solution of CHI-AD [42–44] (Fig. 6). It was shown in this case that a subtle change in the molecular architecture of such CD dimers dramatically changes their ability to form bridges between modified chitosan chains. Whereas β-CD dimers with two spacers (called β-CD duplexes) could significantly enhance the viscosity of a CHI-AD solution at a concentration of 2.5 g/L, dimeric species with only one linkage between the two CD cavities provided negligible thickening effect [42,45]. This is illustrated in Fig. 7 showing the variation of the viscosity as a function of the [CD]/[AD] ratio for dimeric species tethered with octamethylene chains. These different abilities to physically cross-link modified chitosan chains were related to different molecular flexibilities and inclusion properties of the CD dimeric species. Indeed, calorimetry titration measurements demonstrated that the binding of sodium adamantyl acetate (ADAc) by the β-CD duplex (Fig. 7B) is 11 times stronger than that by the dimer having only one C8 chain (Fig. 7A). This was attributed to a strengthening of hydrophobic interactions due to the presence of the two C8 hydrophobic chains connecting the CD cavities.
Schematic representation of dimeric species of β-CD forming physical bridges between hydrophobically modified polysaccharide chains.
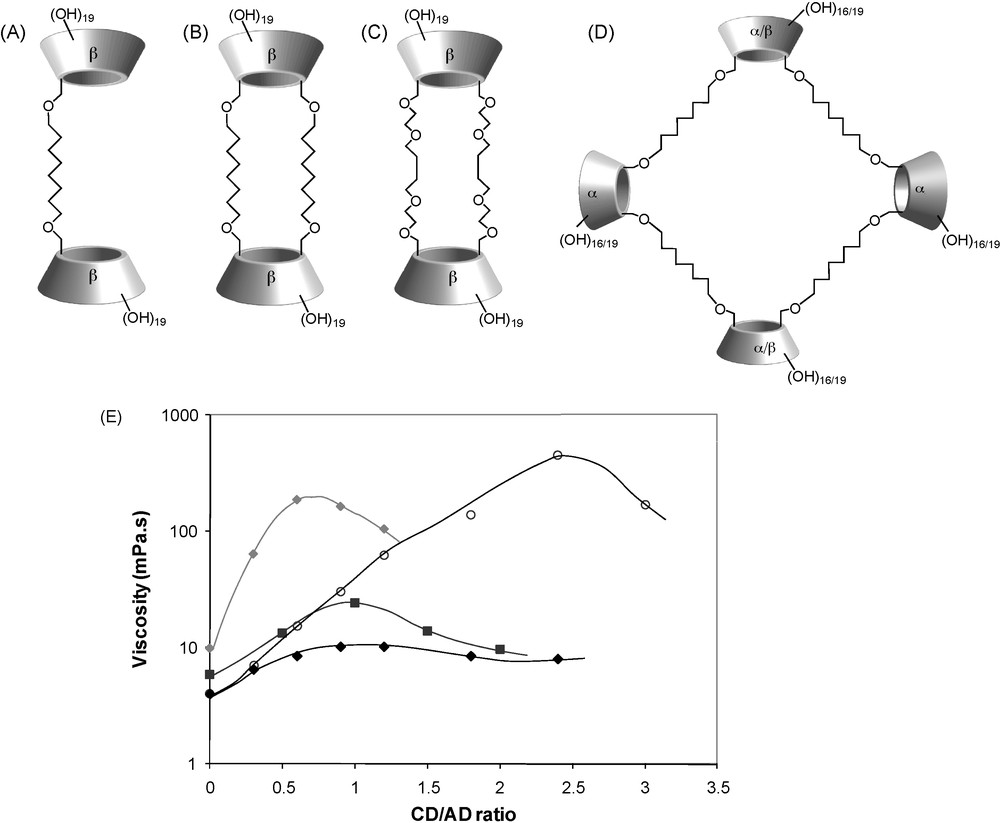
Dimeric and tetrameric species of cyclodextrin forming supramolecular assemblies by complexation with AD-grafted chitosan: (A) dimeric species tethered with one octamethylene chains; (B) duplex tethered with two octamethylene chains; (C) duplex tethered with two oligo(EO) spacer arms; (C) tetraplex; (D) variation of the viscosity of solutions of CHI-AD at a concentration of 2.5 or 3 g/L (0.3 M CH3COOH/0. 03 M CH3COONa, 25 °C) with the [CD]/[AD] ratio (i.e., with increasing concentration of CD oligomeric species) [(♦) tetraplex/CHI-AD 2.5 g/L, (■) duplex with oligo(EO) arms/CHI-AD 3 g/L, () duplex with C8 chains/CHI-AD 2.5 g/L], (♦) dimeric species with a C8 chain/CHI-AD 2.5 g/L (adapted from [44,45]).
Moreover, the duplex with C8 spacers led to higher viscosity values compared to its counterpart doubly linked with hydrophilic arms (Fig. 7C). This was also related to the greater binding strength of the CD cavities of the former toward adamantane than that of the latter and possibly, to the formation of aggregates promoted by inclusion of the octamethylene spacer in the CD cavity. Promiscuity of many CD cavities resulting from aggregation of such duplexes may enhance cross-linking efficiency due to multivalent association with the polymer. As can be seen on Fig. 7E, there is an optimal [CD]/[AD] ratio viscosity beyond which the zero-shear viscosity decreases. Such a decrease can be attributed to a lower probability of effective interchain cross-links as a result of increasing duplex monocomplexation. The same tendency was also observed in a reverse situation, i.e., when CHI-CD was combined with AD-modified poly(ethylene glycol)s [32].
These CD duplexes were synthesized according to an original methodology relying on an efficient regioselective deprotection reaction of perbenzylated CDs and giving access to CDs bearing two [43] or three [46–51] new functionalities on their primary rim in good overall yields. The relevant choice of benzyl protecting groups additionally allows the use of modern efficient chemical reactions in apolar solvents and standard silica-gel flash chromatography. The methodology giving access to difunctionnal CDs also allowed one to prepare a cyclic tetramers β-CD, called β-CD tetraplex (Fig. 7D). Addition of β-CD tetraplex to a solution of CHI-AD lead to a spectacular enhancement of the viscosity, in the same range of magnitude as for duplex with C8 chains, and much higher than with duplex containing oligo(ethylene oxide) (oligo(EO)) spacer arms (Fig. 7). The highest viscosity value observed for tetraplex with respect to the [CD]/[AD] ratio is not quite as high as that obtained with duplex with C8 chains, but it appears at a much lower [CD]/[AD] ratio, reflecting the better efficiency of tetraplex as a cross-linking agent. Similarly to duplexes, the observed decrease of the viscosity beyond the optimal [CD]/[AD] ratio can be attributed to an increase of tetraplex monocomplexation to the detriment of di- or multi-complexation.
All these results thus showed that these supramolecular complexation-based assembly systems are significantly affected by the structures of the small host molecules.
Alternatively, neutral hydrophobically modified polysaccharides (dextrans) were physically cross-linked with synthetic charged host polymers. Isothermal titration calorimetry experiments demonstrated that the two polymers, i.e., dextran modified with adamandane groups (DEX-ADA) and a synthetic polyacid bearing pendant β-CD units (P(MVE-MA)-g-βCD), interact mainly via inclusion complexes between adamatane groups and cyclodextrin cavities [52]. However, additional interaction mechanisms, i.e., hydrogen bonds and electrostatic repulsions, were shown to participate in the cohesion of the networks. These interactions imparted to the networks pH-sensitive properties. Interestingly, when these networks were loaded with an apolar drug, benzophenone (BZ), slow releases were obtained (10–12 days) with slower kinetics at pH 2 than at pH 7. The lower kinetics at low pH was related to the presence of additional interactions between the two polymer partners through hydrogen bonds, leading to a strengthening of the associative network as shown by pH-dependent rheological properties. Analysis of the experiment at pH 7 showed that drug release is controlled both by diffusion in the network and inclusion complex interactions with CD cavities.
2.2 Specific interaction in modified neutral polysaccharide systems
Investigation of the potential of such networks as drug delivery devices were also performed from neutral systems prepared from poly(β-CD-EPH) and dextran grafted with dodecyl side chains (DEX-C12) [53]. The formation of two phases was observed after combining these two polymers in aqueous solutions. At least 90% of the total amount of polymer introduced associated in a viscous phase (named gel), which is at the bottom in the vial shown in Fig. 8. It should be noted that this associative phase separation phenomenon was closely dependent on the molar mass of poly(β-CD-EPH). Indeed, when low molar mass poly(β-CD-EPH) bearing a few CDs per chain was mixed with dextran grafted with alkyl chains, soluble complexes were obtained [54]. Moreover, the extension of the two phase domain was dependent on the alkyl chain length and the grafting ratio [54].
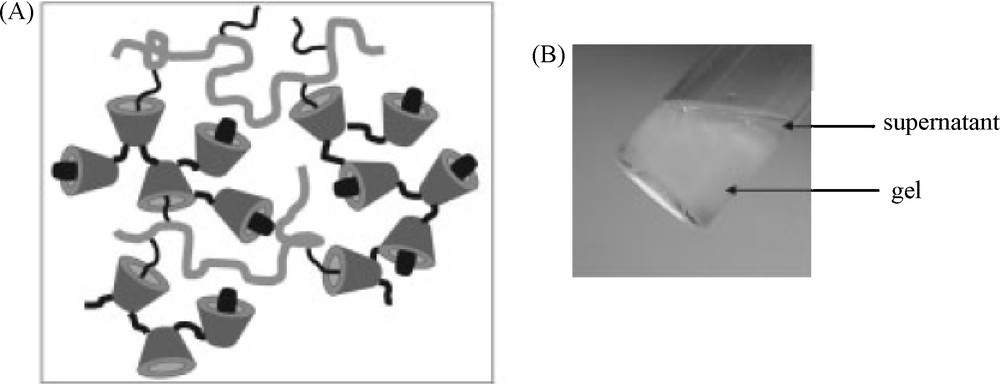
Self-assembling hydrogels based on CD complexation: (A) schematic representation of the formation of DEX-C12/poly(β-CD-EPH) networks (DEX-C12/poly(β-CD-EPH) ratio 50/50 (w/w)). Some alkyl chains of DEX-C12 are complexed into the CD cavities of the poly(β-CD-EPH), leaving also free CDs for the inclusion of hydrophobic drugs; (B) Photographs of the formation of a DEX-C12/poly(β-CD-EPH) gel (adapted from [53]).
Compared to the networks based on CHI and HA, the viscous properties prevailed in these “neutral gels”. Indeed, the latter gels were characterized by a loss modulus higher than the storage modulus over the range of frequencies investigated. The differences observed between the rheological behaviors of these assemblies may be partly related to the strength of the β-CD/AD and β-CD/C12 chain inclusion complexes stabilizing the systems, as mentioned above, and the intrinsic properties of the polysaccharide chains (polymer chain stiffness reflected by the intrinsic backbone persistence length (Lp) and polyelectrolyte character).
Interestingly, observation of the neutral gels by transmission electron microscopy (TEM) after freeze-fracturing suggested that they were composed of microgels consisting of small aggregates of associative particles connected through C12/CD complexes. In view of the possible application of DEX-C12/poly(β-CD-EPH) gels as injectable drug delivery systems, DEX-C12/poly(β-CD-EPH) gels were submitted to oscillatory measurements after their passage throughout a syringe connected to an 18-gauge needle, classically used for intramuscular and subcutaneous injections. Similar rheological behaviors were found for the “injected” and “noninjected” gels, suggesting that the associative network should have the same structure at the site of injection. In addition, the hydrophobic compounds, the sunscreen agent benzophenone (BZ) and the anticancer drug tamoxifen (TM), could be successfully incorporated into these gels with high loading efficiencies (> 90%). The BZ loading was more than 20-fold higher than the TM loading, which was attributed to the stronger affinity of BZ for the CD cavity (Ka(β-CD/BZ) = 1900 M−1) [55] than TM (Ka(β-CD/TM) = 1400 M−1) [56]. Thus, the presence of BZ molecules appeared to modify the network structure by competitive complexation with the CD grafted molecules. Both entrapped compounds were released in a sustained manner in vitro. However, the gels loaded with BZ exhibited a much longer release (complete release achieved after 16 days) than the TM-loaded gels. These phenomena were attributed to drug diffusion coefficients in the gel, competition between drug and alkyl chains for the CD cavities, and partition of the drug between the gel phase and the supernatant.
3 Self-assembled nanogels based on host–guest cyclodextrin inclusion complex
In the past few decades, submicronic polymeric particles have attracted considerable attention as potential drug delivery devices for the controlled release of active molecules and targeting [57–61]. However, the preparation methods of such particles require the use of potentially toxic organic solvents and surfactants, which is often not acceptable, at least for parenteral administration. Therefore, to overcome these technological issues, new self-assembling nanogels/nanoassemblies were developed avoiding the use of organic solvents and surfactants [4]. They consist of a hydrophilic polymer backbone, on which hydrophobic moieties are grafted. Among these associative polymers, hydrophobized polysaccharides, such as pullulan, cellulose derivatives, dextran and chitosan [62] are particularly attractive due to their biocompatibility, low toxicity and additionally biodegradability for some of them, which are advantageous for biological and pharmaceutical applications.
While mixtures of hydrophobically modified dextrans with poly(β-CD-EPH) formed macroscopic hydrogels at high polymer concentrations [41,53–54], Gref et al. showed the ability to obtain self-assembled nanogels by mixing diluted aqueous solutions of dodecyl-modified dextran and poly(β-CD-EPH) (Fig. 9) [63–68]. The feasibility and stability of those nanogels (NG) were found to be dependent on several parameters including the degree of substitution of DEX-C12, the polymer concentration Cp, the molar mass of poly(β-CD-EPH) and the weight ratio between DEX-C12 and poly(β-CD-EPH). In particular, key parameters were: DS ≥ 4%, Cp < 10 g/L, β-CD-EPH Mw > 106 g/mol, and weight ratio DEX-C12/poly(β-CD-EPH) R = 1.
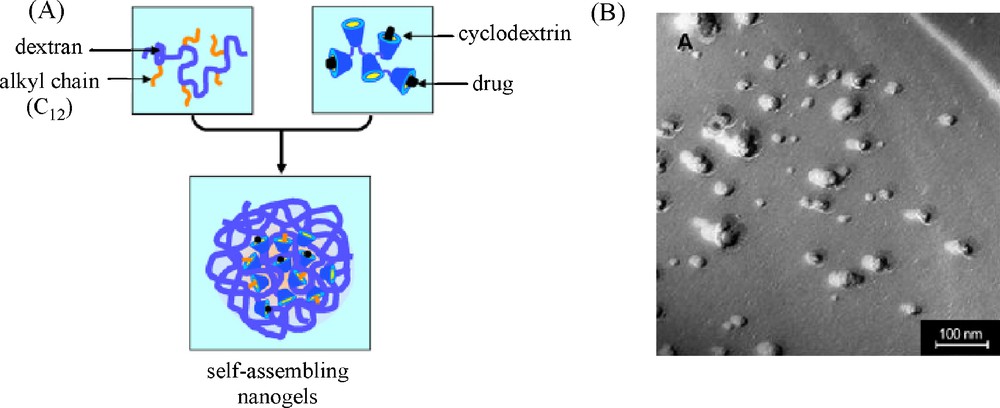
Synthesis of self-assembling DEX-C12/poly(β-CD-EPH) nanogels: (A) NG are spontaneously obtained by mixing the solutions of the two complementary polymers; (B) photomicrographs of a DEX-C12/poly(β-CD-EPH) nanogel suspension (10 g/L) taken by transmission electron microscopy after freeze-fracture 2 h after preparation (adapted from [65]).
Indeed, it was shown by 1H NMR that for such a ratio, all the C12 chains were included in the CD cavities allowing remarkable stability of the nanogels. The latter have a diameter of about 120–150 nm whatever the concentration of the two polymer solutions. However, the diameter tended to increase upon time for high polymer concentrations (10 mg/mL), due to particle aggregation and/or fusion. It should be noted that the stability of the suspensions could be improved by their storage at 4 °C as complex formation is favored at low temperature. Interestingly, nanoassemblies could be directly freeze-dried and reconstituted in water without any change in size, which allows unlimited storage. As for R = 1, all alkyl chains were included in the CD cavities while leaving about half of the latter empty, hydrophobic drugs of appropriate size could be loaded in the nanogels. Tamoxifen and benzophenone were thus successfully incorporated in the nanogels by adding these compounds to the DEX-C12 and poly(β-CD-EPH) solutions before mixing them together [64,66,67]. Although the model drugs competed with the dodecyl chains for the CD cavities, the formation of nanoassemblies was not dramatically impaired. However, the suspensions of nanoassemblies loaded with TM were very unstable. In the first five minutes after their formation, they fused together forming a gel deposit contrary to the nanogel particles containing BZ which remained stable upon storage at room temperature or 37 °C. BZ molecules were retained in the particles upon storage but were quickly released upon dilution of the nanoassemblies in water. This work thus demonstrated potential applications of the nanogels in the cosmetic field, as sunscreen carriers. Based on these results, these nanoassemblies were next exploited for the targeted delivery of galodinium (GdIII) complexes [63]. In order to optimize the entrapment of the GdIII chelate in the DEX-C12/poly(β-CD-EPH) nanoparticles, the former was functionalized by adamantane allowing complexation with the free CD cavities in the nanogels. A sharp enhancement of the relaxivity was observed from 5.2 mM−1 s−1 at 20 MHz and 37 °C for the chelate itself to 48.4 mM−1 s−1 once the adducts are formed with DEX-C12/poly(β-CD-EPH). These supramolecular nanoassemblies with incorporated gadolinium (GdIII) complexes thus show promising applicability as magnetic resonance imaging (MRI) contrast agent.
4 Synthesis of stimuli-responsive nanometric films based on the multilayer assembly of host and guest biopolymers
Recently, our group demonstrated the ability to obtain multilayer films by alternate deposition of host and guest polysaccharides on a guest surface [69]. These assemblies may be sensitive to various stimuli which can act either on the electrostatic interactions between the polymer chains or on the host–guest complexes stabilizing the multilayers. The latter thus show distinct properties compared to multilayers build-up from oppositely charged polyelectrolytes [70]. The study of the latter assemblies has met an important success over the last 10 years due to the ease of their construction and their numerous potential applications. These range from optical coatings, to filtration, biosensors to tissue engineering. These films with controlled thickness (ranging from the nanometer to micrometer scale as a function of the number of deposited layers), constitute ideal substrates for cell adhesion and growth [11].
The multilayers based on host–guest complexation were prepared from the alternate deposition of chitosan derivatives bearing either cyclodextrin or adamantane molecules, on a planar surface functionalized by AD molecules. The stability of these films relies on the multivalent CD/AD complexation process, occurring after each step of the construction. Our results indicated that the film growth is mainly governed by the availability of the complexation sites offered by each layer. Thus, it was limited to three layers (Fig. 10). These results highlight the interest of better understanding the growth mechanism for these films. Taking advantage that complexation between ferrocene molecule (Fc) and β-CD is strongly diminished upon electrochemical conversion of Fc to the ferrocenium cation [71], our group recently investigated the formation of LBL assembly made of Fc-modified chitosan and CHI-CD and their response to the application of an electric field. Our preliminary results demonstrated the ability of these self-assembled films to detach from the surface in response to the electrochemical oxidation of the Fc groups [72]. This demonstrates the potential of using these self-assembled polysaccharide films as sacrificial substrates for applications in tissue engineering.
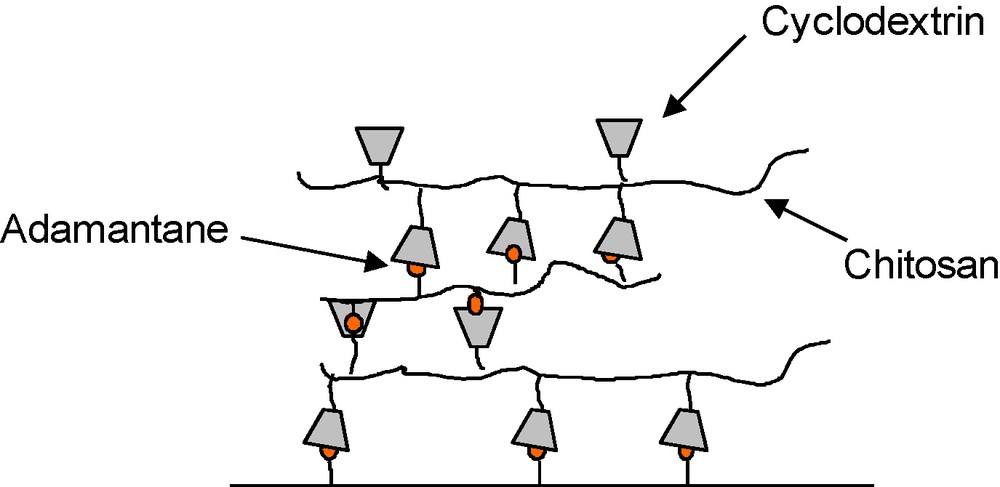
Schematic representation of a planar multilayer film made of host and guest polysaccharides.
5 Conclusion
Self-assembling polysaccharide systems based on cyclodextrin complexation are relatively new materials that have been developed over the past 10 years. Some studies related to drug encapsulation inspired interesting development of these supramolecular biomaterials for drug delivery. Physical hydrogels based on the self-assembly of host and guest biopolymers could be used as promising injectable drug delivery systems for sustained controlled release of drugs. In the case of hyaluronic acid, such assemblies may find useful applications in viscosupplementation, owing to the cumulative effects of transport properties of cyclodextrins for bioactive molecules (anti-inflammatory drugs in particular), viscoelasticity of the resulting network and bioactivity of HA. Moreover, it was shown that under particular conditions, gel nanoparticles could be obtained, opening new potential applications in the cosmetic and pharmaceutical fields. The development of such assemblies on surfaces is an emerging area, which still faces many challenges in future studies. Such systems may impact the area of tissue engineering. Although a number of proof-of-concept studies have been demonstrated, smarter materials designs are expected for a better balance of higher functions and performance.