

1 Introduction
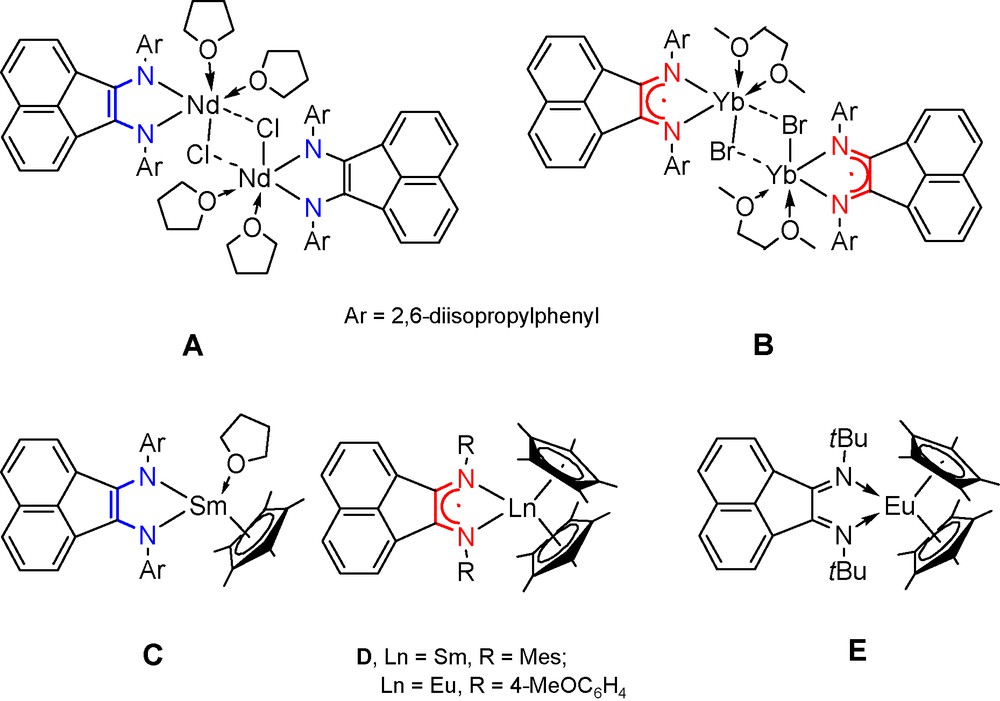
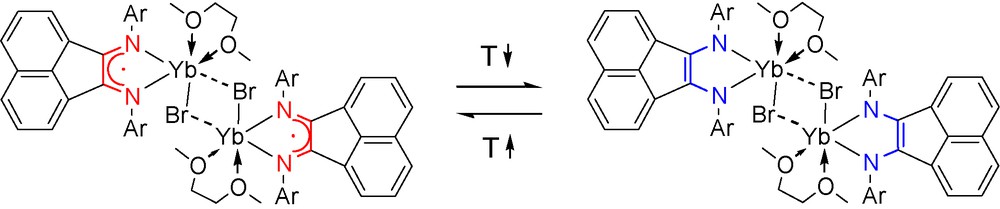
Electronic and spatial features of 1,2-bis(arylimino)acenaphthenes (BIANs) provide for the formation of metal complexes that reveal remarkable molecular structures and reactivity. The most helpful ligand of the 1,2-bis(arylimino)acenaphthene family is 1,2-bis[(2,6-diisopropylphenyl)imino]acenaphthene (dpp-BIAN). The alkali metal reduction of dpp-BIAN results in the formation of its mono-, di-, tri-, and tetra-anions [1]. The redox-activity of dpp-BIAN has been further documented by the preparation of a series of group II [2] and XIII [3] metal complexes with the dpp-BIAN ligand in the radical-anionic and dianionic state. A shielding of the nitrogen lone pairs in dpp-BIAN by isopropyl groups of the phenyl substituents prevents the formation of dimeric structures with bridging dpp-BIAN ligands. This peculiarity of the dpp-BIAN ligand plays a crucial role in the formation of molecular species with direct metal-metal bonds, e.g. Zn–Zn [4], Ga–Ga [5], Zn–Ga [5], Li–Ga [6] and Na–Ga [6]. It is also suggested that the nitrogen lone pairs of the dpp-BIAN may contribute not only in the metal-nitrogen bonding (π-component), but also in the metal-metal bonding. A review concerning development of the coordination chemistry of BIAN ligands with s- and p-block elements has been published recently [7]. By comparison, lanthanide complexes with BIAN ligands have been studied much less and are limited by 13 compounds. Some of them are presented in Scheme 1 showing the structures A [8], B [9], and C–E [10]. Within the lanthanide series, the ytterbium derivative [(dpp-BIAN)Yb(μ-Br)(DME)]2 (B) is a sole example that exhibits a thermally induced reversible metal-to-ligand electron transfer in solution – redox isomerism phenomenon (Scheme 2) [9]. This phenomenon is characterized by existence of two electronic isomers with different charge distribution. The low temperature isomer is always represented by species with a more oxidized metal ion Mn+1 and the high temperature isomer consists of the reduced metal ion Mn. Originally, such electron transfer was observed in the bis(o-benzosemiquinone)cobalt complex [11]. In order to get more insight in the redox-isomeric transformation involving the rare earth element, we started preparation of the ytterbium complex with two dpp-BIAN ligands. Although we have not succeed the preparation of desired neutral ytterbium complex (dpp-BIAN)2Yb, a series of anionic bis(dpp-BIAN)lanthanide complexes have been synthesized and characterized by single crystal X-ray analysis. Indeed, the neutral samarium derivative (dpp-BIAN)2Sm has been isolated and studied by X-ray crystallography.
2 Results and discussion
2.1 Syntheses and characterization of the newly prepared compounds
The reaction of (dpp-BIAN)Yb(DME)2 (1) [9] with dpp-BIAN in DME proceeds slowly at reflux and is accompanied by a color change from red-brown to blue-green. When equimolar amounts of the reagents were used even after attainment of the final blue-green color, a remarkable amount of free dpp-BIAN is still present in the reaction mixture. In toluene, the reaction of 1 with 0.5 molar equivalent of dpp-BIAN completes within 1 h at reflux and affords complex [(dpp-BIAN)2Yb][(dpp-BIAN)Yb(DME)2] (2) as deep blue crystals (Scheme 3).
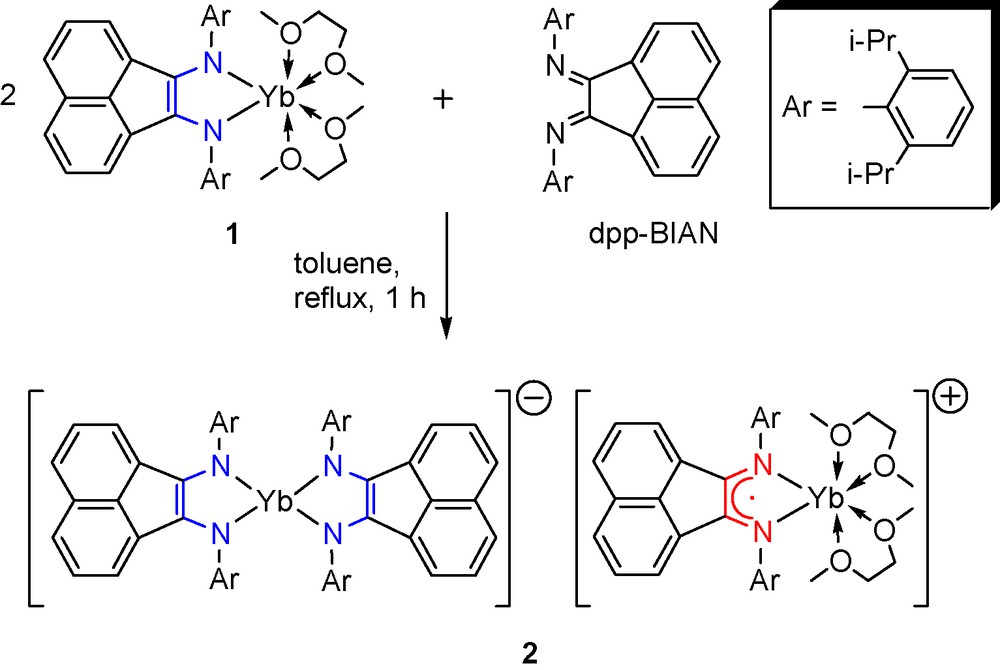
Since in the course of the synthesis of compound 1 from ytterbium metal and dpp-BIAN in DME at reflux the reaction mixture turned first blue-green and then red-brown, we suggest that reduction of dpp-BIAN with ytterbium proceeds via intermediate formation of complex 2, which with an excess of the metal gives compound 1.
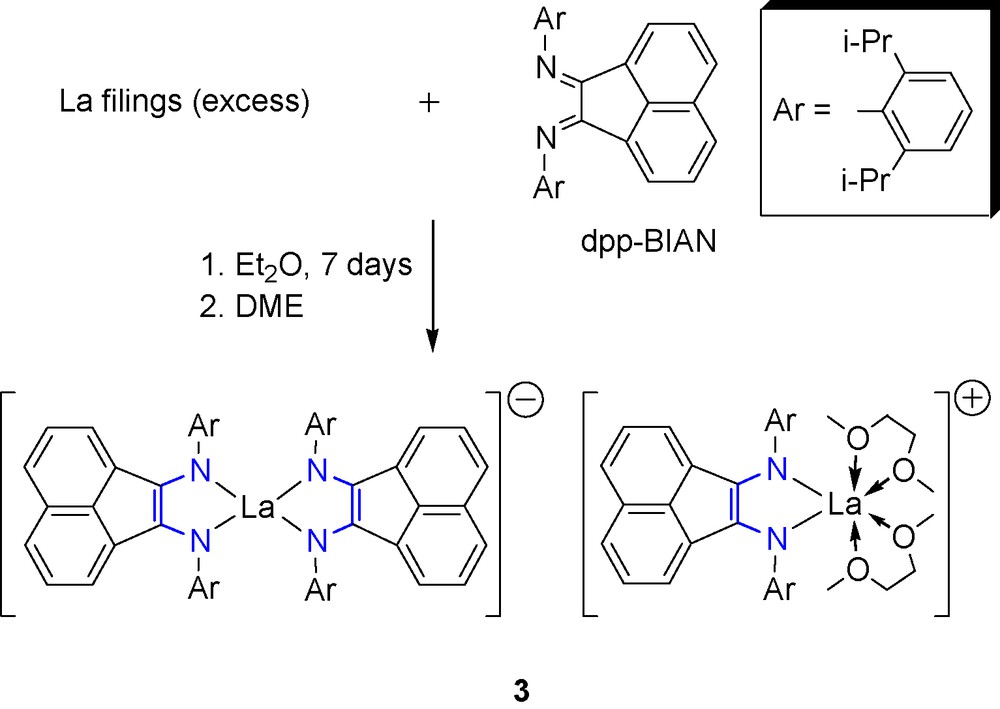
In order to obtain the analogous complex with lanthanum, for which, in contrast to ytterbium, the oxidation state +3 is common, dpp-BIAN was reacted with an excess of lanthanum metal in diethyl ether. At ambient temperature, the reaction proceeds slowly and affords blue microcrystalline powder after stirring the reaction mixture for 7 days. Extraction of the product with DME and crystallization from toluene affords complex [(dpp-BIAN)2La][(dpp-BIAN)La(DME)2] (3) as deep green crystals (Scheme 4).
The presence of the paramagnetic dpp-BIAN radical-anion in complex 2 prevents its characterization by NMR spectroscopy and, on the other hand, the ESR spectroscopy is useless for characterization of complex 2 due to the presence of trivalent ytterbium. Unexpectedly, the 1H NMR spectrum of diamagnetic compound 3 in THF-d8 is also less informative: a broadening of all of the signals may be indicative for dynamic processes in metal coordination spheres. Due to the low solubility of compound 3 in benzene, no useful NMR data could be acquired for complex 3 in C6D6. The IR spectra of 2 and 3 are very much alike, but also do not provide for enough information on their structure. Therefore, the X-ray single crystal analyses of compounds 2 and 3 were carried. The crystallographic data (vide infra) show that despite the same stoichiometry the oxidation states of the metals and the reduction states of the dpp-BIAN ligands in the cationic parts of 2 and 3 are different.
In order to prepare a lanthanum complex with the lanthanum to dpp-BIAN ratio one-to-one LaI3(Et2O)n was reacted with 1 molar equivalent of dpp-BIAN and 3 molar equivalents of potassium metal. In this way, we intended to prepare the lanthanum derivative with lanthanum-lanthanum bond. In diethyl ether at rigorous stirring at ambient temperature potassium dissolved completely within 48 h. Simultaneously, a green microcrystalline solid precipitated. Extraction with Et2O allowed separation of the green product from KI. Crystallization from Et2O afforded complex [(dpp-BIAN)2La][K(Et2O)4] (4) as green needle-like crystals (Scheme 5).

The composition of the complex 4 does not correspond to the reaction stoichiometry since potassium is not completely eliminated as KI. Probably some amount of LaI3(Et2O)n is left unreacted in the mixture. In other words, the reaction of complex 4 with LaI3(Et2O)n does not proceed, or occurs too slowly. The 1H NMR spectrum of the diamagnetic compound 4 in C6D6 (Fig. 1) reveals four signals of methine protons of the isopropyl substituents (septets centered at δ 4.12 (2 H), 4.04 (2 H), 3.85 (2 H), 2.94 (2 H) ppm). In the free dpp-BIAN, the protons of methyl groups in a given isopropyl substituent are not equivalent due to the restricted rotation around C(methine)-C(ipso) bond. Therefore the 1H NMR spectrum of the free dpp-BIAN consists of one septet (4 methine protons) and two doublets (each 12 methyl protons). When four non-equivalent methine protons are observed, one may expect the appearance of the eight doublets of the methyl protons. In fact in the 1H NMR spectrum of compound 4 only three individual doublets are observed. Other five doublet signals corresponding to the methyl groups are positioned in the narrow range 1.2–1.0 ppm. Moreover, these five doublets overlap with the signals of diethyl ether.

1H NMR spectrum of complex 4 in C6D6 (200 MHz, 293 K).
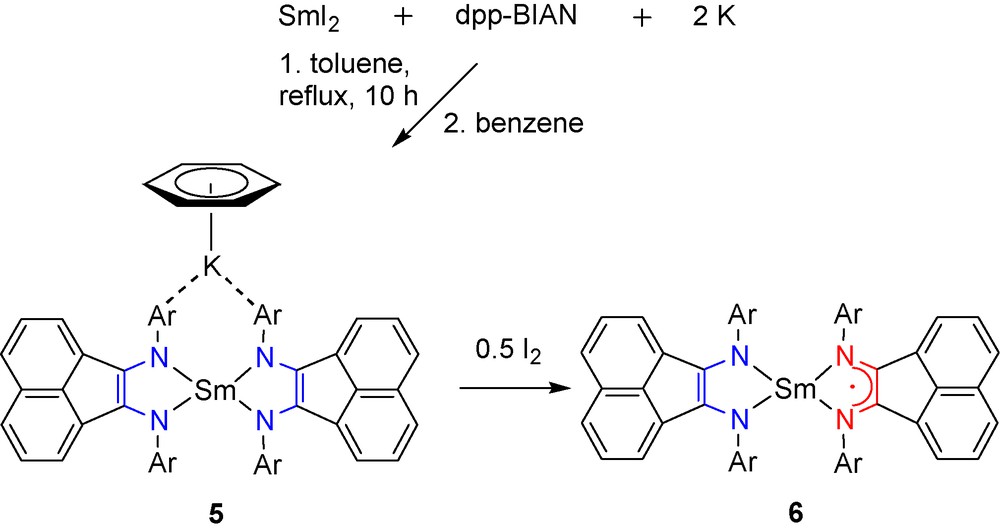
The one-pot reaction of solvent-free SmI2 with potassium (2 molar equivalents) and dpp-BIAN (1 molar equivalent) in toluene at reflux within several hours is accompanied with a color change to deep blue. Crystallization of the crude product from benzene affords compound [(dpp-BIAN)2Sm][K(C6H6)] (5) as deep blue almost black crystals (Scheme 6). Again, the potassium is present in the isolated product, although the starting reagents were taken in an exact stoichiometry. Despite the presence of the paramagnetic Sm(III) center in complex 5, the 1H NMR spectrum is valuable for its characterization. As for complex 4, the 1H NMR spectrum of 5 consists of four methine proton septets, although these are located in a rather wide range (δ 10.99 (2 H), 8.72 (2 H), –6.18 (2 H) and –11.87 (2 H) ppm). The paramagnetic influence of the samarium center causes the appearance of 12 signals of the methyl protons (see experimental section). The benzene molecule present in the complex 5 is probably exchanged with the C6D6 and thus its coordination to potassium could not be observed in the 1H NMR spectrum. Oxidation of complex 5 with 0.5 molar equivalent of iodine affords neutral and solvent-free bis(diimine)samarium complex (dpp-BIAN)2Sm (6) (Scheme 6) isolated from toluene as plate-like deep blue crystals.
2.2 Molecular structures of 2–6
The molecular structures of compounds 2, 3, 4, 5 and 6 were determined by single crystal X-ray diffraction and are depicted in Figs. 2–6, respectively. Although the data for 4 are poor the geometric parameters of molecules 4 are used in discussion since these parameters are rather conform to those of other complexes described here. In the solid state complexes 2 and 3 consist of separated anions [(dpp-BIAN)2Ln] and cations [(dpp-BIAN)Ln(DME)2] (Ln = Yb, 2; Ln = La, 3).
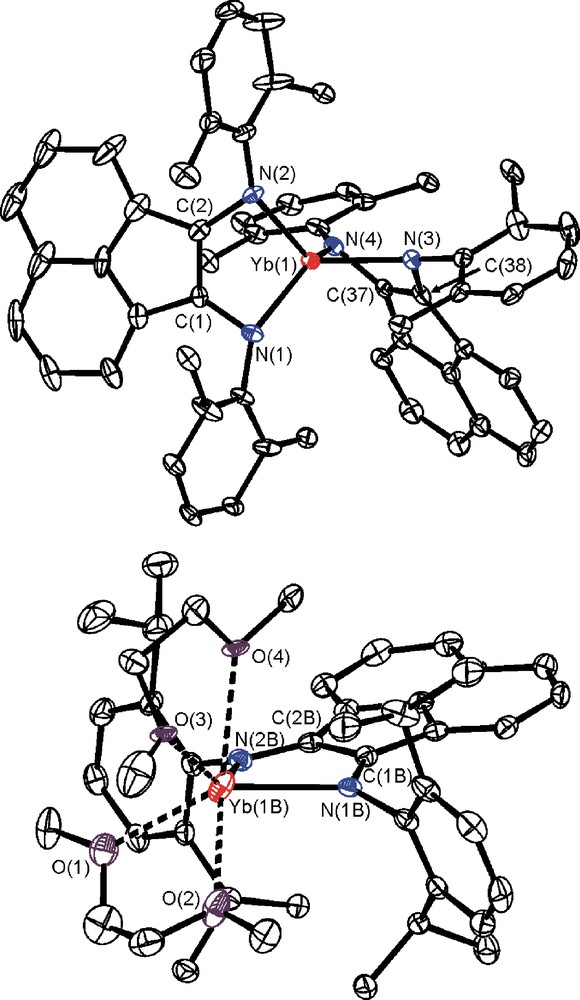
Molecular structure of 2: top – anion [(dpp-BIAN)2Yb], bottom – cation [(dpp-BIAN)Yb(DME)2]. The hydrogen atoms as well as the CH3 groups in the anion are omitted for clarity. Selected bond lengths [Å]: Yb(1)–N(1) 2.194(3), Yb(1)–N(2) 2.198(3), Yb(1)–N(3) 2.191(2), Yb(1)–N(4) 2.206(2), Yb(1)–C(1) 2.614(3), Yb(1)–C(2) 2.609(3), Yb(1)–C(37) 2.624(3), Yb(1)–C(38) 2.629(3), N(1)–C(1) 1.386(4), N(2)–C(2) 1.401(4), N(3)–C(38) 1.412(3), N(4)–C(37) 1.405(4), C(1)–C(2) 1.410(5), C(37)–C(38) 1.397(3), Yb(1B)–N(1B) 2.387(2), Yb(1B)–N(2B) 2.403(2), Yb(1B)–O(1) 2.479(3), Yb(1B)–O(2) 2.421(3), Yb(1B)–O(3) 2.520(3), Yb(1B)–O(4) 2.546(3), Yb(1B)–C(1B) 3.127(3), Yb(1B)–C(2B) 3.135(3), N(1B)–C(1B) 1.333(3), N(2B)–C(2B) 1.342(4), C(1B)–C(2B) 1.449(4).
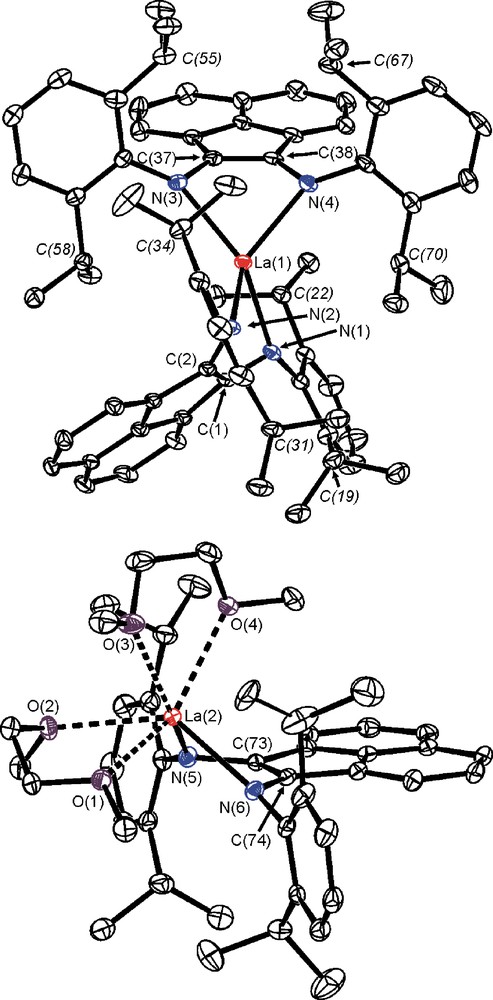
Molecular structure of 3: top – anion [(dpp-BIAN)2La], bottom – cation [(dpp-BIAN)La(DME)2]. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å]: La(1)–N(1) 2.396(2), La(1)–N(2) 2.386(2), La(1)–N(3) 2.403(2), La(1)–N(4) 2.382(2), La(1)–C(1) 2.775(3), La(1)–C(2) 2.792(3), La(1)–C(37) 2.792(2), La(1)–C(38) 2.790(3), N(1)–C(1) 1.397(3), N(2)–C(2) 1.393(4), N(3)–C(37) 1.401(3), N(4)–C(38) 1.393(3), C(1)–C(2) 1.420(4), C(37)–C(38) 1.401(4), La(2)–N(5) 2.307(2), La(2)–N(6) 2.300(2), La(2)–C(73) 2.773(2), La(2)–C(74) 2.757(2), La(2)–O(1) 2.6379(19), La(2)–O(2) 2.599(2), La(2)–O(3) 2.6125(19), La(2)–O(4) 2.5547(19), N(5)–C(73) 1.410(3), N(6)–C(74) 1.405(3), C(73)–C(74) 1.402(3).

Molecular structure of 4. The hydrogen atoms as well as cation [K(Et2O)4]+ are omitted for clarity. Selected bond lengths [Å]: La(1)–N(1) 2.375(6), La(1)–N(2) 2.391(6), La(1)–N(3) 2.385(5), La(1)–N(4) 2.391(5), La(1)–C(1) 2.820(8), La(1)–C(2) 2.818(7), La(1)–C(37) 2.807(6), La(1)–C(38) 2.796(7), N(1)–C(1) 1.407(9), N(2)–C(2) 1.379(9), N(3)–C(37) 1.408(8), N(4)–C(38) 1.407(8), C(1)–C(2) 1.439(10), C(37)–C(38) 1.402(9).

Molecular structure of 5. The hydrogen atoms as well as the CH3 groups of isopropyl substituents are omitted for clarity. Selected bond lengths [Å]: Sm(1)–N(1) 2.3110(10), Sm(1)–N(2) 2.2976(9), Sm(1)–C(1) 2.6634(11), Sm(1)–C(2) 2.6828(10), K(1)–C(25) 3.1705(11), K(1)–C(26) 3.2166(11), K(1)–C(30) 3.4217(12), K(1)–C(1S) 3.3153(16), K(1)–C(2S) 3.3198(15), K(1)–C(3S) 3.3205(16), N(1)–C(13) 1.3936(15), N(1)–C(1) 1.4007(13), N(2)–C(2) 1.3906(15), C(1)–C(2) 1.4092(16).

Molecular structure of 6. The hydrogen atoms as well as the CH3 groups of isopropyl substituents are omitted for clarity. Selected bond lengths [Å]: Sm(1)–N(1) 2.2495(11), Sm(1)–N(2) 2.2589(9), Sm(1)–N(3) 2.4411(10), Sm(1)–N(4) 2.4329(9), Sm(1)–C(1) 2.6655(11), Sm(1)–C(2) 2.6892(10), Sm(1)–C(37) 2.9963(11), Sm(1)–C(38) 2.9868(11), N(1)–C(1) 1.4036(16), N(2)–C(2) 1.3865(16), C(1)–C(2) 1.4053(18), N(3)–C(37) 1.3383(16), N(4)–C(38) 1.3383(16), C(37)-C(38) 1.457(2).
A ligand arrangement in the anions [(dpp-BIAN)2Ln]− in both cases is very similar: metal atoms coordinate in a distorted tetrahedral fashion four nitrogen atoms of two dpp-BIAN ligands. The dihedral angles between planes N(1)-Ln-N(2) and N(3)-Ln-N(4) in 2 and 3 are 72 and 88°, respectively. A coordination number of four is rather unusual for the rare earth elements because of their large ionic radii and the presence of vacant 5d-, 6s- and 6p-orbitals. A lack of coordinated solvent molecules in anions [(dpp-BIAN)2Ln]− is explained by effective shielding of the metal coordination spheres by bulky 2,6-diisopropylphenyl groups. The distances between La(1) and four methine protons attached to the carbon atoms C(22), C(34), C(58) and C(70) lie in the range 2.85–3.01 Å. In the anions [(dpp-BIAN)2Ln]−, the Yb-N and La-N distances fall into narrow ranges (2: 2.191(2)–2.206(2); 3: 2.382(2)–2.403(2) Å). The difference in Yb–N(anion) and La–N(anion) distances (0.2 Å) corresponds well to the difference in the ionic radii of La(III) and Yb(III) (1.032 and 0.868 Å for six-coordinate metal ions, respectively) [12]. In contrast to other rare earth metal complexes of the dpp-BIAN dianion, the metal atoms in [(dpp-BIAN)2Ln]− deviate significantly from the planes formed with atoms N(1)-C(1)-C(2)-N(2) and N(3)-C(37)-C(38)-N(4) (2: av. 1.26 Å; 3: 1.49 Å), cf. (dpp-BIAN)Yb(DME)2 (0.43 Å) [9] and [(dpp-BIAN)NdCl(THF)2]2 (0.43 Å) [8]. An alteration of the C-C and C-N bond lengths within the metallacycles in [(dpp-BIAN)2Ln]− (Ln = Yb, 2; Ln = La, 3) is indicative of the dianionic character of both dpp-BIAN ligands in these complexes: the N(1)-C(1), N(2)-C(2), N(3)-C(37) and N(4)-C(38) bonds (2: av. 1.401 Å; 3: 1.396 Å) are elongated compare to those bonds in neutral dpp-BIAN (both 1.282 Å) [13] as well as in its radical-anion coordinated to a metal, e.g. [(dpp-BIAN)YbBr(DME)]2 (av. 1.346 Å) [8].
Cations [(dpp-BIAN)Yb(DME)2]+ and [(dpp-BIAN)La(DME)2]+ in 2 and 3 represent a six-coordinate species. A first glance at their geometries reveals that the deviation of ytterbium atom from the diimine plane is not so pronounced as for lanthanum atom. Thus, the lanthanum atom is positioned 1.29 Å out of the plane N(5)-C(73)-C(74)-N(6), whereas deviation of the ytterbium atom from the plane N(1B)-C(1B)-C(2B)-N(2B) is only 0.53 Å. Furthermore, the La-N distances in [(dpp-BIAN)La(DME)2]+ (2.307(2) and 2.300(2) Å) are much shorter than the Yb-N distances in [(dpp-BIAN)Yb(DME)2]+ (2.387(2) and 2.403(2) Å). On contrary, the La–O(DME) distances are significantly longer compare to Yb-O(DME) bond lengths. One may suggest that oxidation states of metal atoms in [(dpp-BIAN)Yb(DME)2]+ and [(dpp-BIAN)La(DME)2]+ are different: Yb(II) vs. La(III). The Yb-N bond lengths in [(dpp-BIAN)Yb(DME)2]+ (av. 2.395 Å) are longer than in isostructural six-coordinate ytterbium complex of dpp-BIAN dianion (dpp-BIAN)Yb(DME)2 (2.355 Å) [9], but shorter than in [(dpp-BIAN)YbBr(DME)]2 (2.428 Å), which consists of dpp-BIAN radical-anion [9]. Thus, we conclude that cation [(dpp-BIAN)Yb(DME)2]+ in 2 consists of divalent ytterbium and the dpp-BIAN radical-anion, whereas the isostructural cation [(dpp-BIAN)La(DME)2]+ in 3 is composed from trivalent lanthanum and dpp-BIAN dianion. The radical-anionic character of the diimine ligand in 2 is further documented by an alteration of the bond distances within the diimine fragment. Thus, the N(1B)–C(1B) (1.333(3) Å) and N(2B)–C(2B) (1.342(4) Å) distances are shorter than corresponding distances in the anion [(dpp-BIAN)2Ln]− (av. 1.401 Å) and in the neutral (dpp-BIAN)Yb(DME)2 (av. 1.385 Å) [9], but close to those in complex [(dpp-BIAN)YbBr(DME)]2 (av. 1.346 Å) [9]. It is also worth mentioning that the interaction of the anionic dpp-BIAN ligands (radical-anionic as well as dianionic) to the metal in the cationic species is stronger than in neutral/anionic complexes. For example, despite of a higher coordination number of the metal in the cation [(dpp-BIAN)La(DME)2]+ the La-N bond distances are shorter (2.303 Å) than those values in the anion [(dpp-BIAN)2La]− (2.392 Å). Complex 4 consists of separated ions [(dpp-BIAN)2La]− and [K(Et2O)4]+. The geometry of the anion [(dpp-BIAN)2La]− in complex 4 is similar to that in complex 3.
The molecule of compound 5 (Fig. 5) consists of anion [(dpp-BIAN)2Sm]− and potassium cation. In contrast to compound 4 the potassium cation in 5 is coordinated to the anion. The molecule of 5 is located on the crystallographic axis, which runs via samarium and potassium atoms. Thus, the dpp-BIAN ligands in 5 are crystallographically equivalent. As expected, the Sm–N distances in 5 (av. 2.30 Å) are shorter than La–N distances in complex 3 (av. 2.39 Å) but longer than Yb–N distances in complex 2 (av. 2.20 Å). A diainionic nature of both dpp-BIAN ligands in 5 is confirmed by the elongated C–N and shortened C–C bonds in the diimine fragment. The potassium cation is incorporated into the anion [(dpp-BIAN)2Sm]− via coordination to two phenylaryl substituents of the dpp-BIAN ligand. Furthermore, potassium coordinates benzene molecule in a η6-fashion.
The neutral samarium complex 6 is isostructural with anions [(dpp-BIAN)2Ln]− (Ln = Yb, 2; La, 3 and 4; Sm, 5). For samarium, two oxidation states (+2 and +3) are known. On the other hand, in its complexes dpp-BIAN may be present either as neutral, radical-anionic or dianionic ligand. Therefore, one can imagine an existence of three redox-isomers of complex 6: (dpp-BIAN)2−Sm2+(dpp-BIAN)0, (dpp-BIAN)1−Sm2+(dpp-BIAN)1− and (dpp-BIAN)2−Sm3+(dpp-BIAN)1−. A significant difference in the samarium-nitrogen distances between two dpp-BIAN ligands in complex 6 (2.2495(11)/2.2589(9) and 2.4411(10)/2.4329(9) Å) is already indicative of a different reduction states of the dpp-BIAN ligands. For this reason, the redox-isomer (dpp-BIAN)1−Sm2+(dpp-BIAN)1− can be excluded from consideration. Since the bonds Sm(1)–N(1) and Sm(1)–N(2) (av. 2.25 Å) are even shorter than those in complex 5 (av. 2.30 Å), the corresponding dpp-BIAN ligand in 6 must be a dianion. Further, the Sm(1)–N(1) and Sm(1)–N(2) distances in 6 are well compared with respective bonds in the six-coordinate Sm(III) complex of the dpp-BIAN dianion, [(dpp-BIAN)SmBr(DME)]2 (2.263(2) and 2.259(2) Å) [14]. On the other hand, the Sm(1)–N(3) and Sm(1)–N(4) distances (av. 2.44 Å) in 6 correspond well to those distances in the Sm(III) complex with dpp-BIAN radical-anion (dpp-BIAN)SmBr2(DME) (av. 2.44 Å) [14]. Based on these structural facts we conclude that complex 6 consists of Sm(III) coordinated by one dianionic and one radical-anionic dpp-BIAN ligand.
Since every reduction state of the dpp-BIAN ligand has its own structural fingerprint the electronic distribution in complex 6 can be further evaluated by the inspection of the bond alteration in the diimine fragments. In the dianionic ligand the N(1)–C(1) and N(2)–C(2) bonds (1.4036(16) and 1.3865(16) Å) correspond well to those bonds in dpp-BIAN dianions in the complexes 2, 3, 4 and 5, whereas the bonds N(3)–C(37) and N(4)–C(38) (both 1.3383(16) Å) are much shorter and indicate the radical-anionic character of the corresponding ligand.
3 Summary
In summary, we have shown that despite the presence of bulky N-2,6-diisopropylphenyl substituents in dpp-BIAN, the formation of bis(diimine)lanthanides takes place in various reactions: (i) in the reaction of Yb(II) complex of dpp-BIAN dianion with free dpp-BIAN; (ii) in the course of reduction of dpp-BIAN with metallic lanthanides; and (iii) in the alkali metal salt elimination reactions starting from the lanthanide halides. Despite of the low coordination number of the rare earth metal ions in [(dpp-BIAN)2Ln]− ether solvents do not coordinate to the metal due to its shielding by the isopropyl groups of the dpp-BIAN. The redox-activity of coordinated dpp-BIAN has been illustrated by oxidation of one of the dianionic ligand in [(dpp-BIAN)2Sm]− to the radical-anionic state to afford neutral complex (dpp-BIAN)2−Sm3+(dpp-BIAN)1−. The cations [(dpp-BIAN)Yb(DME)2]+ and [(dpp-BIAN)La(DME)2]+ illustrate that the use of the redox-active ligands in the coordination chemistry of the lanthanides allows formation of their isostructural compounds with the lanthanide metals being in different oxidation states.
4 Experimental section
General: all manipulations were carried out in vacuum or under nitrogen by using Schlenk techniques. The solvents were distilled from sodium/benzophenone prior to use. 1,2-bis[(2,6-diisopropylphenyl)imino]acenaphthene (dpp-BIAN) was prepared by condensation of acenaphthenequinone with 2,6-diisopropylaniline (both from Aldrich) in refluxing acetonitrile. Compound (dpp-BIAN)Yb(DME)2 (1) was prepared by reduction of dpp-BIAN with ytterbium metal [9]. Solvent-free SmI2 was prepared from SmI2(THF)2 by removal of coordinated THF in vacuum at 200 °C during 6 h. The melting points were measured in sealed capillaries. The IR-spectra were recorded on Vertex 70 spectrometer, the 1H NMR spectra, on Bruker DPX 200 and Bruker III Avance 400 instruments. The magnetic moment of compound 2 was determined by the Evans method [15] in DME. The CH-analyses were obtained using original apparatus, the CO2 and H2O were determined gravimetrically after combustion of the sample. The yields of the products are calculated from the amount of the dpp-BIAN used in the syntheses.
4.1 [(dpp-BIAN)2Yb][(dpp-BIAN)Yb(DME)2] (2)
To a solution of (dpp-BIAN)Yb(DME)2 (1) (prepared in situ from 0.25 g (0.5 mmol) of dpp-BIAN) in DME (40 mL) 0.125 g (0.25 mmol) of dpp-BIAN was added at stirring. The solvent was evaporated in vacuum and the residual solid was dissolved in toluene (40 mL). In the course of heating at 95 °C within an hour the color of the reaction mixture turned from brown to deep blue. From the concentrated toluene solution the product crystallizes at ambient temperature as deep blue crystals. - Yield 0.47 g (85%). - M.p. = 294 °C (dec.). - μ (293 K) = 4.71 μВ. - IR (ν/сm−1): 1584 (s), 1507 (w), 1494 (w), 1412 (m), 1358 (w), 1302 (s), 1251 (s), 1239 (s), 1207 (w), 1188 (m), 1156 (w), 1098 (s), 1051 (s), 1002 (w), 930 (m), 918 (w), 887 (w), 855 (s), 818 (s), 798 (s), 775 (s), 760 (s), 697 (m), 686 (w), 625 (s), 540 (w). - UV-VIS (DME, 293 K): 672 nm. - (2 + 2 C7H8) (2212.75): calcd. C 70.56, H 7.11; found C 69.22, H 7.01.
4.2 [(dpp-BIAN)2La][(dpp-BIAN)La(DME)2] (3)
Suspension of 0.50 g (1.0 mmol) of dpp-BIAN in Et2O (30 mL) was added to lanthanum filings (ca. 10 g). A mixture was stirred at room temperature during 170 h. After evaporation of the solvent a product was dissolved in DME (30 mL) and the formed solution was decanted from metal. The crude product left after evaporation of DME was dissolved in toluene (70 mL) and the toluene solution was filtered off. Compound 3 was isolated from toluene as deep green rhombic crystals. - Yield (0.2 g, 27%). - M.p. = 210 °C. - IR (ν/сm−1): 2723 (w), 1911 (w), 1846 (w), 1783 (w), 1611 (w), 1582 (s), 1354 (w), 1303 (s), 1248 (w), 1186 (w), 1113 (m), 1055 (m), 999 (w), 926 (m), 855 (s), 816 (s), 796 (m), 769 (s), 756 (s), 681 (m), 626 (m), 619 (m), 594 (w), 581 (w), 545 (w). - (3 + 3 C7H8) (2236.56): calcd. C 73.57, H 7.39; found C 73.01, H 7.17.
4.3 [(dpp-BIAN)2La][K(Et2O)4] (4)
A solution of iodine (0.38 g, 1.5 mmol) in Et2O (50 mL) was added to lanthanum filings (ca. 5 g). During a stirring of the mixture for 24 h at room temperature the color of iodine disappeared completely. A white suspension of LaI3(Et2O)n was separated from metal by decantation. From LaI3(Et2O)n/Et2O mixture the solvent was condensed under vacuum to the metal left and the formed white suspension was decanted again. This procedure was repeated several times. To the obtained suspension of LaI3(Et2O)n in Et2O 0.5 g (1.0 mmol) of dpp-BIAN and 0.12 g (3.0 mmol) of potassium were added. A stirring of the mixture at ambient temperature for 48 h resulted green microcrystalline solid. Extraction of this solid with Et2O (3 × 40 mL) afforded 0.54 g (73%) of compound 4 as green crystalline powder. - M.p. = 149 °C. - 1H NMR (200 MHz, C6D6, δ, ppm): 7.42-6.70 (m, 20 H, arom.), 6.57 (d, 2H, J = 7.0 Hz, arom.), 6.28 (d, 2H, J = 6.8 Hz, arom.), 4.12 (spt, 2H, J = 6.8 Hz, CHMe2), 4.04 (spt, 2H, J = 6.8 Hz, CHMe2), 3.85 (spt, 2H, J = 6.8 Hz, CHMe2), 3.25 (q, 16 H, J = 7.0 Hz, O(CH2Me)2), 2.94 (spt, 2H, J = 6.8 Hz, CHMe2), 1.40 (d, 3 H, J = 7.0 Hz, CHMe2), 1.26-0.99 (m, 15 H, CHMe2), 1.11 (q, 24 H, J = 7.0 Hz, O(CH2Me)2), 0.95 (d, 3 H, J = 7.0 Hz, CHMe2), 0.37 (d, 3 H, J = 7.0 Hz, CHMe2). - (4) (1475.89): calcd. C 71.61, H 8.19; found C 70.40, H 7.97.
4.4 [(dpp-BIAN)2Sm][K(C6H6)] (5)
A mixture of SmI2 (0.40 g, 1 mmol), dpp-BIAN (0.50 g, 1.0 mmol) and potassium metal (0.08 g, 2.0 mmol) in toluene was stirred at 100 °C. In 10 h a deep blue solution was filtered off and toluene was evaporated in vacuum. Crystallization of the crude product from benzene gave compound 5 as deep blue prismatic crystals. - Yield 0.58 g (73%). - M.p. 110 °C. - IR (ν/сm−1): 3034 (w), 1614 (w), 1586 (s), 1476 (w), 1423 (m), 1325 (s), 1248 (m), 1180 (w), 1147 (w), 1105 (m), 1054 (w), 1034 (m), 1000 (w), 971 (w), 926 (s), 903 (w), 881 (w), 856 (s), 818 (s), 800 (m), 793 (m), 770 (s), 752 (s), 721 (m), 694 (s), 678 (vs), 629 (m), 619 (m), 596 (w), 577 (w), 548 (w), 509 (w). - 1H NMR (200 MHz, C6D6, δ, ppm): 10.99 (br spt, 2H, J = 4.9 Hz, CHMe2), 8.71 (br spt, 2H, J = 4.9 Hz, CHMe2), 7.95-7.85 (m, 4 H, arom.), 7.75 (d, 2H, J = 7.3 Hz, arom.), 7.64 (d, 2H, J = 7.3 Hz, arom.), 7.44 (pst, 2H, J = 7.3 Hz, arom.), 7.22–6.96 (m, 38H, arom.), 6.91 (d, 2H, J = 7.3 Hz, arom.), 6.42 (d, 2H, J = 7.3 Hz, arom.), 6.17 (d, 2H, J = 7.3 Hz, arom.), 5.46 (d, 2H, J = 7.3 Hz, arom.), 3.32 (d, 6H, J = 4.9 Hz, CHMe2), 2.90 (d, 6H, J = 4.9 Hz, CHMe2), 2.83 (d, 6H, J = 4.9 Hz, CHMe2), 1.20 (d, 6H, J = 4.9 Hz, CHMe2), –0.09 (s, 6H, CHMe2), –0.76 (s, 6H, CHMe2), –2.36 (s, 6H, CHMe2), –5.74 (s, 6H, CHMe2), –6.18 (s, 2H, CHMe2), –11.87 (s, 2H, CHMe2). - (5 + 4 C6H6) (1581.39): calcd. C 77.47, H 7.01; found C 76.72, H 6.92.
4.5 (dpp-BIAN)2Sm (6)
To a solution of 5 (0.31 g 0.2 mmol) in toluene (25 mL) a solution of 0.025 g (0.1 mmol) of iodine in toluene was added slowly at stirring. A deep blue solution was filtered off from KI. Compound 6 was isolated (0.2 g, 87%) as deep blue plate-like crystals. - M.p. = 310 °C. - IR (ν/сm−1): 3056 (w), 1613 (w), 1588 (m), 1482 (w), 1430 (s), 1360 (m), 1325 (vs), 1254 (m), 1241 (s), 1205 (w), 1188 (m), 1179 (w), 1159 (w), 1109 (m), 1080 (w), 1055 (w), 1038 (m), 1001 (w), 943 (m), 930 (m), 866 (w), 849 (m), 820 (s), 798 (s), 772 (s), 758 (vs), 729 (s), 695 (m), 623 (m), 538 (w). - (6 + C7H8) (1243.93): calcd. C 76.28, H 7.13; found C 75.77, H 6.98. The presence in 6 of dpp-BIAN radical-anion prevents acquisition of the informative NMR spectroscopic data.
4.6 X-ray crystallographic studies of 2–6
The intensity data were collected at 100 K on Bruker SMART APEX diffractometer using graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation. SADABS [16] was used to perform area-detector scaling and absorption corrections. The structures were solved by direct methods using SHELXS-97 [17] and by full-matrix least squares techniques against Fo2 using SHELXL-97 [18]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed in idealized positions and their Uiso values were set to ride on the Ueq values of the parent carbon atoms (Uiso(H) = 1.5 Ueq for methyl carbons and 1.2 Ueq for other carbons).
2: C116H140N6O4Yb2 × 2C7H8, Triclinic, P-1, lattice constants a = 14.2305(10), b = 20.5465(14), c = 20.7717(14) Å, α = 109.328(1), β = 92.132(2), γ = 104.312(1), V = 5506.0(7) Å3, Z = 2, μ(Mo-Kα) = 1.745 mm−1, θmax = 26.00°, 13494 [Rint = 0.0622] independent reflections measured with I > 2 σ(I); max. residual electron density 1.861 and –1.623 e/A−3; 1432 parameters, R1 (I > 2 σ(I)) = 0.0535; (all data) = 0.1096.
3: C116H140N6O4La2 × 3C7H8, Monoclinic, P2(1)/n lattice constants a = 15.4460(5), b = 28.5354(8), c = 29.2589(8) Å, β = 104.4200(10), V = 12489.8(6) Å3, Z = 4, μ(Mo-Kα) = 0.728 mm−1, θmax = 26.50°, 19769 [Rint = 0.0393] independent reflections measured with I > 2 σ(I); max. residual electron density 1.952 and –1.644 e/A−3; 1225 parameters, R1 (I > 2 σ(I)) = 0.0489; (all data) = 0.1415.
4: C88H120KLaN4O4, Monoclinic, P2(1)/n lattice constants a = 12.2933(9), b = 18.9838(14), c = 36.002(3) Å, β = 93.239(2), V = 8388.5(11) Å3, Z = 4, μ(Mo-Kα) = 0.608 mm−1, θmax = 25.00°, 11235 [Rint = 0.0853] independent reflections measured with I > 2 σ(I); max. residual electron density 2.163 and −2.201 e/A−3; 883 parameters, R1 (I > 2 σ(I)) = 0.1198; (all data) = 0.3537.
5: C78H86KN4Sm × 4C6H6, Monoclinic, C2/c lattice constants a = 25.2344(9), b = 14.7959(5), c = 24.3385(8) Å, β = 112.2580(10), V = 8410.1(5) Å3, Z = 4, μ(Mo-Kα) = 0.798 mm−1, θmax = 27.50°, 8643 [Rint = 0.0304] independent reflections measured with I > 2 σ(I); max. residual electron density 1.140 and −0.281 e/A−3; 727 parameters, R1 (I > 2 σ(I)) = 0.0267; (all data) = 0.0649.
6: C72H80N4Sm × C7H8, Triclinic, P-1 lattice constants a = 13.4837(4), b = 15.4143(4), c = 17.7745(5) Å, α = 72.3870(10), β = 89.3260(10), γ = 67.4520(10), V = 3229.05(16) Å3, Z = 2, μ(Mo-Kα) = 0.956 mm−1, θmax = 27.50°, 13416 [Rint = 0.0142] independent reflections measured with I > 2 σ(I); max. residual electron density 1.665 and −1.190 e/A−3; 727 parameters, R1 (I > 2 σ(I)) = 0.0275; (all data) = 0.0693.
Positional parameters, hydrogen atom parameters, thermal parameters, bond distances and angles have been deposited as supporting information. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication no. CCDC-759437 (2), -759438 (3), -759439 (4), -759440 (5) and -759441 (6). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: +(44)1223-336-033; email: deposit@ccdc.cam.ac.uk).
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Project No. 10-03-00430).