

1 Introduction
Molecules may become particularly stable when the electron count at a central atom reaches a magic number. Two well-known examples are Lewis’ octets and Langmuir's 18-electron principle, corresponding to filling s + p- and s + p + d-like shells. For the history and actual interpretation of the 18e principle, see Pyykkö [1].
If, in addition to singly occupied s + p + d shells, an f- or f-like shell at the central atom would be occupied, one would obtain systems with the magic number 32. Obviously, the central atom would then have to be an actinide, with a relatively diffuse and energetically available 5f shell. Our first suggestions [2] were of the type . None of them have yet been prepared. Our second suggested 32e-system is U@C28 [3]. This molecule is experimentally known and forms spontaneously in the vapour phase. Its magnetic properties had been analyzed but the 32e nature of the bonding system had not been noticed before.
We now return to the An@M12 series, adding the case M = Sn. We also now report on the heats of formation, IR and UV/Vis spectra of both systems.
2 Theoretical calculations
The geometries of the systems (assuming Ih symmetry) were optimized with the TURBOMOLE program package [4] using the hybrid B3LYP functional. The TZVP basis sets were employed for Pu, Sn and Pb elements associated with energy consistent effective core potentials (ECP) that take into account scalar relativistic effects [5–9]. The use of ECPs results in 32, four and four explicitly handled electrons for Pu, Sn and Pb, respectively.
Energy decomposition analysis (EDA) and estimations of thermodynamic properties were carried out using the ADF 2009.01 program package [10,11] at the TURBOMOLE geometries with the hybrid B3LYP functional. Scalar relativistic effects were considered using the zero-order regular approximation (ZORA). Slater-type orbitals (STOs) were employed as basis functions in SCF calculations. The TZ2P ZORA relativistic basis sets used have triple-ζ quality augmented by two sets of polarization functions. Sixteen, four and four explicit electrons were retained for Pu, Sn and Pb, respectively, within the ADF calculations. The others were treated by the frozen core approximation. The energy decomposition scheme of the ADF program package is based on the work by Morokuma [12,13] and Ziegler and Rauk [14]. The interaction energy ΔEint is decomposed into electrostatic, Pauli repulsion and orbital mixing components:
Supplementary information on chemical bonding was provided with an electron localization function (ELF) analysis of the ADF results with the DGrid 4.5 program package [15].
3 Results and discussion
3.1 Electronic structure of the Pu@Sn12 cluster
The stannaspherene cage has been established to be a highly stable species due to its delocalized π bonding with a closed electron shell and its spherical Ih symmetry [16]. The plumbaspherene cage is analogous to with a π bonding and a Ih symmetry [17]. Its diameter of 629 pm is slightly larger than the stannaspherene one (607 pm). Thanks to their large size, both cages can trap an atom to form new endohedral clusters that have been pointed out both experimentally and theoretically [18,19]. These clusters are pseudo-icosahedral and stable compounds.
The Pu@Sn12 cluster is expected to be energetically more stable than its parts. The central actinide ion will stabilize the surrounding cage. Geometric and energetic parameters are provided in Table 1. The icosahedral geometry is preserved after including the ion. Furthermore, similarly to the Pu@Pb12 cluster, the expansion of the cage, when it is filled by the Pu2+ ion, is small, only 19 pm. The ion can still be easily trapped in the stannaspherene cluster although the cage has a smaller radius than the one by 12 pm. The third important point is that the HOMO-LUMO gap is large, and close to 2 eV in the endohedral system.
Bonding Energy (BE) analysis starting from the (M = Sn, Pb) and Pu2+ fragments.
| Pu@Sn12 B3LYP | Pu@Pb12 B3LYP | |
| r(Pu-M) (pm) | 322.1 | 333.3 |
| Sn12/Pn12 Free cage radius (pm) | 303.0 | 315.1 |
| HOMO-LUMO (eV) | 1.97 | 1.93 |
| BE | −26.19 | −26.76 |
| Pauli repulsion | 20.38 | 18.15 |
| Electrostatic | −21.59 | −21.57 |
| Steric | −1.21 | −3.43 |
| Orbital | −24.98 | −23.34 |
A comparison of the analysis of the electronic bonding energy of the Pu@Sn12 and Pu@Pb12 clusters is given in Table 1. Both systems are very stable with a bonding energy around 26 eV, compared to the divalent ions. The Pu@Pb12 cluster is slightly more stable by 0.6 eV than the Sn one. Due to the smaller size of the cage, the Pauli repulsion contribution is larger in the stannaspherene cluster, leading to a steric term consisting of electrostatic plus repulsion contributions, that is found smaller for Pu@Sn12. The orbital interactions are large, in absolute value 87 to 95% of the total bonding energy. This is a strong indication of chemical bonding, involving both the cage and plutonium orbitals. The orbital energy diagrams for the Pu@Pb12 and Pu@Sn12 clusters are compared in Fig. 1. The diagrams are very similar for both systems, with an “ns block” of occupied orbitals for the cage, and a valence block, corresponding to the interaction between the plutonium and the cage. The nature of the valence molecular orbitals was investigated through the Symmetrized Fragment Orbital (SFO) analysis of the ADF results. For Pu@Sn12, it is clear that Pu 7s, 7p, 6d and 5f valence orbitals interact with the cage ones. This is highlighted by a strong participation of the atomic plutonium orbitals in the 16 doubly occupied valence molecular orbitals, involving 32 electrons: ag, gu, hg, t1u (26e stannaspherene 5p band) and t2u (6e Pu2+ 5f shell). Noticeably, the 5f atomic orbital of Pu are strongly involved in the t2u HOMO with a participation larger than 60%.
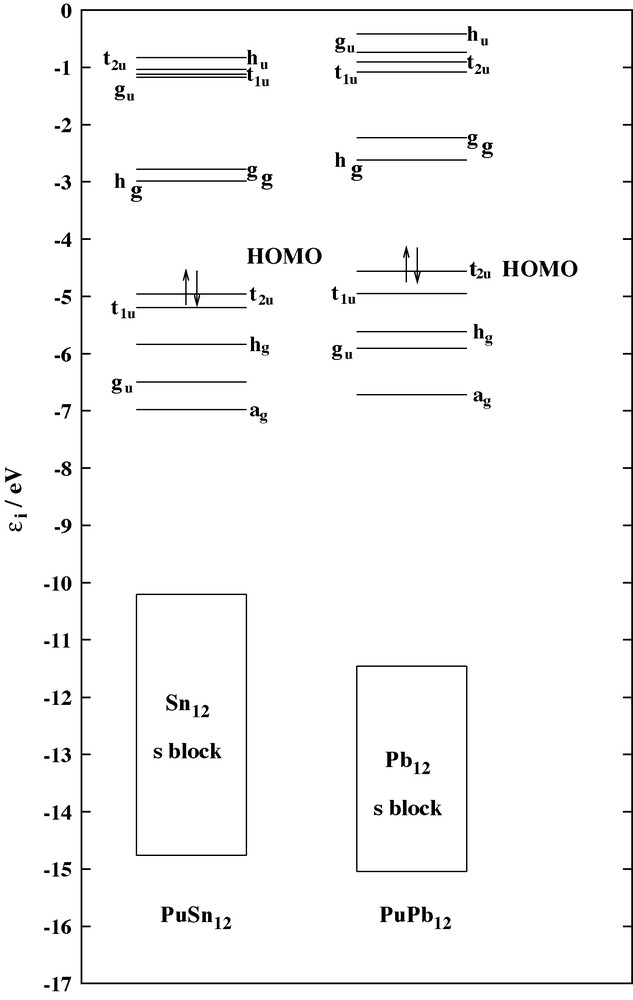
Comparison of the valence molecular orbitals between the Pu@Sn12 and Pu@Pb12 clusters.
The electron localization function provides a “picture” of the electronic structure showing the regions of the molecular space where the electrons localize. The local maxima of ELF define localization domains. The Fig. 2 (left side) displays the isosurface ELF = 0.45 for Pu@Sn12. In this picture, between the valence core Pu and Sn basins, we may find a Pu-Sn bonding basin also pointed out in the cut plane representation (right size, red color). These elements reinforce our previous conclusions about the formation of a strongly bonded Pu@Sn12 system.

ELF 0.45-localization domain (Pu-Sn).
To conclude, in addition to our previously Pu@Pb12 32-electron system, we now report theoretical evidence for a stable stannaspherene cluster Pu@Sn12 with very similar 32-electron bonding system.
3.2 Estimation of the thermodynamic stabilities of the Pu@Sn12 and Pu@Pb12 clusters
The reaction enthalpies and reaction entropies for Pu2+ encapsulation in Pb12 or Sn12 cage are estimated from standard statistical thermodynamics (ideal gas assumed) after calculating DFT/B3LYP harmonic vibrational frequencies. The calculated enthalpy (ΔrH) and Gibbs free energy (ΔrG) are provided in Table 2 (T = 298.15 K and P = 1 atm). The reactions of encapsulation of the plutonium ion in the stannaspherene and plumbaspherene cages are exothermic. These large values confirm the high thermodynamic stability of the two clusters, the Pu@Pb12 compound being slightly more stable. These results are in agreement with the electronic stabilization discussed above due to the large orbital interaction between the Pu ion and the cage.
DFT/B3LYP reaction enthalpies and reaction Gibbs free energies (kJ/mol) for Pu2+ encapsulation in the Pb12 and Sn12 cages (T = 298.15 K, P = 1 atm.).
| Reaction | ΔrH | ΔrG |
| −2097 | −2051 | |
| −2137 | −2102 |
In order to locate thermodynamically the Pu@Sn12 and Pu@Pb12 gas-phase clusters with respect to the solid phase, we have used the experimental standard enthalpy of formation at 298.15 K (349 kJ/mol for Pu [20], 195.2 kJ/mol for Pb [21], 301.2 J/mol for Sn [21]), the entropy at 298.15 K (177.2 J/K.mol for Pu [20], 175.4 J/K.mol for Pb [21], 168.5 J/K.mol for Sn [21]) and the ADF calculated gas-phase data.
Pu@Sn12 and Pu@Pb12 are evaluated respectively at about 1950 kJ/mol and 650 kJ/mol for the enthalpy and at about 1550 kJ/mol and 250 kJ/mol for the Gibbs free energy above Pu(s) + 12 Pb(s). The existence of the present cluster compounds is made more likely by the experimental observation of the mixed metallic phases Pu3Pb, Pu5Pb3, Pu5Pb4, Pu4Pb5, PuPb2, PuPb3 in the bulk [20].
3.3 Spectroscopic properties
3.3.1 Vibrational
The DFT/B3LYP computed harmonic vibrational frequencies are given in Table 3 for the and 12 cages and the corresponding Pu@M12 clusters. Due to the Ih symmetry, only the T1u vibrational modes are IR active. The cage-deformation modes are calculated to be 126 cm−1 and 90 cm−1 for the Sn12 and Pb12 cages respectively. The inclusion of Pu2+ ion in the cage slightly increases the frequencies to the values of 132 cm−1 and 107 cm−1 respectively, reflecting the rigidity of the cage and a weak vibrational-mode coupling with the central atom. The intra-sphere motion of Pu atom could be viewed as a particle-in-a box translation with very small vibrational frequencies of 16 cm−1 and 30 cm−1 for Pu@Sn12 and Pu@Pb12 respectively. This situation is very different from our previous smallest and more strongly bounded An@C28 clusters for which we have systematically observed combined motions of the cage and of the central atom in the cage.
TM602. Calculated harmonic vibrational frequencies in cm−1 for Pu@M12 (M = Sn, Pb) clusters and the M cage.
| Pu@Sn12 B3LYP | Pu@Pb12 B3LYP | |
| Pu intra-sphere motion mode | 16.32 (2.22) | 30.35 (1.12) |
| Pu@M12 Cage-deformation mode | 132.33 (8.54) | 107.22 (3.24) |
| Sn12/Pn12 Cage-deformation mode | 126.23 (0.19) | 90.06 (0.00) |
3.3.2 Electronic transitions
Time-dependent density functional theory (TD-DFT) was used to compute electronic vertical singlet excitation energies with the B3LYP functional and the TURBOMOLE program package. Our goal is not to produce an extensive analysis of the electronic spectra, but rather to find the position of the main absorption bands in order to obtain an idea of the optical properties of the Pu@M12 clusters. In the Ih point group symmetry, only the T1u symmetry excitations are electric-dipole-allowed. The results for the Sn12/Pb12 cages and Pu@M12 clusters are displayed in Figs. 3a and b respectively. In Fig. 3, the band shapes are approximated with the modified Gauss function proposed by L. Antonov for electronic absorption spectra [22]:
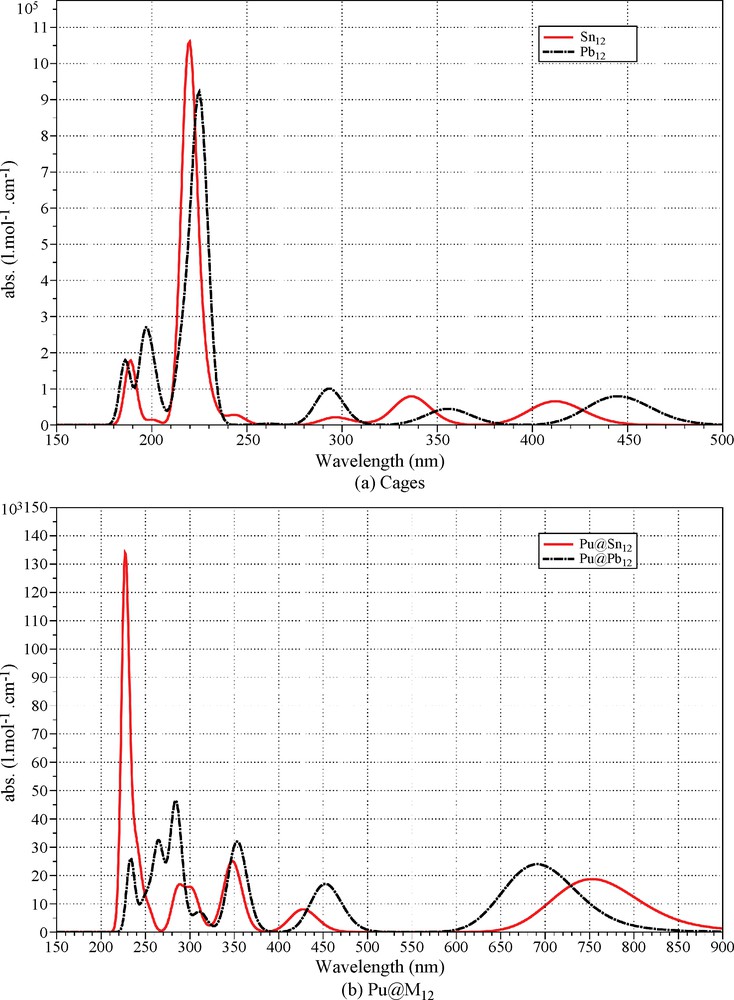
Electronic absorption spectra calculated by TD-DFT with a B3LYP functional.
The basic parameters in this formula are the position of the band maximum λmax, the maximal intensity given as ϵmax (in l mol−1 cm−1), the band width at half-maximal intensity . The values of the molar extinction coefficient ϵmax were calculated by means of the usual equation [23]:
For the and cages, the calculated absorption spectra are similar. The absorptions are mainly in the ultraviolet with a strong absorption at about 220–230 nm and weak absorptions between 290–480 nm.
The modifications in the absorption spectra induced by the encapsulation Pu2+ ion are essentially the introduction of an absorption in the red (700–750 nm) with a blueshift from Sn12 to Pb12. The major MOs involved in this excitation are t1u [6p(Pu)/5p(Sn) or 6p(Pb)] to hg [6d(Pu)/5p(Sn) or 6p(Pb)].
We have not discussed here the role of spin-orbit coupling. In our previous work [2], we have found these effects small on the ground state molecular properties. For the Pu@Pb12 cluster, the spin-orbit coupling did not invalidate the conclusions about the 32-electron principle. Both SO-components of each orbital were filled and the net effects on the energy or the HOMO-LUMO gap were small. As tin is lighter than lead and the SO effects scale as Z2, the effects should, if anything, be even smaller for tin. Of course we may expect a role of the spin-orbit coupling on the electronic spectra with a shift of the absorption bands. We have tried to investigate these effects with the ADF program package. Unfortunately, it is not possible to converge the process without including a level shift which gives incorrect results, and hence cannot be used to calculate excitation energies.
4 Conclusions
The Pu@Sn12 cluster has been revealed to be chemically stable. The strong mixing of the Pu atomic orbitals and the molecular orbitals of the Pu@M12 cluster for M = Pb is also found to take place for the stannaspherene cage, M = Sn. Compared to the C60 fullerene, the present cage is only slightly smaller, and still allows the encapsulation of an nd or nf transition-metal atom. Concerning optical properties of the present systems, the visible absorption strongly dependends on the nature of the encapsulated ion. Therefore one may anticipate in the future tunable optical properties by changing the central ion in the stannasphere or plumbasphere cages. The new Pu@Sn12 cluster is a further example of a 32-electron system, following the earlier Pu@Pb12 cluster and the An@C28 series.
Acknowledgements
P.P. belongs to the Finnish Center of Excellence (CoE) in Computational Molecular Science (2006–2011). This work was carried out in the framework of a collaboration between the French CEA–Direction des Sciences de la Matière and the University of Helsinki. We thank the international program of the École Polytechnique (Palaiseau, France) for supporting a one-month stay in France by P.P. This work was granted access to the HPC resources of [CCRT/CINES/IDRIS] under the allocation i2009086146 made by GENCI (Grand équipement national de calcul intensif) and to the Center of Scientific Computing (CSC, Espoo, Finland).