

1 Introduction
The cyclodextrins (CDs) are undoubtedly amongst the most studied macrocyclic molecules. Easy access from natural sources, together with their unique ability to form inclusion complexes in water with a large variety of hydrophobic molecules have attracted chemists working in many different areas [1,2]. However, only few studies have dealt with multisubstituted CDs, in which a CD unit bears more than one substituent [3], in stark contrast with other macrocycles — amongst other calix[4]arenes [4] — that are much easier to functionalise in a regioselective manner. Native CDs (Fig. 1) can be qualified as “conformational straightjackets” because of the existence of a hydrogen-bond network involving secondary hydroxyl groups belonging to adjacent glucose units, which makes the cyclic structure very rigid and, therefore, not the most versatile preorganisation platform [5].
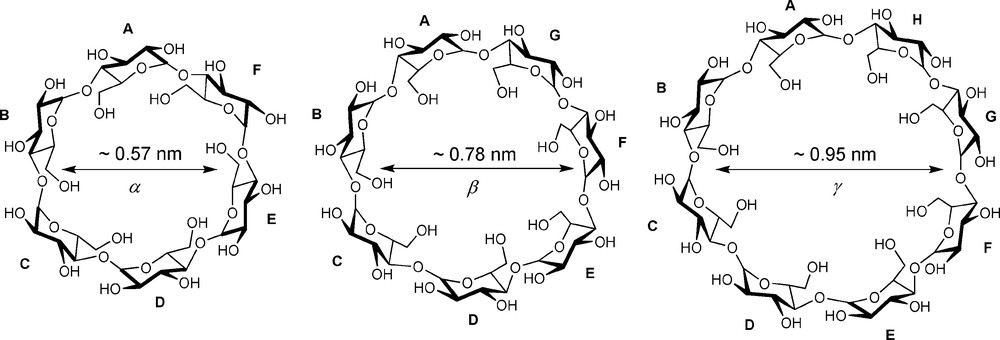
Native CDs of various sizes.
On the other hand, fully methylated CDs are much more flexible, yet rigid enough to maintain the familiar torus shape and, to a certain extent, the supramolecular properties of their unsubstituted analogues in water [6]. In other words, the good compromise between flexibility and conformational stability makes them good candidates for the synthesis of multidentate podands suited for chelation of transition metal centres. The present account examines the way the regiofunctionalisation methods developed in the authors’ group allows the preparation of valuable multidendate chelating ligands based on methylated CDs. It further shows how the latter are able to position a metal centre in close proximity to the CD cavity and incidentally promote non-covalent interactions between the cavity interior and the metal first coordination sphere [7]. The implications of such metal encapsulation [8] on some industrially relevant catalytic reactions will also be discussed. Note that chelate complexes derived from monosubstituted CDs will not be considered in this review since the CD scaffold does not act as a structuring unit in these molecules.
2 CD regiofunctionalisation methods based on the bulky supertrityl protecting group (sTr) and difunctional analogues — easy access to methylated multisubstituted CD derivatives
Most functionalisation methods have so far focused on the so-called persubstitution and monosubstitution. In the former, all hydroxyl groups at one site (either 2-, 3- or 6- positions) are modified, whereas in the latter only one single hydroxyl group, whether primary or secondary, is affected (Fig. 2).
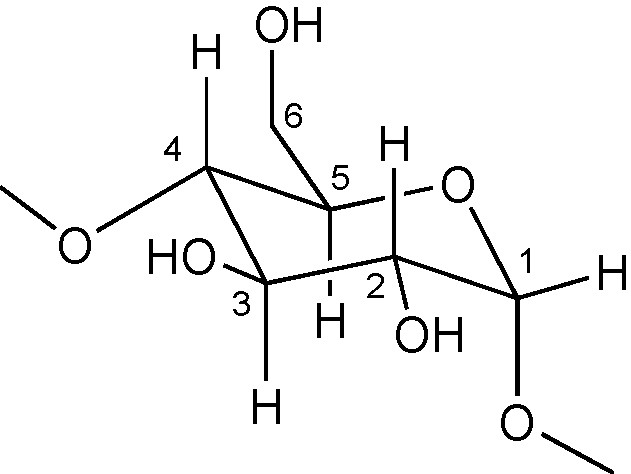
Glucose unit with numbering of the C atoms.
The corresponding derivatives are of limited interest for the preparation of chelating ligands as the number of coordinating sites is either too large in the case of the persubstituted CDs, so that a single chelate complex cannot be isolated, or too small in the second type of podands for maintaining the metal in a rigid manner with respect to the cavity. Only recently, chemists started to address the more challenging task of differentiating between hydroxyl groups of the same type [9]. Early methods relied on the use of small electrophilic agents, meaning that all hydroxyl groups react indiscriminately to give a mixture of products [3]. The desired compound has to be then painstakingly separated from other regioisomers and analogues by chromatographic methods, although in some rare cases a single species can be obtained in good yields by recrystallisation [10]. With the use of difunctional reagents like rigid arenedisulfonyl chlorides, the regioselectivity of disubstitution or tetrasubstitution can be improved to a certain degree as capped species with minimum cavity distortion tend to be formed [11,12]. A far more versatile and far-reaching method consists in deprotecting persubstituted CDs in a regioselective manner. Sinaÿ and Sollogoub have demonstrated that DIBAL-H-promoted de-O-alkylation of perbenzylated [13,14] or permethylated cyclodextrins [15] occurs with remarkably high regioselectivity, giving access to a wealth of multidifferentiated CD derivatives with a degree of functionalisation ranging from 2 to 6. Alternatively, bulky alkylating agents like trityl chloride can also be used to discriminate between CD primary hydroxyl groups [16–18]. After substituting the non-alkylated hydroxyls with inert residues like methyl groups, the protecting groups can be easily removed, if desired, using acidic hydrolysis to produce valuable starting material for multidentate ligand synthesis. Reacting a native α− or β-CD with an excess (3.3 eq.) of analogous but larger alkylating agent like tris(4-tert-butylphenyl)methyl chloride (1), also designated as supertrityl chloride (sTrCl) resulted in increased selectivities for, respectively, AD and AC disubstituted regioisomers 2a/2b and 3a/3b in the presence of triethylamine and 4-dimethylaminopyridine (DMAP) in DMF [19].
In the case of α-CD, replacing the solvent and triethylamine with pyridine leads to the formation of only one tetrasubstituted species, namely the ABDE regioisomer, together with some trialkylated species if a large excess of 1 (6 eq.) is used [20]. For purification purposes and in order to increase solubility in organic solvents, the remaining hydroxyl groups are usually methylated as in 5 (Scheme 1). As mentioned in the introduction, methylation also renders the CD macrocycle more flexible as a result of the disappearance of the hydrogen bond-network involving the secondary hydroxyl groups. Compared to tritylated derivatives, the presence of sTr groups on the CD macrocycle leads to larger difference of Rf values between regioisomers, hence facilitating their chromatographic separation.
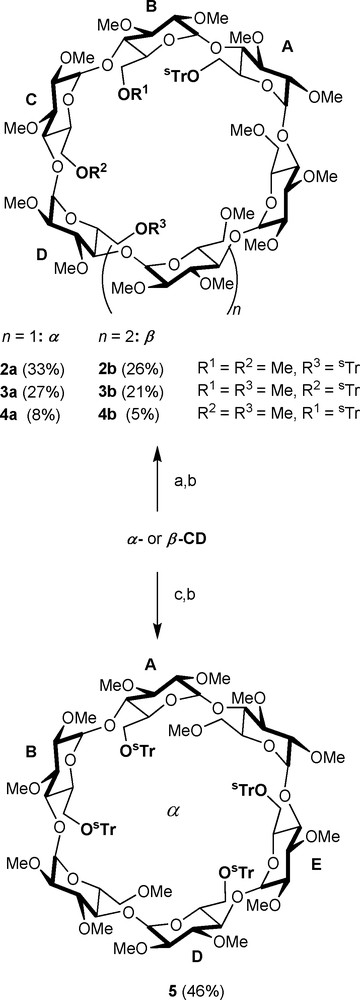
Multifunctionalisation of α − and β-CD using sTrCl (1). Reagents and conditions: (a) 3.3 sTrCl, DMAP, Et3N, 50 °C, 16 h; (b) 36 NaH, rt, 1 h, then 30 MeI, rt, 16 h; (c) sTrCl, DMAP, pyridine, 70 °C, 16 h.
To gain access to the highly desirable AB disubstituted regioisomers in both α-CD and β-CD series, modified electron-rich areneditrityl chlorides such as 6 and 7 were used to bridge adjacent glucose units in a selective manner (Scheme 2) [21].
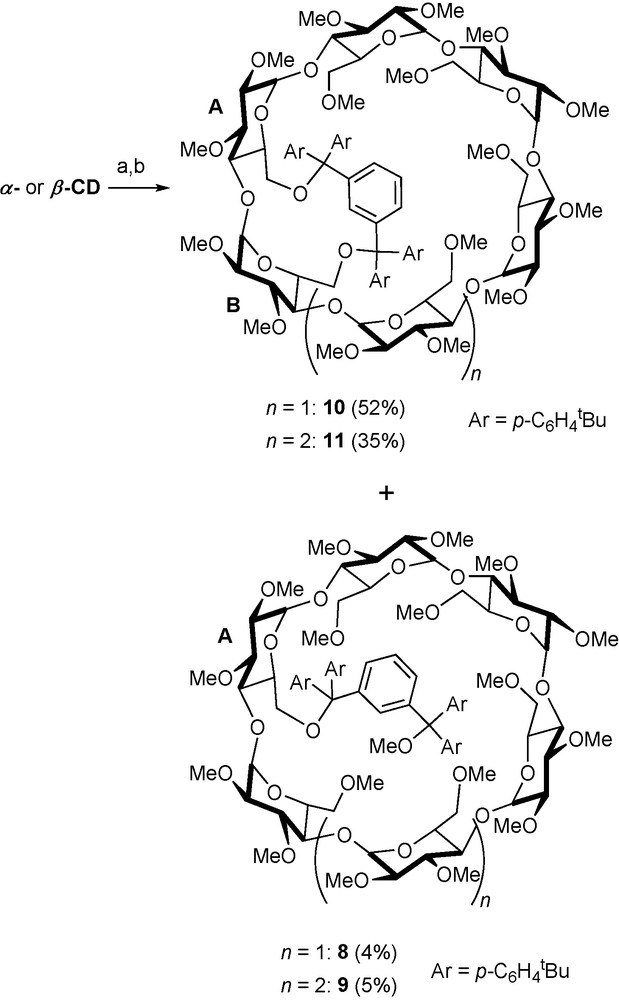
Bridging adjacent glucose units with dichloride 7. Reagents and conditions: (a) 7 (1 equiv.), DMAP, pyridine, 70 °C, 13 h; (b) 36 NaH, rt, 1 h, then 30 MeI, rt, 16 h.
Although the presence of small amounts of monosubstituted species such as 8 and 9 could not be avoided due to the lability of the bonds between the quaternary carbon atoms of the modified trityl unit and the sugar oxygen atoms, yields as high as 55 and 35% of AB disubstituted species 10 and 11, respectively, could be reached. To sum up, functionalisation methods based on the use of trityl-derived reagents are particularly suited to the preparation of multisubstituted methylated CDs as the protecting groups are stable under the alkaline conditions used for methylation and can be easily removed by acidic hydrolysis.
3 Chelating multidentate ligands derived from methylated cyclodextrins
The range of multifunctional CDs that have been made available can easily be turned into multidendate chelating ligands by further modification. In this respect, several strategies were devised to force a metal centre to be located close to the CD cavity [22]. The first one consisted in bridging the macrocycle with a capping coordinating subunit (X) so that coordination leads to a complex in which the metal centre is positioned directly above the cavity entrance. Alternatively, the same feature can be achieved with podands able to give rise to large chelate complexes. However, the coordinating arms have to be properly designed to avoid the formation of unwanted cyclic oligomers and polymeric material [23]. A third strategy, which combines the two former involved the double bridging of the molecular cavity with endo-oriented donor atoms so that the coordinating unit is rigid enough to allow the exclusive formation of chelate complexes (Fig. 3).
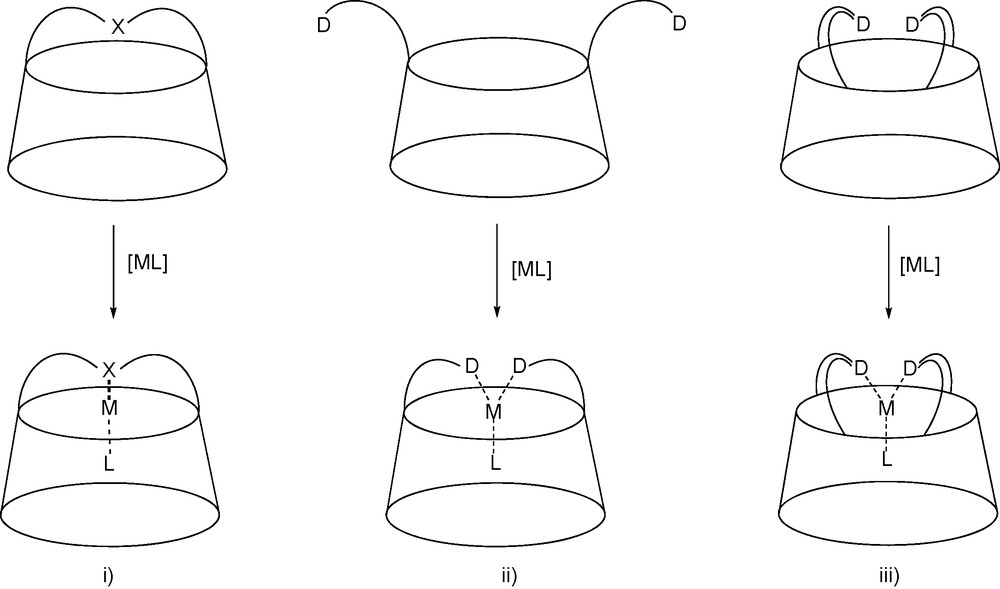
The three strategies used for positioning a metal centre near the entrance of a molecular cavity: i) coordinating single handle, ii) chelating podand, iii) chelating double handle. “D” designates a donor atom.
3.1 Nitrogen chelators
Diamines 13a and 13b [24], which are respectively prepared from difunctionalised CDs 2a and 2b (Scheme 3) via the key intermediates 12a and 12b, are versatile starting materials for a range of nitrogen containing CD ligands.
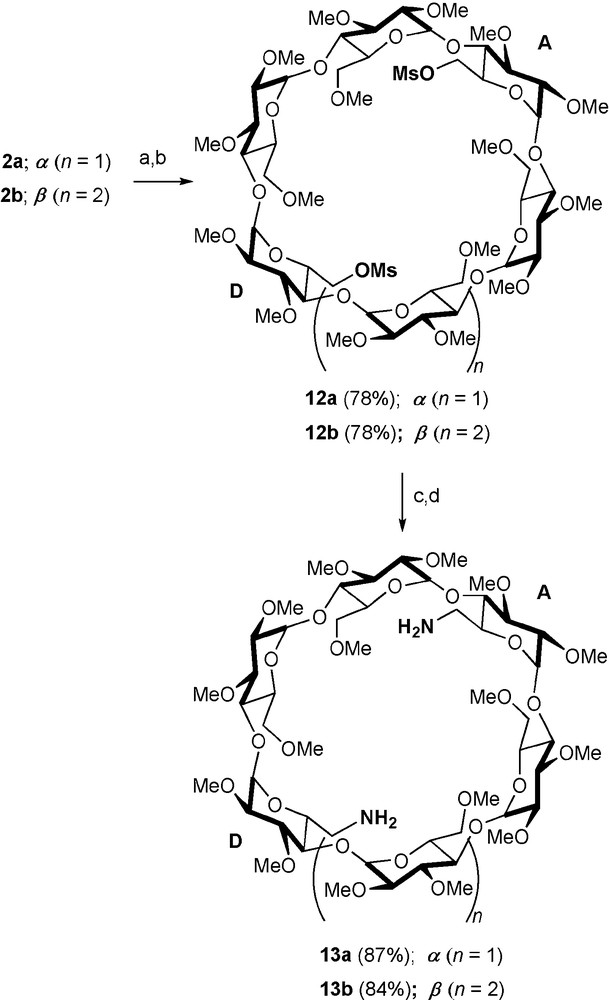
Synthesis of diamines 13a/13b. Reagents and conditions: (a) aq. HBF4 (34%), MeCN, rt, 0.5 h; (b) MsCl, DMAP, pyridine, rt, 6 h; (c) NaN3, DMF, 65 °C, 18 h; (d) PPh3, dioxane, 1 h, then aq. NH4OH (35%), rt, 16 h.
They behave themselves as chelators although significant amounts of metallo-oligomeric material is also being produced upon metal complexation. For example, 13a is able to give rise to the chelate complex cis-[PtCl2•13a] in 17% yield despite the strong distortion imposed in the complex on the methylated CD scaffold (Fig. 4).
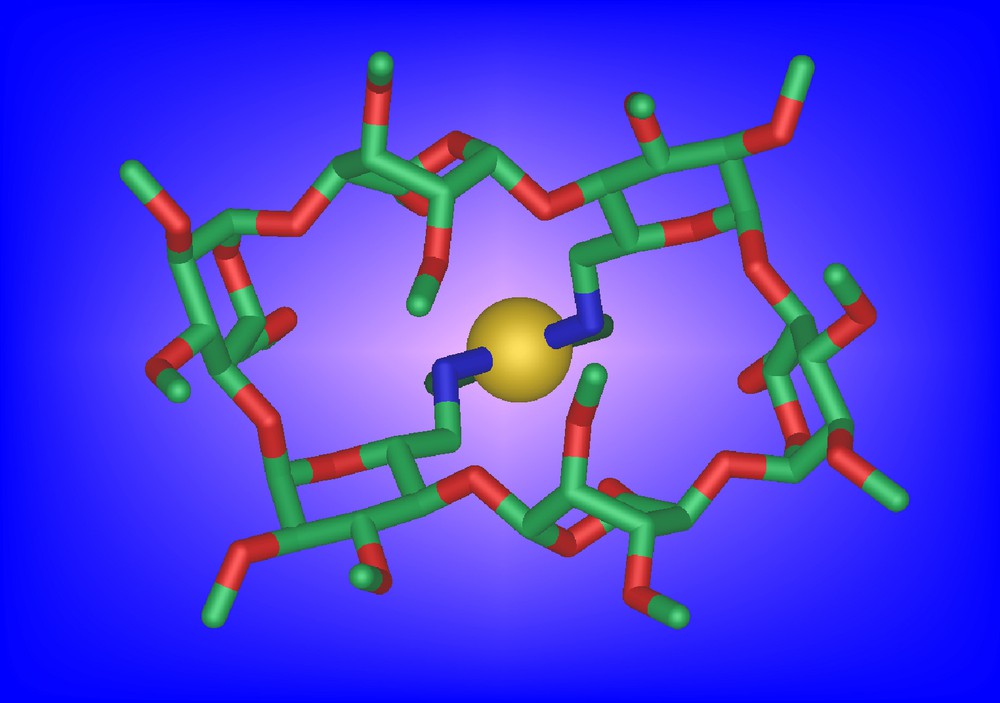
Molecular structure of cis-[PtCl2•13a]•H2O•CHCl3. For clarity, the chloroform and water molecules are not shown.
Diamines 13a and 13b can be rigidly capped to afford “handle-type” chelating ligands, in which the nitrogen coordinating unit forces the metal centre to be positioned at the apex of the cavity upon coordination. In ligand 14 (Scheme 4) [25], the endo-oriented 2,2′-bipyridyl capping fragment acts as a chelator for metal complexes with square planar geometries as in [PdCl•14]. It can also promote the reversible coordination of weakly binding methoxy groups as in the fluxional tetrahedral complex [Ag•14H]BF4, which shows that the CD torus itself can act as a first sphere of coordination towards transition metals. On the other hand, when the nitrogen donors atoms are exo oriented as in 15, heterotopic octahedral complexes such as [Ru(bpy)2•15][PF6]2 can be synthesised [26].
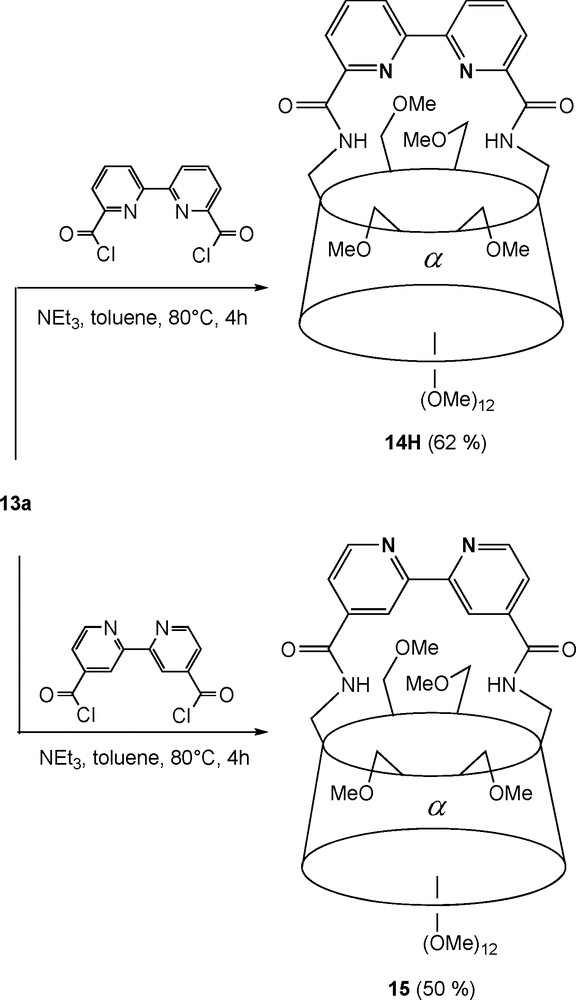
Synthesis of bipyridyl-capped CD ligands 14H and 15.
This complex was shown to form a 1:1 inclusion complex with 1,4-benzoquinone (BQ). A vectorial photo-induced electron-transfer between the included guest and the Ru(bpy)3 moiety was shown to take place within the CD cavity upon irradiation of the photoactive unit.
Rigidly-capped CDs such as 16a and 16b can also offer steric protection for unusual coordination species as in trigonal bipyramidal ethylene polymerisation precatalysts [FeCl2•16a] and [FeCl2•16b], which would otherwise give rise to octahedral N6Fe(II) complexes (Scheme 5) [27].
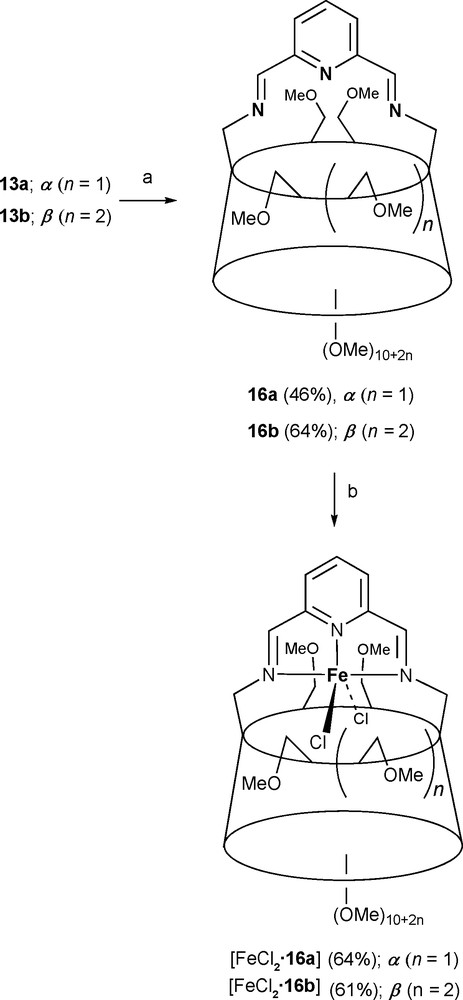
Synthesis of precatalysts [FeCl2•16a] and [FeCl2•16b]. Reagents and conditions: (a) 2,6-diformylpyridine, MeCN, molecular sieves (4 Å), rt, 2 h; (b) anhydrous FeCl2, n-butanol, 80 °C, 30 min.
3.2 Oxygen chelators
Aiming at the development of novel synthetic analogues of natural siderophores, Coleman et al. described the first example of a chelating ligand involving a permethylated cyclodextrin unit [28]. The hexadentate podand 17, which is made of three catecholate groups grafted to a C3-symmetrical, methylated CD platform, is able to bind a single metal centre via oxygen donor atoms only. Formation of the octahedral complex [Fe•17]3− was shown to take place in aqueous solution at pH values higher than 4 by UV-vis spectroscopy. At even higher pH, coordination of an Al(III) ion could also be evidenced by 1H NMR spectroscopy, upfield shifts of the aromatic protons as well as B,D,F methoxy signals being detected upon complexation. In addition, the water-soluble chelate complex [Al•17]3− acts as a metallo-receptor towards amphiphilic guests like the p-nitrophenolate anion.
3.3 Phosphorus chelators
Being a softer donor atom than nitrogen and oxygen, phosphorus, in particular in its P(III) oxidation state, is well suited for the synthesis of multidentate CDs able to bind a variety of transition metals. A P(III) atom can be incorporated in a CD, simply by reaction of the strongly nucleophilic phosphide anion with a CD derivative bearing suitable leaving groups or by Stelzer's palladium-catalysed cross coupling reaction [29] if iodoaryl groups have been previously grafted onto the CD macrocycle as in 18 and 19 (Scheme 6) [30].
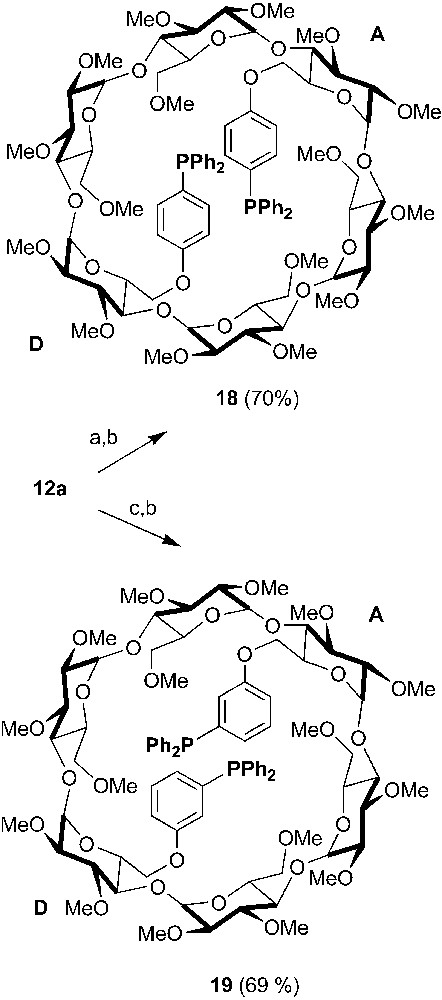
Synthesis of chelating ditriaryphosphine 18 and 19 starting from 12a. Reagents and conditions: (a) 4-iodophenol, K2CO3, DMF, 75 °C, 20 h; (b) PPh2H, Pd(OAc)2, AcONa, DMA, 130 °C, 3 h; (c) 4-iodophenol, K2CO3, DMF, 75 °C, 20 h.
In these ligands, the rigid coordinating arms match the cavity diameter and therefore facilitate metal chelation without cavity deformation. Indeed, both ligands afforded significant amounts of a mixture of equilibrating cis and trans chelate complexes ([MCl2•18/19]) upon treatment with [MCl2(PhCN)2] (M = Pd or Pt), although some oligomeric material could also be detected. Unfortunately, the cavity is far too remote from the metal centre for it to act as a first or even a second sphere of coordination.
Because methylated CDs are quite flexible, shortening the coordinating arms does not affect the chelating ability of the bidentates. Thus, regioisomeric diphosphine ligands 21 and 22, which were prepared respectively by treatment of dimesylates 12a and 20 with the strongly nucleophilic diphenylphosphide anion (Scheme 7), behaved predominantly as chelating ligands when opposed to metals having either square planar or octahedral coordination spheres [31,32]. In stark contrast with [MCl2•18/19], only trans-P,P arrangements are present in the resulting chelate complexes [PtCl2•21/22], [PdCl2•21/22], and [RuCl2(CO)2•21].
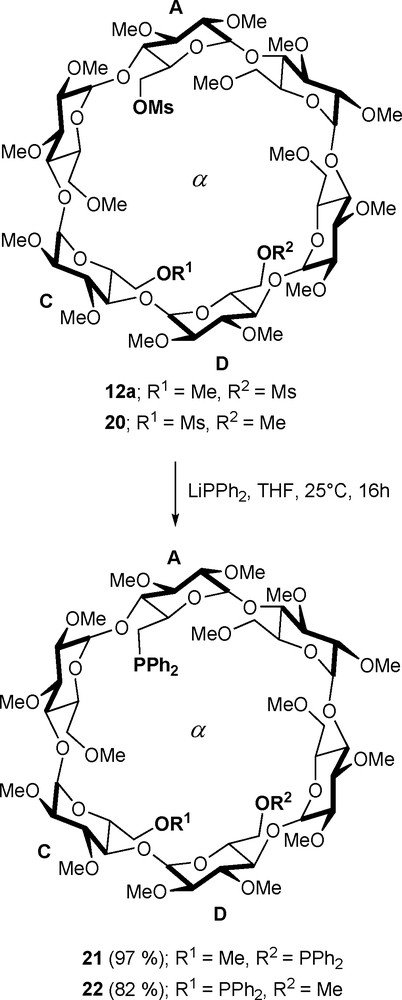
Synthesis of diphosphines 21 and 22.
When the two phosphorus atoms are located on adjacent glucose units and therefore much closer to each other as in 23, chelate complexes with a cis geometry are being formed [33], e.g. [Rh(COD)•23]BF4 and [PtCl2•23].
In all these complexes, the metal centre is also nearer to the primary face opening, allowing the CD cavity to play the role not only of first, but also of second sphere of coordination. Indeed, in all the chlorido complexes derived from α-CD, weak Cl•••H-5 interactions maintain the MCl bond inside the CD cavity. This is particularly revealing in complexes having hetero-dicoordinated metal centres, such as [RhCl(CO)•21/22] or [PdClMe•21/22], in which the chloride anion is entrapped in the cavity, the other ligands, CO or methyl, being located outside [31]. In the case of the octahedral complex [RuCl2(CO)2•21], the cavity entraps both chlorido ligands (and no CO), thus illustrating the high affinity of the cavity for halide anions [32,34]. Interestingly, treatment of [RhCl(CO)•21/22] with NaBH4 causes the replacement of the chlorido ligand with a hydrido one, which in turn leads to the confinement of the ligand of higher affinity for the cavity, in this case the CO ligand ([RhH(CO)•21/22]).
As in complexes [Ag•14H]BF4, the CD torus acts as a first sphere of coordination in the fluxional silver (I) complexes [Ag•21]BF4 (Scheme 8) [35]. All four primary face methoxy groups behave as labile ligands and are involved in the coordination of the Ag+ cation in a dynamic way, each of them being bound successively to the metal centre. Being weakly bound to the metal, the coordinated methoxy group of [Ag•21]BF4 can be easily replaced by one or even two acetonitrile ligands depending on the amount of acetonitrile being added. It was shown that the cavity plays an active role in stabilizing the otherwise highly unstable tetrahedral [AgP2(CH3CN)2]+ species by making acetonitrile dissociation less favourable. On the other hand, only one benzonitrile ligand, which is larger than acetonitrile, fits in the cavity resulting in the exclusive formation of the trigonal complex [Ag(PhCN)•21]BF4, even in the presence of a large excess of benzonitrile. Clearly, the cavity is able to control the coordination process by means of steric interactions between its inner walls and the included ligand.
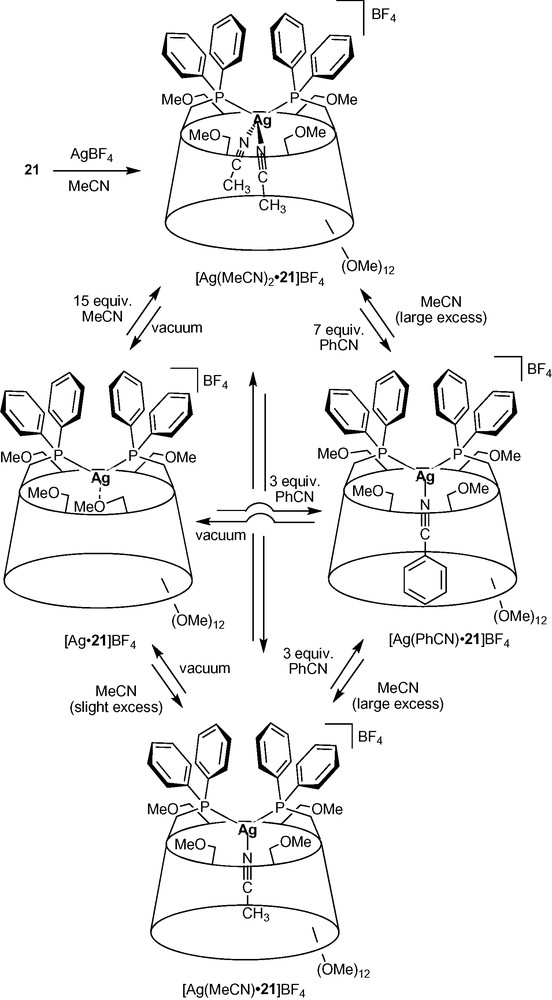
Nitrile ligand-exchange reactions occurring inside a CD cavity.
Other CD-based P(III) bidentate chelators were also synthesized. Unlike their phosphine counterparts, which give rise to chelate complexes regardless of the metal reagent being used, whether cationic or neutral, the diphosphites 24 and 25 (Scheme 9) produced chelate complexes ([Rh(NBD)•24]PF6 and [Rh(NBD)•25]BF4) only when reacted with cationic complexes having highly labile ligands, e.g. [Rh(NBD)(THF)2]+. This behaviour was accredited to the poorer preorganisation of their coordinating arms [36].
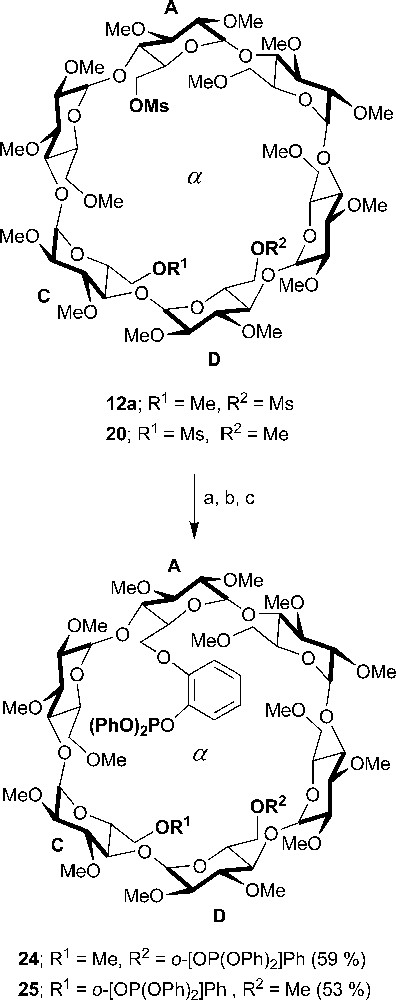
Synthesis of ditriarylphosphites 24 and 25. Reagents and conditions: (a) 2-benzyloxyphenol, NaH, DMF, rt, 16 h; (b) Pd/C, H2 (1 atm), EtOH, 48 h; (c) chlorodiphenylphosphite, CH2Cl2, Et3N, 1 h.
ABDE-Tetrafunctionalised α-CD 5 is ideally suited for getting access to tetraphosphine 27 (α-TEPHOS) via the key intermediate 26 (Scheme 10). The tetradendate ligand 27 behaves as a bis-chelator when opposed to highly reactive cationic metal complexes to afford dinuclear complexes such as [Rh2(NBD)2•27][BF4]2. The bis-chelation process is regiospecific as only proximal glucose units are capped. Conversely, a mixture of chelate and oligomeric complexes was produced with less labile, neutral metal reagents such as [PtCl2(PhCN)2] [20].
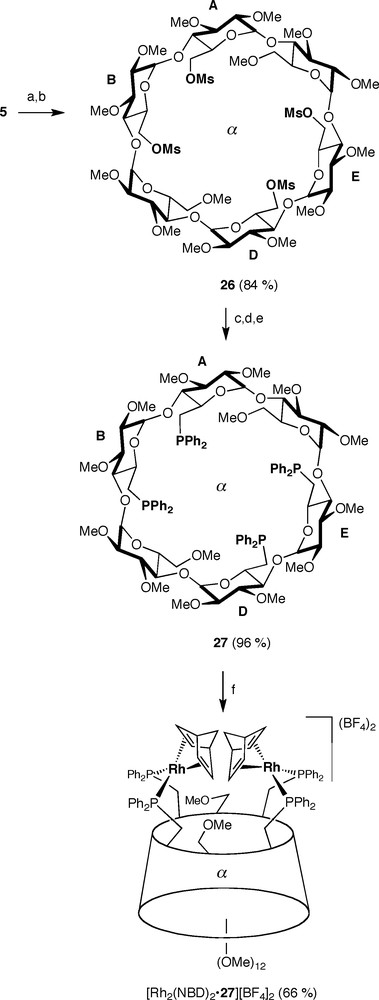
Synthesis of tetraphosphine 27 and bis-chelate complex [Rh2(NBD)2•27][BF4]2. Reagents and conditions: (a) aq.HBF4 (34%), MeCN, rt, 0.5 h; (b) MsCl, DMAP, pyridine, rt, 6 h; (c) 6 HPPh2, n-BuLi, THF, −78–−5 °C, 12 h; (d) BH3•THF, THF, 0–−5 °C, 14 h; (e) Et2NH, reflux, 16 h; (f) [RhCl (NBD)]2, 2 AgBF4, THF/CH2Cl2, rt, 1.5 h.
The same tetramesylate 26 reacts with two equivalents of phenyl phosphide in a fully regio- and stereoselective way to give quantitatively diphosphine 28 (Scheme 11). Fortunately, both phosphorus lone pairs point towards the cavity centre making it a very rigid and introverted chelator [37].
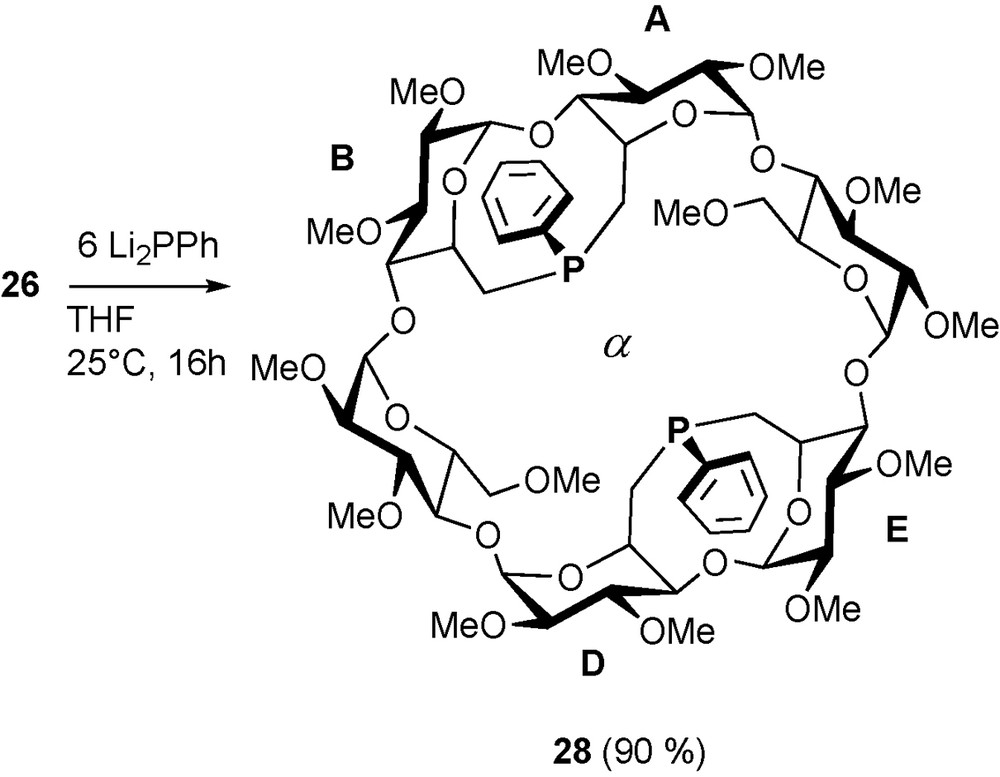
Synthesis of diphosphine 28.
Unlike all other aforementioned P(III) ligands, 28 gives rise to trans chelate complexes such as [PtCl2•28], [PdClMe•28] and [RhCl(CO)•28] in a quantitative way when opposed to d8 transition metal ions [38]. Although trans spanning ligands have been sought after for more than a century [39], this goal has so far remained elusive as of all the bidentate ligands claimed to be trans chelators, none of them have been able to preclude the formation of either cis chelates or bimetallic and higher oligomers.1 It is believed that the trans chelating properties of 28 arise from its rigid nature so that both phosphorus atoms are kept at a distance unsuitable for cis geometries. Furthermore, the ligand cannot bind a metal centre in a divergent way because of its introverted nature, which explains the absence of oligomers. Last but not least, the cavity is thought to play an active role in controlling the geometry of the metal centre because only one small enough ligand such as a halide can be included in the CD cavity at a time. As in chlorido complexes derived from 21 and 22, downfield chemical shifts up to 0.9 ppm for some inner cavity H-5 protons are detected upon halide inclusion (Fig. 5). For some of the halido complexes, the protection provided by the cavity is so high that the encapsulated halide cannot be substituted by nucleophiles as strong as MeLi. Thus, treating the bromido complex [NiBr2•28] with excess methyllihium produces exclusively the unusually stable unsymmetrical species [NiBrMe•28].
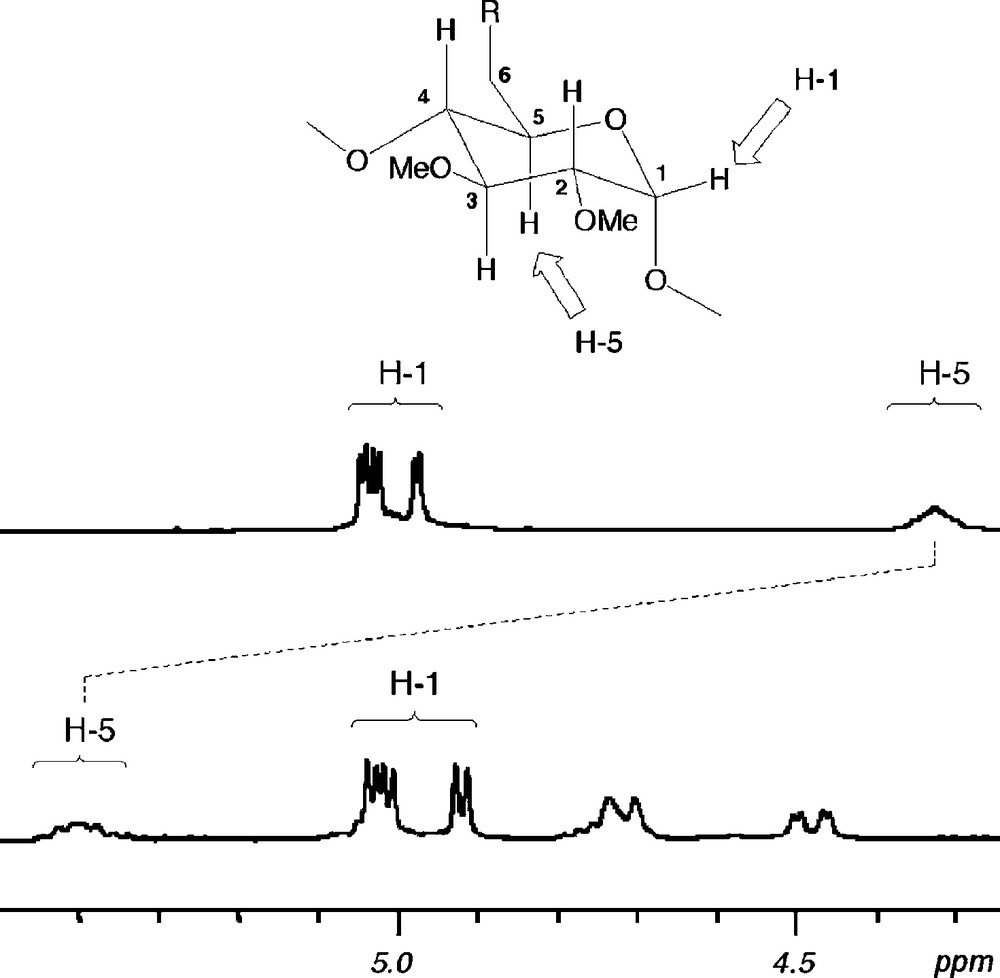
Parts of 1H NMR spectra of ligand 28 (top) and complex [PdClMe•28] (bottom) recorded in CDCl3 at 300.1 MHz.
Ligand 28 is flexible enough to accommodate a metal centre having a trigonal geometry, however with an unusually large P-M-P angle (143°) as in the silver complexes [AgCl•28]. In this case, the CD cavity acts as a receptor not only for halido ligands as expected, but also for other small enough anions such as tetrafluoroborate (in [AgBF4•28], Fig. 6) and nitrate (in [AgNO3•28]). Weak interactions between inner cyclodextrin CH bonds and atoms of the anion, together with conventional coordination of the anion to the silver cation are thought to be responsible for this synergistic inclusion phenomenon [34].
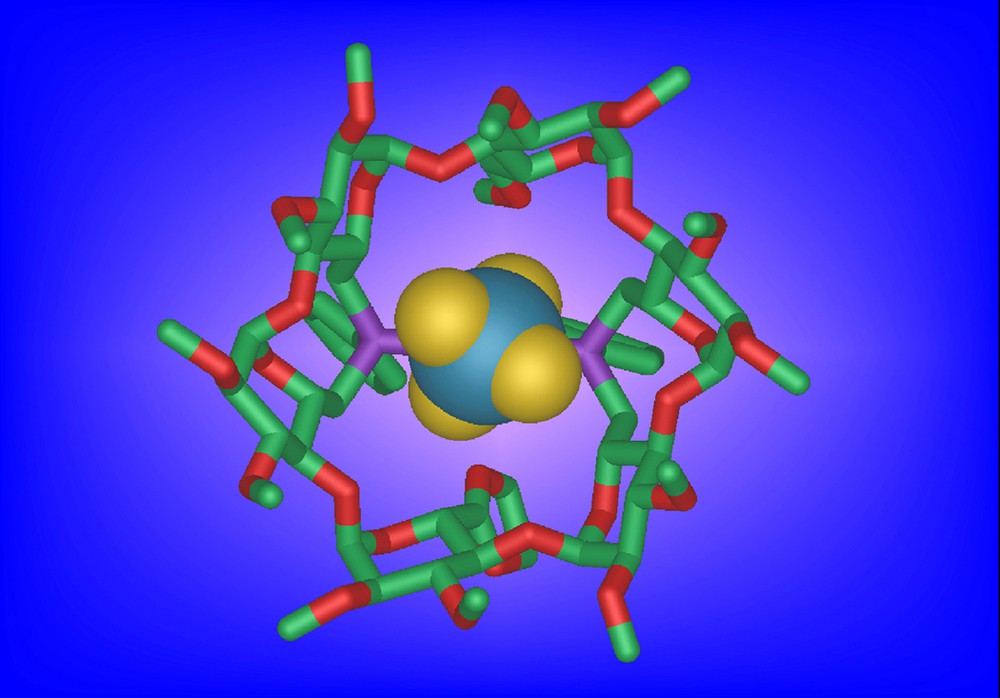
Molecular structure of [AgBF4•28] showing the included BF4 counterion.
3.4 Sulfur chelators
Sulfur like phosphorus is another element perfectly suited for late transition metal coordination, albeit sulfur-based ligands tend to be more labile than their phosphorus counterparts [40]. In addition, the two lone pairs of a prochiral sulfur atom add a degree of complexity when such a donor atom is associated with a chiral entity like a CD macrocycle. In others words, the presence of two diastereotopic lone pairs can lead to inseparable mixtures of diastereomeric complexes if the coordination process is not controlled. The bidentate ligand 29 comprises two lone pairs on each sulfur atom, which lie in two clearly distinct environments: one of them is directed towards the cavity centre, whereas the other points outwards. It was obtained via a highly regioselective but, unlike 28, not specific double capping reaction between tetramesylate 26 and an excess of sulfide dianion [41] (Scheme 12). The formation of the other possible regioisomer 30 is much less favourable because its macrocyclic structure is strongly distorted as revealed by the widespread NMR signals of the H-1 protons [42] (Fig. 7). The two regioisomers can be separated conveniently by column chromatography.
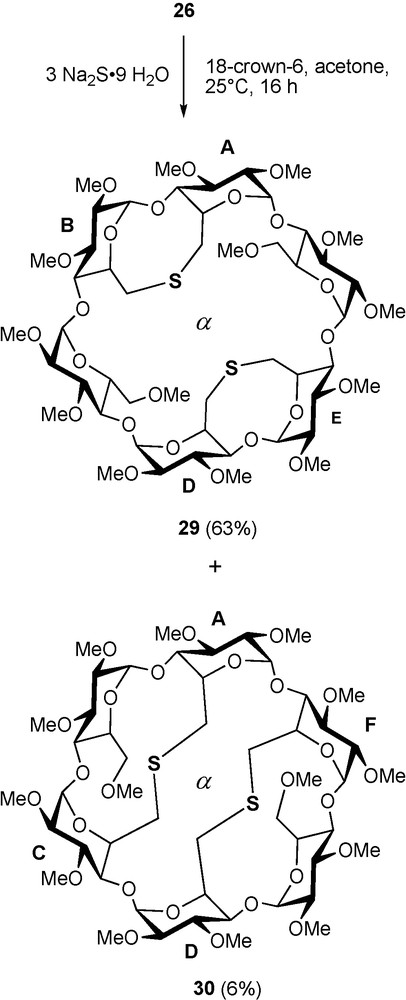
Synthesis of dithioethers 29 and 30.
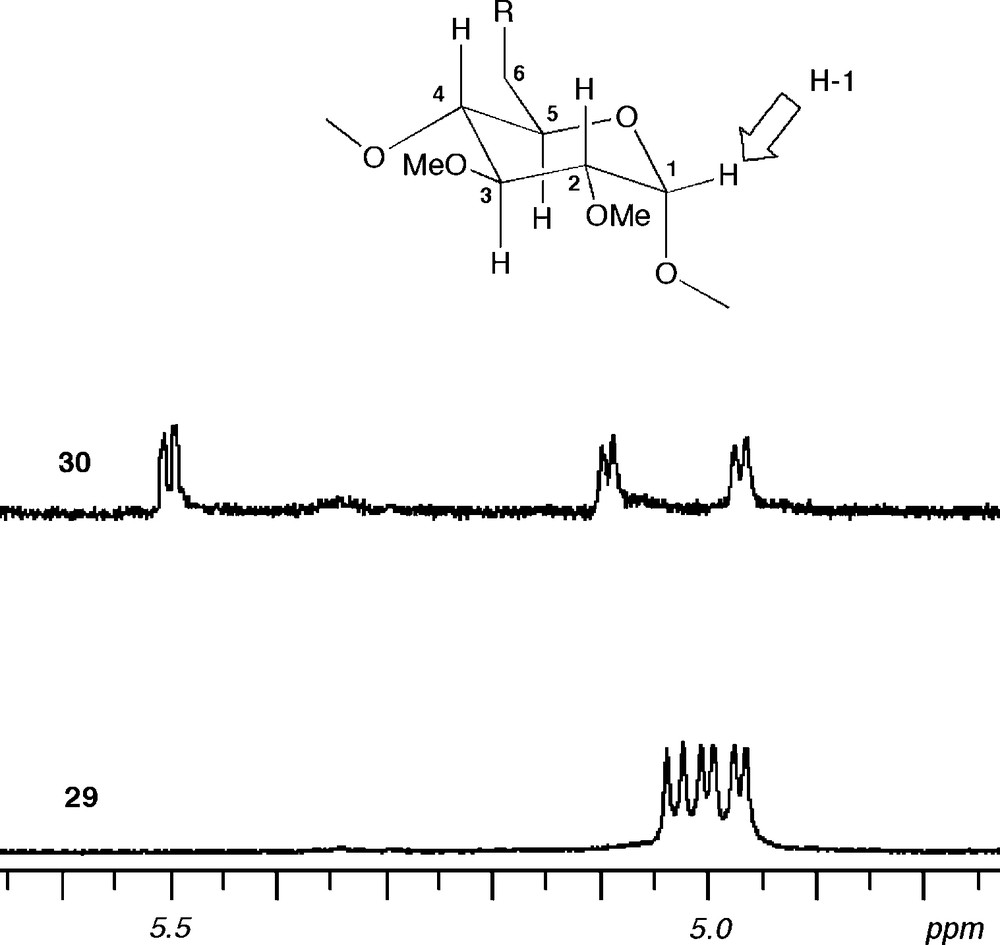
Parts of 1H NMR spectra recorded in CDCl3 at 300.1 MHz showing the H-1 signals of C2-symmetrical 29 (bottom) and 30 (top). In the strongly distorted macrocycle 30, the H-1 protons resonate within a widespread frequency band (Δδmax = 0.53 ppm), whereas in the overall circular CD 29, the same signals lie in a narrow range (Δδmax < 0.07 ppm).
Not surprisingly, when 29 is involved in metal coordination, neutral dimeric species like [{PdCl2•29}2] and [{PtCl2•29}2] are being mainly produced. However, as noticed before, reactions with cations tend to favour chelation rather than oligomerisation. Ligand 29 is no exception as it behaves as a perfect chelator towards Ag+ in [Ag(butanone)•29]BF4. Here, the metal centre is coordinated to the endo rather than to the exo lone pair because the butanone ligand is hosted by the CD cavity.
4 Catalytic properties of chelate complexes derived from methylated CDs
Early studies on CD-containing metal catalysts focused on metallo-enzyme mimics [43–46] and not surprisingly, dealt with biologically relevant catalytic reactions such as hydrolysis of esters. It is only in the mid 1990s that Reetz associated a CD unit with metallo-organic fragments designed to perform industrially important catalytic reactions such as hydrogenation and hydroformylation of olefins (Fig. 8) [47].
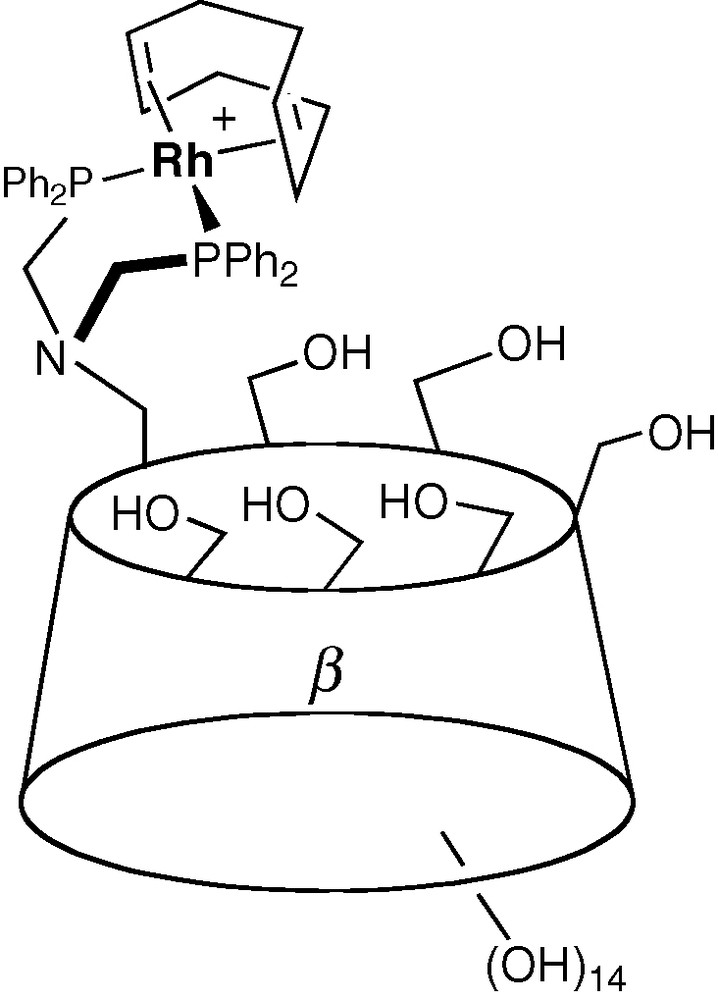
One of Reetz's supramolecular catalyst based on a CD skeleton.
As for enzyme mimics, the idea was to exploit the unique ability of CDs to form inclusion complexes with lipophilic substrates in water in order to direct them to the catalytic centre where they are transformed [48,49]. Here, the catalytic process can qualify as a supramolecular one as attractive weak forces rather than repulsive interactions control access of the substrate to the metal centre. Although these systems showed significant rate of acceleration compared to CD-free analogues for reactants the size and shape of which are compatible with the CD cavity, few of them reached the efficacy of natural enzymes both in terms of selectivity and turnover, as most failed to address the key issue of stabilizing the transition states and intermediates of the catalytic transformation. In the case of metal-capped methylated CDs, it is not so much supramolecular catalysis that was sought after but rather the control of the metal first sphere of coordination via non covalent attractive and steric repulsive interactions so as to induce unusual reactivity at the metal centre which will hopefully alter the catalyst performances in a beneficial way. In addition, stereoselectivities of asymmetric catalytic reactions are expected to improve when the metal centre is brought into close contact with the chiral CD unit.
As expected, when using a CD catalyst, in which the metal centre is far remote from the cavity, the outcome of the catalytic reaction differs little from that of classical, cavity-free ligands such as PPh3. For example, a typical TOF value of 70 mol(aldehydes) mol(Rh)−1 h−1 and l:b ratio of 2.3 were found for the hydroformylation of 1-octene in H2O-MeOH using [RhCl(CO)•19] as catalyst (Scheme 13) [30].
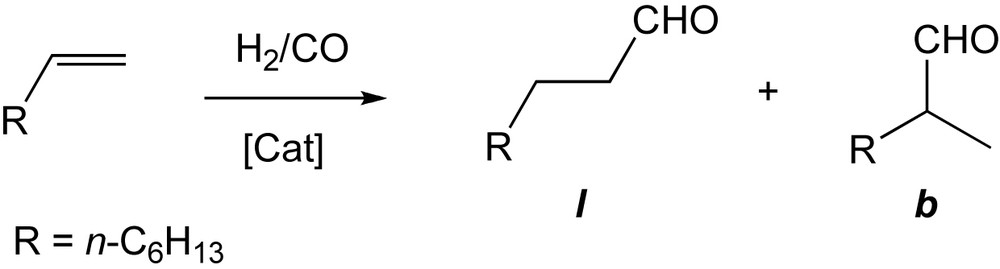
Hydroformylation of 1-octene.
Moreover, the lack of water-solubility prevents such chelate complexes from behaving as a supramolecular catalyst. However, asymmetric induction can still be effective, even if the metal centre is not located in the immediate vicinity of the chiral macrocyclic unit. This could notably arise when the coordinating arms are rigid enough to transmit the chiral information to the chelated catalytic centre. Ee values as high as 84% ((R)-isomer) were obtained with cis chelate complex [Rh(NBD)•25]BF4 in the asymmetric hydrogenation of prochiral dimethyl itaconate although the observed TOF value (3.5 mol(product) mol(Rh)−1 h−1) is rather low. Interestingly, the TOF value is next to zero in the more sterically demanding AD regioisomer [Rh(NBD)•24]BF4 where access of the substrate to the metal centre is severely restricted [36]. Remarkably, very similar ee values (62 and 92% for the hydrogenation of dimethyl itaconate and itaconic acid respectively) were obtained in the presence of the β-CD complex [Rh(COD)•23]BF4, however with an inverted enantioselectivity ((S)-isomer) [33]. Clearly, the mode of enantiodifferentiation must be very different in the latter system, the active site of which is much closer to the CD chiral unit. [Rh(COD)•23]BF4 is also significantly more active than their α-CD counterparts [Rh(NBD)•24/25]BF4, probably because the larger β-CD cavity and AB capping allows better access of the substrate to the metal centre. Dinuclear complex [Rh2(NBD)2•27][BF4]2 [50] proved to be a very poor hydrogenation catalyst despite the fact it contains metal caps similar to those of [Rh(COD)•23]BF4. Again, poor access to the metal centre was blamed for this lack of reactivity. In spite of their trans stereochemistry, nickel complexes of diphosphine 28 ([NiBr2•28], [NiBrMe•28]) were shown to catalyse the dimerisation of small olefins after activation with excess methylaluminoxane (MAO) [38]. The TOF values up to 43,000 mol(C2H4) mol(Ni)−1 h−1 in the case of ethene are not far off those obtained with conventional cis chelate complex [NiBr2(DPPE)] (DPPE = Ph2PCH2CH2PPh2) [51]. In the case of the more sterically demanding propene, an activity of 14 000 mol(C3H6) mol(Ni)−1 h−1 was observed. The distribution of dimerisation products for both reactions is comparable to that of the standard DPPE nickel catalyst. However, the CD precatalysts require larger amounts of MAO than [NiBr2(Ph2PCH2CH2PPh2] to be operational, but once activated, they are remarkably stable as their drop of activity remains small over time. Unusual intermediates with a trigonal-bipyrimidal (TBP) or a sawhorse structure (Fig. 9) were postulated to account for the insertion step, which classically occurs within square-planar [Ni(PR3)2(alkyl)(olefin)]+ complexes with a cis configuration [52,53]. The unusual stability of the catalysts might be related to the strong chelating properties of the ligand.
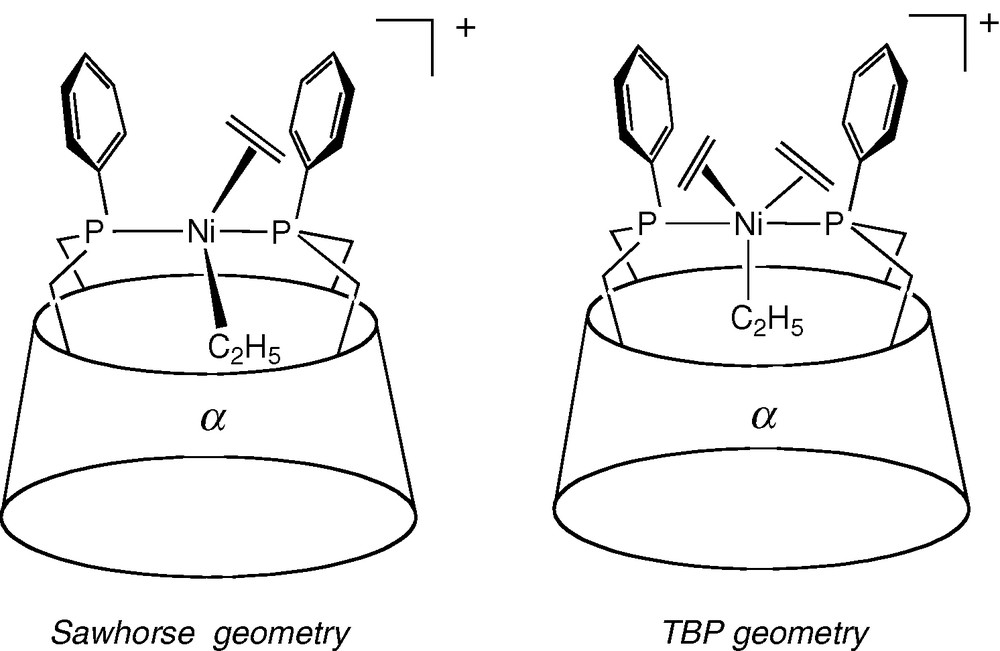
Proposed intermediates in the dimerisation of ethene with [NiBr2•28]/MAO.
MAO activation is also effective for both polymerisation precatalysts [FeCl2•16a] and [FeCl2•16b] [27]. However, only when the cavity is large enough as in [FeCl2•16b], does the activated iron complex catalyse the polymerisation of ethene. The fact that the observed TOF values (125 mol(C2H4) mol(Fe)−1 h−1) are 1000-fold lower than those observed for 31 [54], which also comprises the 2,6-bis(imino)pyridyl core, but with substituted aryl rings instead of a CD cavity as steric protection groups, proves that the latter again restricts somehow access of the substrate to the metal centre. The presence of numerous ether oxygen atoms on the CD macrocycle that might interact unfavourably with the MAO-generated cationic iron(III) active species is another explanation for this lower catalytic rate. In terms of molecular weight, melting temperatures and crystallinity, the polyethylene generated by [FeCl2•16b] is very similar to that obtained with the conventional system 31 [55]. Overall, [FeCl2•16b] constitutes the first example of a molecular polymerization catalyst in which chain growth takes place within a tubular environment.
5 Conclusion and outlook
In this review, we have shown how new derivatisation methods, based on the bulky supertrityl groups, provide easy access to a wealth of primary face functionalised methylated cyclodextrins. These flexible, yet conformationally stable macrocycles constitute versatile preorganisation platforms for the synthesis of chelating ligands capable of positioning a metal centre in such a way that the CD ligand is able to act both as a first and second sphere of coordination towards the chelated ion. Confinement of a metal within a CD cavity has proven beneficial to the stabilisation of catalytic active sites and allows shape and size-selective reactions to take place. Methylated CDs can also be used as chiral ancillary groups provided the catalytic site is fixed with regard to the cavity. The fact that naturally-occurring CD scaffolds can generate not only high, but also inverted enantioselectivities in the hydrogenation of prochiral functional olefins is a clear indication of the potential of such chiral synthons in the design of transition metal catalysts for asymmetric transformations. Current investigations are directed towards water-soluble versions of the aforementioned complexes, which should behave as supramolecular catalysts, and chelate complexes derived from the larger β− or γ-CD, which are likely to display higher catalytic rates.
Acknowledgements
Special thanks go to all the coworkers whose names appeared in the references related to the authors’ work.
1 The definition of cis and trans complexes refers to square planar MX2L2 complexes of the group 10 metal ions.