

1 Introduction
λ3-Phosphinine is an analogue of benzene and that accounts for a wide range of properties, including its π-complex chemistry, but the P-lone pair causes a distinct difference as it qualifies the P-heterocycle as a σ-ligand at the same time [1]. For the late transition metals, like iron for example, the σ-ligand property dominates strongly and [(η1-C5H5P)5Fe] can be isolated from the reaction of an appropriate Fe-source and unsubstituted phosphinine C5H5P [2]. No trace of Fe-π-complexes has been observed for that ligand. On the other hand, even in the absence of shielding o-substituents, sandwich complex [(η6-C5H5P)2V] is formed as the only isolable product of vanadium vapor and C5H5P again [3]. Depending on both, the size of o-substituents and the specific transition metal, a delicate balance of the two principal ligand properties of phosphinine and its derivatives can be stated.
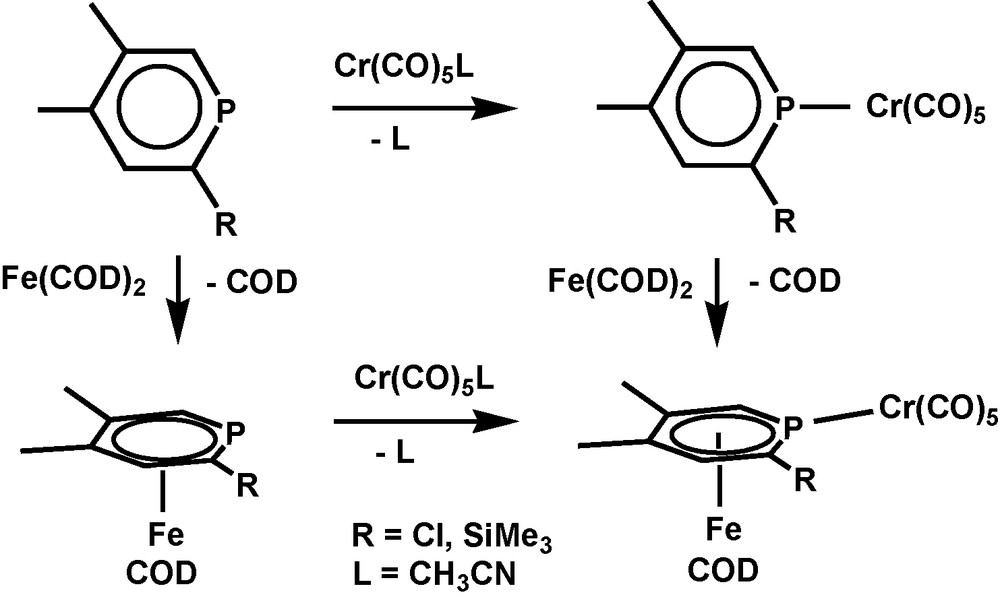
In the case of iron, the coordination mode of the phosphinine depends on the co-ligands as well and σ- and π-ligand properties are almost independent from each other. (1,5-cyclooctadiene)Fe fragments form only [(1,5-COD)(η6-phosphinine)Fe] complexes, for o-Cl or SiMe3 substituted phosphinines irrespective of additional σ-coordination [4] (Scheme 1).
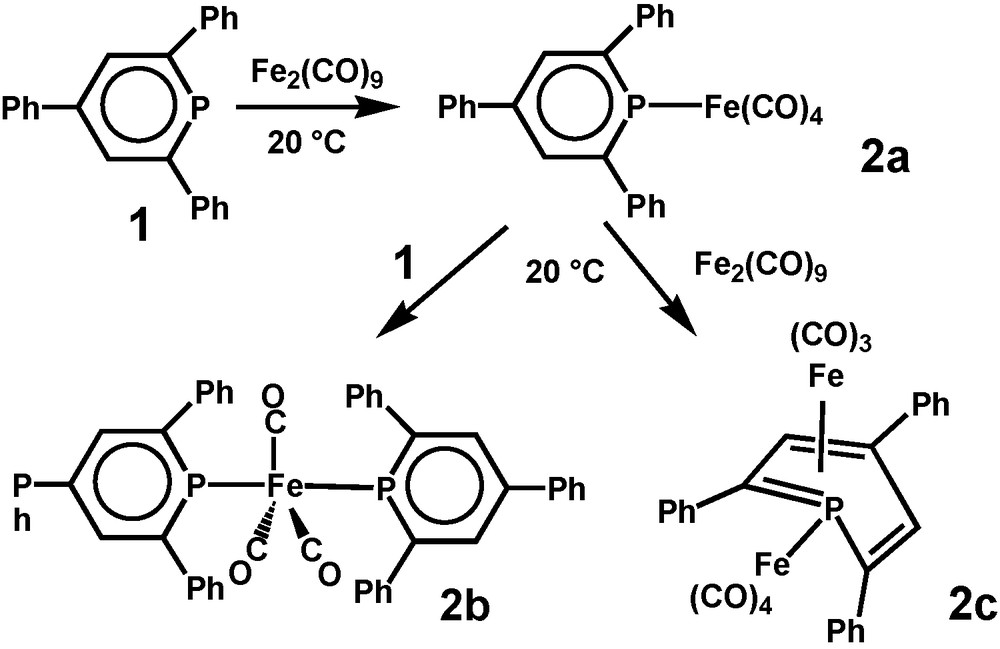
[(Phosphinine)nFe(CO)m] complexes offer more structural options. σ-Coordination dominates for 2,4,6-triphenylphosphinine (1) as one or two may substitute CO ligands to form (2,4,6-triphenylphosphinine)Fe(CO)4 (2a) and (2,4,6-triphenylphosphinine)2Fe(CO)3 (2b), respectively, but η4-bonding adds to that in case of an occupied phosphorus lone pair. (μ,η4-2,4,6-triphenylphosphinine)Fe2(CO)7 (2c) was identified as an unexpected binuclear reaction product. 2a proved to be an intermediate for the formation of 2b as well as for 2c. Neither a [(η6-phosphinine)Fe(CO)2] derivative, nor its dinuclear [(μ,η6-phosphinine)Fe2(CO)6] version has been observed [5] (Scheme 2).
If compared to hydrocarbons as arenes, (η4-arene)Fe complexes have been observed for naphthalene derivatives to form stable [(η4-naphthalene)FeL3] complexes [6], (L = P(OR)3; L3 = η6-arene) but monocyclic arene ligands like benzene or toluene have been identified as η4-ligands with respect to iron only in the case of highly reactive [(η6-arene)(η4-arene)Fe] species which decompose in the range of −50 to −60 °C [7,8]. The phosphorus atom of 2c thus causes a stabilization effect that equals that of a non-coordinated naphthalene ring with its aromatic π6-electron system. The molecular structure of 2c in the crystalline state is depicted in Fig. 1.
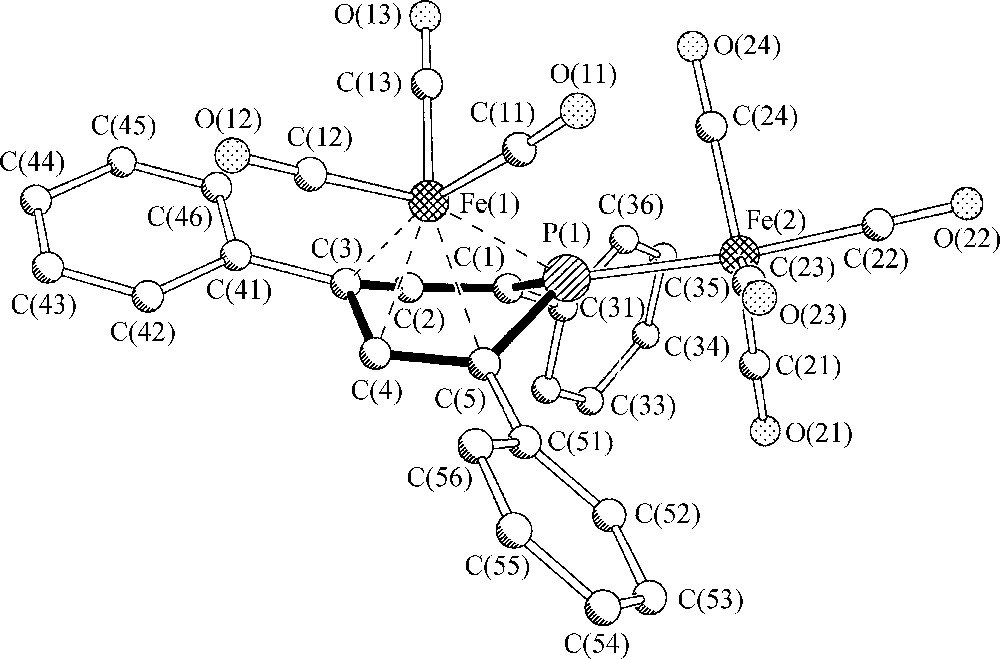
Molecular structure of (μ,η4-2,4,6-triphenylphosphinine)Fe2(CO)7 2c in the solid state. Hydrogen atoms have been omitted for clarity.
It documents a folding of the phosphinine ligand by 46.4° along the line P(1)-C(3), a value slightly above the respective folding angles of (η4-napthalene)FeL3 (L = P(OMe)3, L3 = η6-hexamethylbenzene) complexes (34° and 41°) [6]. This increase may be a consequence of the specific σ,π-bonding situation of the phosphorus atom and the repulsive interaction between a Fe(CO)4 fragment and two o-phenyl phosphinine substituents. Significant π-electron localization within the heterocycle is indicated by alternating CC and PC intraring bond lengths [5].
Only one λ3,λ3-diphosphinine transition metal complex family appeared in the literature. It concerns [(η6-1,3-diphosphinine)(η4-1,3-diphosphete)Fe] complex class 4, which has been formed by a [2 + 2 + 2] cyclic addition reaction of two moles of tert-butylphosphaalkyne (3), one mole of a terminal alkyne, and [(η4-1-methylnaphthalene)(η6-toluene)Fe] (5) as a highly reactive Fe(0) source [9] (Scheme 3). Functional alkyne substituents R′ with alcohol, ester, or ether groups are tolerated. Targeted attempts to prepare W(CO)5 complexes of 1,2-diphosphinines failed. Only (1,2-dihydro-1,2-diphosphinine)W(CO)5 derivatives have been obtained instead [10].

Based on the spectroscopic and structural features of 4, the (η6-1,3-diphosphinine)Fe complexes can be regarded as prototypical (heteroarene)Fe π-complexes, but their thermal stability with a decomposition temperature around 300 °C is remarkable. The compounds are almost as stable as ferrocene or its oligophospha analogues, but exceed the stability of most (η6-arene)Fe complexes with hydrocarbon π-ligands significantly. On the other hand, redox reactions of 4 result only in short lived ions, whereas the oxidation of its toluene complex analogue [(η4-2,4-di-tert-butyl-1,3-diphosphete)(η6-toluene)Fe] (6) yields a stable complex radical cation 6+ [11]. A preparative useful oxidative ligand decomplexation reaction of 4 by FeCl3 or CCl4 allowed the isolation of the respective free 1,3-diphosphinines as stable organophosphorus compounds. X-ray and PES spectroscopic investigations as well as MO calculations qualified them as planar benzene analogues with a significantly reduced HOMO-LUMO gap [12]. These results encouraged us to try investigating the chemistry of (1,3,5-triphospinine)Fe complexes.
2 2,4,6-Tri-tert-butyl-1,3,5-triphosphinine iron interactions
Cyclotrimerization of P-alkyne 3 should lead preferably to 2,4,6-tri-tert-butyl-1,3,5-triphosphinine (7) as the proposed thermodynamically favored aromatic P3C3R3 valence isomer. Some attempts of that sort appeared in the literature and resulted in surprisingly different structural motifs for the obtained complexes, including open and spirocyclic P3C3R3 moieties, depending on the metals used and the conditions applied [1]. Binger et al. reported about two independent routes to free 7 by cyclotrimerization of 3 in the coordination sphere of suitable (COT)Hf and amidovanadyl fragments and decomplexation of the heterocycle [13]. The first η6-arene complex of 7 was obtained by reaction of an excess of P-alkyne 3 with scandium atoms. A triple decker complex was formed in small yield with two terminal (η5-3,5-di-tert-butyl-1,2,4-triphospholyl)Sc units and a μ,η6:η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine middle deck [14]. The availability of free 7 allowed ligand displacement reactions of some Mn, Mo, W, and Ru (η6-arene)M complexes with hydrocarbon ligands to yield the related [(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)MLn] complexes. They are stable compounds [15]. PES spectroscopy and DFT calculations of free 7 as well as a series of [(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)M(CO)3] (M = Cr, Mo, W) complexes gave clear hints on the impact of the three phosphorus atoms on the electronic properties of the benzene ring system. Main difference of 7 and benzene is a significantly diminished HOMO – LUMO gap for 7, which is even smaller than that of 1,3-diphosphinine. The effect concerns the π*-orbitals much more strongly than the occupied π-orbitals. As a consequence, 7 acts as a much better π-acceptor ligand than benzene, but its π-donor capability is almost the same [12,16]. Potassium metal reduction of 7 has been reported to remove one phosphorus atom from 7 to form the related aromatic 2,4,5-tri-tert-butyl-1,3-diphospholyl anion, but electrochemistry revealed a quasi-reversible reduction of 7 at −2.315 V vs. Cp2Fe+/Cp2Fe in THF [17]. As no EPR data for the postulated anion 7− were reported, we reinvestigated the electrochemistry of 7 and determined the EPR spectroscopic properties of 7−. A reversible reduction of 7 was observed at a potential of −1.94 V vs. SCE at room temperature in DME as the solvent. A reason for the differing potentials has not been found, but the redoxcouple 7/7− was stable under the conditions of the experiments of this study. The experimental reduction potential of 7 accounts for a dramatic anodic shift of +1.48 V, if compared to that of benzene (−3.48 V vs. SCE) [18]. It agrees very well with the calculated orbital energy reduction of the π*-orbitals of free 1,3,5-triphosphinine P3C3H3 (7a) by just over 2 eV with respect to benzene [16]. X-band EPR spectroscopy of potassium reduced 7− in toluene proved a clean electron transfer process with the alkaline metal at low temperature too, as only one single species appeared in the spectrum with an AB2 31P hyperfine structure (Fig. 2).
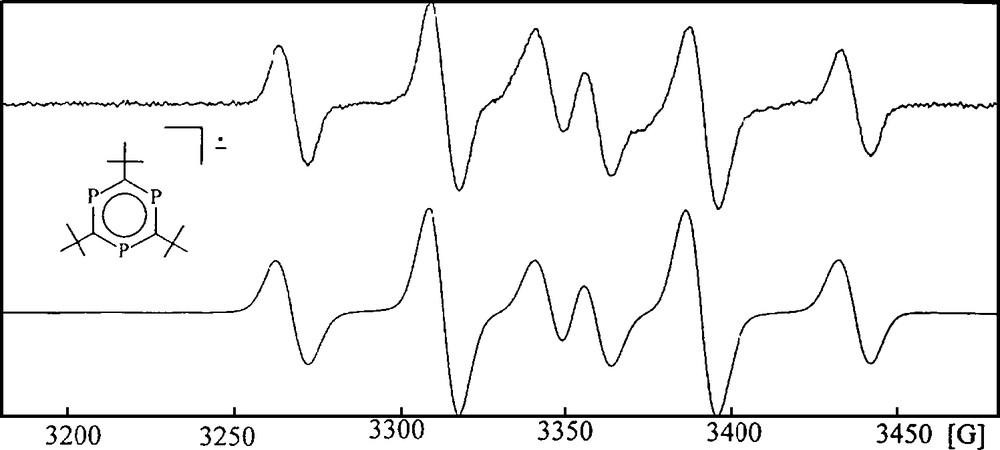
X-band EPR spectrum of 7−K+ in toluene at −60 °C (top); 〈g〉 = 2.0163. Simulation: AB2 31P hyperfine structure, aA(31P) = 77.9 G, aB(31P) = 45.7 G (bottom).
The 31P hyperfine structure of 7− agrees with a single occupation of one of the degenerate 2e″ π*-orbitals [16]. A static Jahn-Teller distortion leads to the distinct differentiation of the A and B2 31P nuclei and causes loss of its C3-symmetry.
Highly reactive 5 (Tdecomp ∼ 0 °C) [6] was reacted with 3 in the absence of alkynes to form [(η4-2,4-di-tert-butyl-1,3-diphosphete)(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)Fe] (8) in analogy to the formation of 1,3-diphosphinine complex 4 as the target product. 8 did not appear in the reaction mixture, but cyclodimerization product 6 and pentaphosphaferrocene derivative 9, a valence isomer of 8. They represent the strongly dominating main products of the reaction [11] (Scheme 4). This was a surprise, as the formation of the two five membered ring ligands of 9 requires breaking of a PC triple bond of 3 even below room temperature. All attempts to solve the problem by variation of reaction conditions or other reactive Fe(0) complexes as starting materials failed to produce 8, but in case of an excess of 3, novel free and iron-complexed Pn(CR)m species appeared in the reaction mixture, most of them with cage structure [19]. [P6C6R6Fe] complex 10 with its bycyclic P4C4R4 ligand is formed in reasonable yield, but the heptamer iron complex 11 is a side product. It represents the highest oligomer of 3 reported in the literature.

18-valence electron (VE) sandwich complexes 6 and 9 are diamagnetic and robust compounds which are not easily oxidized by air contact. In contrast to that, 16 VE complexes 10 and 11 are open shell triplet species, but comparably resistant towards oxidation. Prolonged air contact of 11 resulted even in a chemoselective oxidation of the two equivalent P-atoms of the six membered ring of the cage ligand outside the 2-phospaallyl moiety to form O = PR3 units without iron decomplexation [8].
Highly reactive 16 VE species [(1,5-COD)2Fe] was reacted with 7 to yield [(1,5-COD)(2,4,6-tri-tert-butyl-1,3,5-triphosphinine)Fe] (12) as the target product. In spite of a long reaction time of three weeks, the reaction has to be done at −40 °C, because the diamagnetic reaction product decomposes at room temperature to regenerate 7 and form iron slurry. 31P NMR spectroscopy of the cold reaction mixture revealed complete conversion of 7 into a ligand with an AB2 spin system and the parameters δA = 1.8, δB = 5.4, and JAB = 32.8 Hz. The data point to a mirror symmetric, unsaturated P3 π-ligand of 12 which is incompatible with a (η6-1,3,5-triphosphinine)Fe fragment and all complexed valence isomers of 7 reported to date in the literature. It is remarkable to observe the same AB2 spin system for the three P-atoms of diamagnetic 12 and free radical 7−.
With the aim of enhancing the crystallization properties of 12 by forming a binuclear FeCr σ,π-complex [(CO)5Cr(THF)] was added at −40 °C after finishing the π-complexation step of the reaction. A stable paramagnetic reaction product 13 was isolated in small yield after chromatographic workup and recrystallization (Scheme 5). Due to non-interpretable 1H and missing 31P and 13C NMR spectra of paramagnetic 13, only X-ray crystallography allowed identification of the binuclear complex. Besides the σ-bound Cr(CO)5 fragment, compound 13 contains all atoms of 12, however, neither 1,5-COD, nor 7 were observed as ligands of [(η5-trihydropentalenyl)Fe(μ,1-3-η-4,5,6-trihydro-1,3,5-triphosphinine)Cr(CO)5] (13) (Fig. 3).
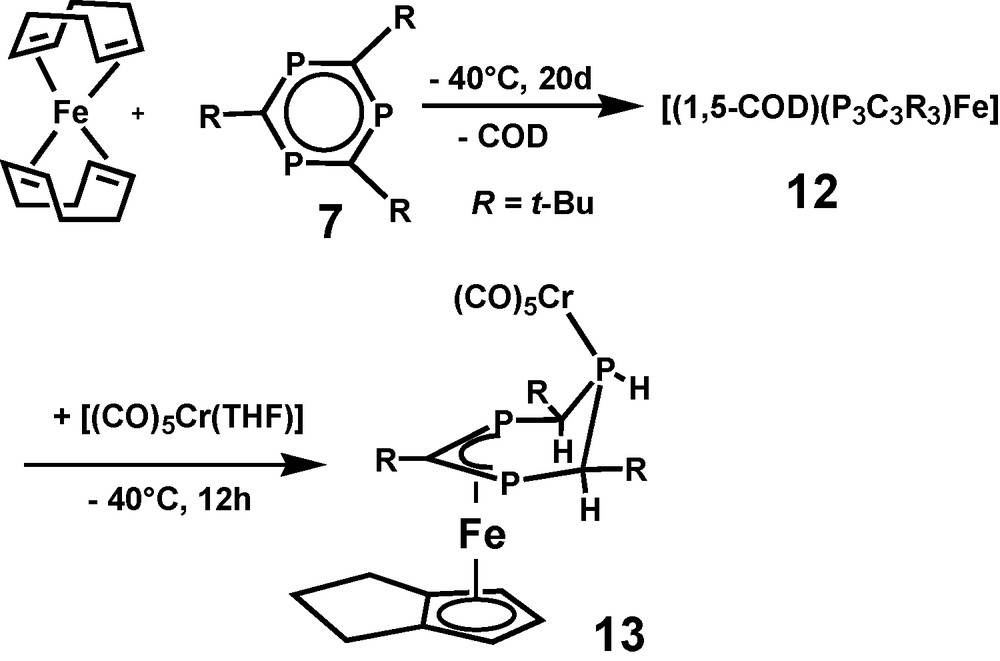
Molecular structure of [(η5-trihydropentalenyl)Fe(μ,1-3-η-4,5,6-trihydro-1,3,5-triphosphinine)Cr(CO)5] 13 in the solid state. Hydrogen atoms are omitted for clarity; C(1), P(1), and C(3) carry hydrogen atoms at the ring side of the iron atom.
Binuclear 13 has an electron count of 16 VE for the central Fe atom. Its paramagnetism equals that of 10 and 11. Besides substantial differences in their coordination spheres, all three share 1,3-diphosphaallyl ligand moieties. As the ring atom connectivity of the 4,5,6-trihydro-1,3,5-triphosphinine ligand of 13 is the same as that of 1,3,5-triphosphinine 7, there is no doubt about its origin from 7. With the exception of the Cr(CO)5 unit, 13 appears to be a rearrangement product of thermo labile 12. Related rearrangement processes which transform 1,5-COD by dehydrogenation into bicyclic pentalene and its derivatives have been reported in the literature. A quantitative dehydrogenation reaction of 1,5-COD to form pentalene is catalyzed at Pt-surfaces. As for the observations of this study, the process requires no heating [20]. Dehydrogenation of a cyclooctenyl ligand to form the same η5-trihydropentalenyl ligand of 13 was reported for [(η3-cyclooctenyl)(η4-1,5-COD)Co] in the presence of free 1,5-COD as an intermolecular hydrogen acceptor [21]. As a consequence of forming an aromatic η5-trihydropentalenyl ligand out of the isolated diene 1,5-COD, a thermodynamic driving force is assumed for that part of the process which leads to 13. The same does not account for the threefold reduction of 7 in the coordination sphere of the iron atom, which transforms an aromatic π6-system into a cyclic π4-1,3-diphosphaallyl anion. As the iron atom is oxidized at the same time to form Fe2+, no simplifying models can be applied to analyze the reaction which leads to 13.
3 Density functional theory (DFT) calculations
Density functional theory (DFT) calculations of the compounds of this study were performed using Gaussian 98 [22] with the B3LYP hybrid functional [23]. In the case of the molybdenum complexes, the LanL2DZ basis set was used [24]. The basis set was augmented with a set of d-functions with exponents of 0.6 and 0.34 on C and P, respectively, and p-functions with exponents of 0.072 on Mo (LanL2DZP) [25]. In the case of the iron complexes, the Schäfer-Horn-Ahlrichs split valence basis set was used [26]. The basis set was augmented with a set of d-functions with exponents of 0.6 and 0.34 on C and P, respectively, and p- as well as f-functions with exponents of 0.098 and 1.05 on Fe and 0.086 and 0.87 on Cr (SHAsvp) [25]. Due to the observation of singlet and triplet ground states for the iron complexes, both have been calculated. In the case of triplet ground states they were calculated on the basis of the unrestricted formalism. As B3LYP is not very precise in determining energy differences between singlet and triplet states, the structures were reoptimized with the B3LYP* functional of Reiher, Salomon, and Hess [27] using the SHAsvp basis set described above. To reduce the complexity of calculations, the P-heterocycles with their C-tert-butyl units were represented in most cases by their respective C–H analogues.
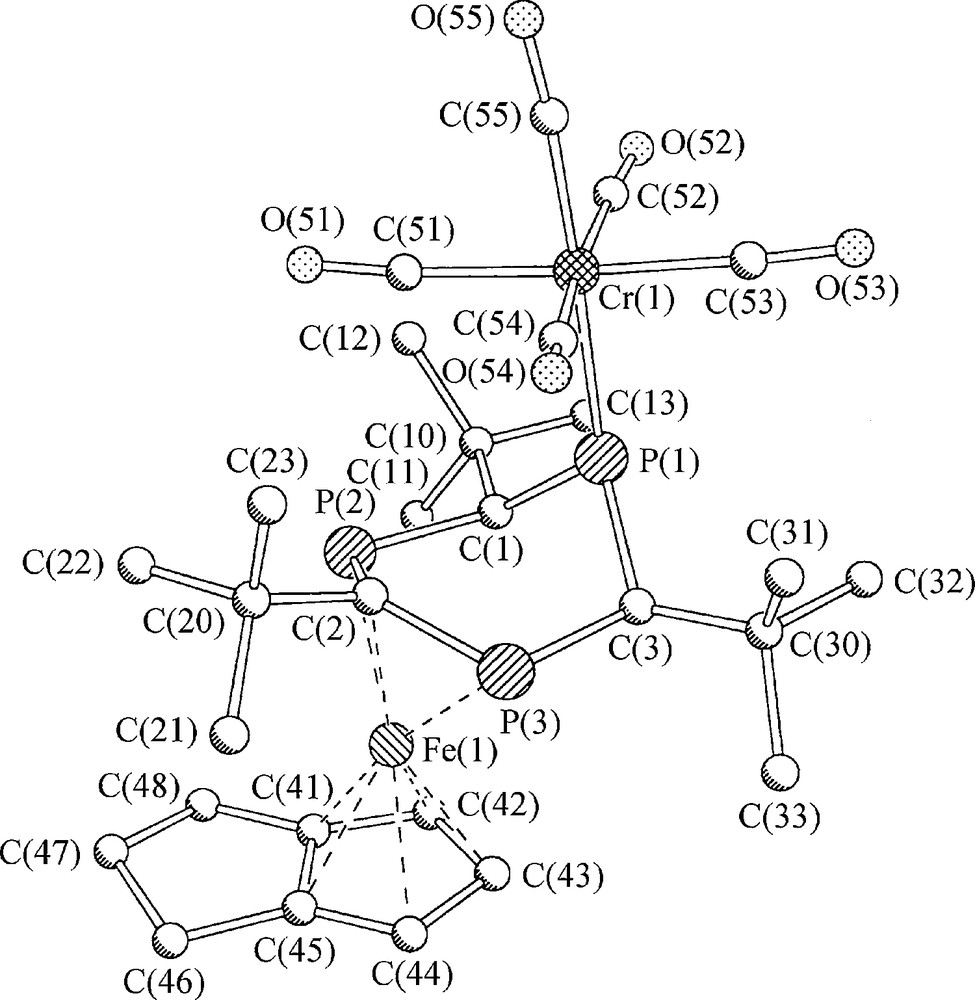
Geometry optimization of the CH version 12a of the proposed reaction product [(η4-1,5-COD)(1,3,5-triphosphinine)Fe] 12 did not result in a minimum structure with a planar C3 symmetric triphosphinine ring. An alternative minimum structure 12b with a boat shaped triphosphinine ligand was found instead. An intramolecular Fe(0) → 1,3,5-triphosphinine two-electron transfer led to two localized PC double bonds within a non-planar, boat-shaped ring and the formation of the (η6-1,3,5-triphosphacyclohexa-2,5-dine-1,4-diyl)Fe fragment of valence isomer 12b (Fig. 4).
B3LYP/SHAsvp calculated schematic structures and energies of 12a, 12b, and 13a.
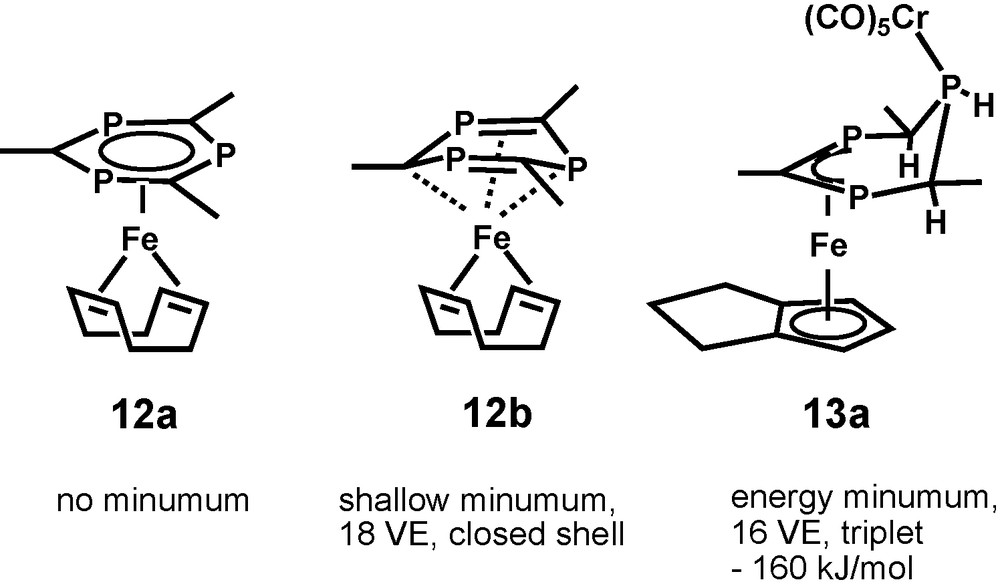
A choice of calculated structural parameters of 12b is given in Fig. 5. Diamagnetism, symmetry reduction of the 1,3,5-triphosphinine ligand, and the small stabilization energy of 12b are compatible with the observation of a well-resolved AB2 31P NMR spectrum and thermolability of 12 at room temperature. 12 is thus believed to be [(η4-1,5-COD)(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphacyclohexa-2,5-dine-1,4-diyl)Fe]. In the light of that result, identity of the 31P spin systems of diamagnetic 12 and radical 7− is not accidental, but reflects the single respectively double occupancy of analogous orbitals, which cause the distortions of 7− and 12.
B3LYP/SHAsvp calculated structural parameters of 12b.
This finding represents a strong contrast to the formal Ru-analogue of 12 [(η4-1,5-COD)(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)Ru] (14) [15]. As for other (η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)Ru complexes [29], 14 appears to be a classical (η6-1,3,5-triphosphinine)M(0) complex with a planar and C3-symmetric heteroarene ligand. As a consequence, the iron atom of a (η4-1,5-COD)Fe fragment has to be regarded as significantly richer in electron density than its Ru-analogue. The incoming 1,3,5-triphosphinine ligand with its excellent π-acceptor properties and its low reduction potential oxidizes the iron atom to form 12 as a Fe2+-complex with the dianionic [P3C3R3]2− ligand, but ruthenium complex 14 is composed of a Ru(0) central metal and neutral ligand 7.
Model complex 13a appeared to be the global energy minimum among all of its isomers which were investigated. If the contribution of the Cr(CO)5 fragment is eliminated, a stabilization energy of −152 kJ/mol can be estimated. This is a strong driving force for the observed rearrangement process 12 → 13. In agreement with the experimental observations with 13, model complex 13a has triplet ground state with the maximum of the spin density in the vicinity of the metal atom and the diphosphaallyl-unit. (Fig. 6). A mechanism for the highly complicated rearrangement process 12 → 13 cannot be suggested in the moment, but an intramolecular mechanism is likely as all three incoming hydrogen atoms of the trihydro triphosphinine ligand are located at Fe-complexed side of the ring. The role of the Cr(CO)5 moiety might be related to rehybridization of the σ-complexed P-atom and strong repulsive interactions with the two adjacent o-tert-butyl substituents of the heterocycle. Both favor ring deformation processes which are crucial for the observed rearrangement.

B3LYP/SHAsvp calculated structure and spin density distribution of triplet complex 13a.
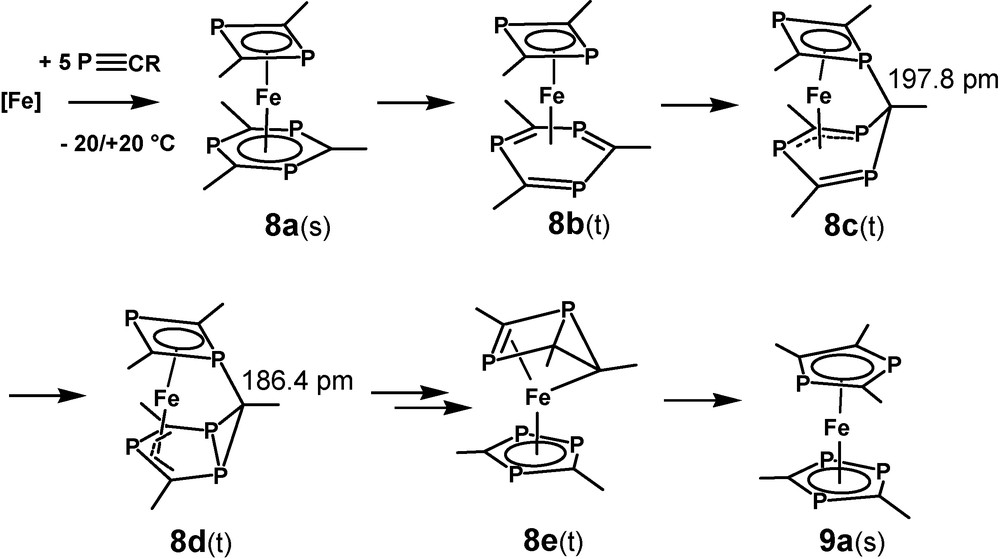
Theoretical evidence for a lack of stability of [(η4-1,5-COD)(η6-1,3,5-triphosphinine)Fe] 12a and the related experimental observations for 12 led directly to the question about the stability of the non-observed species 8 and to the even more interesting point, if there might exist a low-activated rearrangement process between 8 and its pentaphosphaferrocene valence isomer 9 [28]. Unlike 12b, the C–H model compound [(η4-1,3-diphosphete)(η6-1,3,5-triphosphinine)Fe] (8a) forms a distinct energy minimum for a singlet species 8a(s), which is stabilized by −63.6 kJ/mol (B3LYP*/SHAsvp) with respect to the closest triplet state. This should be enough for isolating such a compound at room temperature, however, the influence of the tert-butyl substituents on the stability of 8 is not negligible and not easily to be estimated. Detailed studies which include the substituents are underway, but results are not yet available.
8a(s) forms a sandwich complex with a slightly boat-like distorted η6-1,3,5-triphosphinine ligand but ring bending and bond length alternations are much smaller as in the case of 12a. (Fig. 7). In a chemical sense this can be related to the coligand 1,3-diphosphete. In contrast to 1,5-COD, it is an excellent π-acceptor ligand and competes with the triphosphinine for the iron 3d-electrons. This reduced iron electron density of 8a(s) is incompatible with the specific Fe-triphosphinine interaction of 12 and the central iron atom of 8a(s) remains in a formal zero redox state. Three related triplet structures which form local minima have been identified. One of them is structure related to the crucial intermediate for a reaction, which might be viewed as a model for a low-activated ring element exchange reaction between the two ligands of 8. This concerns 8b(t) with a η4-triphosphinine ligand, a structural motif which is closely related to η4-phosphinine complex 2c. 8b(t) is a candidate for significant substituent influence on the rearrangement process as the space available for them increases through ring folding. A nucleophilic attack of one of the P-lone pairs of the 1,3-diphosphete ligand on the outer carbon atom of the coordinated diene part of the η4-triphosphinine ligand leads to the formation of a single P-C-bond of 197.8 pm between the two rings of triplet species 8c(t) (Fig. 7).
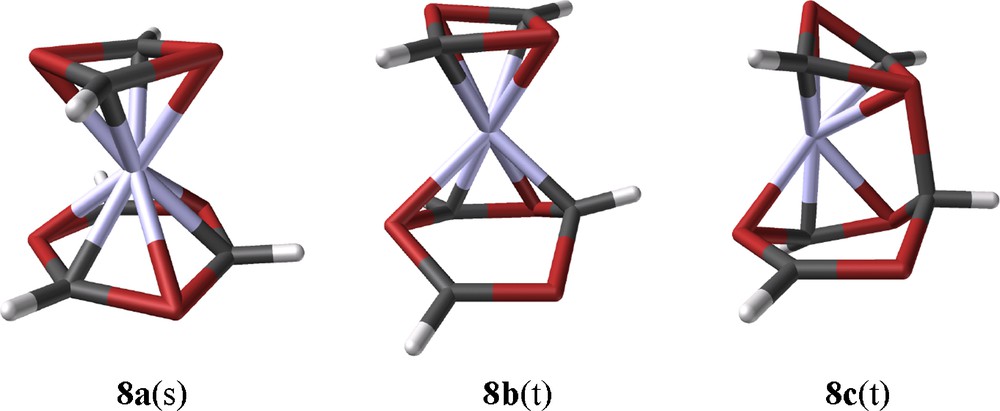
B3LYP*/SHAsvp calculated ground state structures of singlet ground state [(η4-1,3-diphosphete)(η6-1,3,5-triphosphinine)Fe] 8a(s), intermediate triplet state 8b(t) (Erel: 79.6 kJ/mol), and the rearrangement triplet species 8c(t) (Erel: 78.7 kJ/mol).
The calculated molecular structure of 8c(t) seems to be somewhat strange; however, it is isoenergetic with 8b(t) and there exists a case of precedence. A reversible interligand bond forming process of that kind has been observed between two of the π-ligands of tris(2,4-di-tert-butyl-1,3-diphosphete)Mo (15) [30]. This specific reactivity was rationalized as a consequence of a pronounced ambiphilicity of π-bound unsaturated P-heterocycles. DFT calculations of methyl derivative 15a yielded a Cs-symmetric structure as the ground state with an interligand P-C bond length of 201 pm. The calculated structural data of 15a are closely related to the low-temperature X-ray data of 15. (Fig. 8). C2v-symmetric structure 15a′ with its three independent 1,3-diphosphete ligands represents the transition state for a reversible bond formation between the top 1,3-diphosphete ligand and one of the two other ones. This type of structure was assumed as the ground state of 15, when the compound was prepared for the first time [31]. 15a is stabilized by −14.6 kJ/mol (B3LYP/LanL2DZP) with respect to transition state 15a′. Calculations with hydrogen substituents of the 1,3-diphosphete ligands, led to the same principal structures, but the activation energy is reduced to only 2 kJ/mol for the exchange process. Methyl substituent added thus more than 12 kJ/mol to the energetic differentiation of the two states 15a and 15a′, the contribution of the tert-butyl substituents of 15 is definitely bigger.
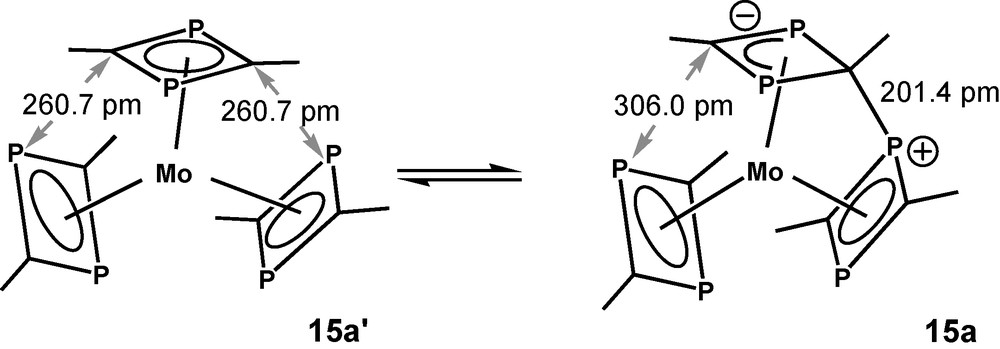
Schematic drawings of the B3LYP/LanL2DZP calculated ground state of 15a and the transition state 15a′ for the reversible interligand bond formation reaction of tris(2,4-dimethyl-1,3-diphosphete)Mo 15a.
Next suggested step for the rearrangement of 8a(s) leads to 8d(t) by formation of a bicyclic P3C3H3 ligand. (Fig. 9). It exhibits a π-bound 2-phosphaallyl moiety and a shortened interligand PC bond length of 186.4 pm. Formation of a bicyclic ligand releases some of the strain of 8c(t). This is reflected by gaining −13.8 kJ/mol relative energy with this step. Additional −21.4 kJ/mol are released when 8d(t) is transformed into 8e(t) by breaking two PC bonds of the three membered ring of 8d(t) and forming the bicyclic P3C2H2 ligand of 8e(t). This part of the rearrangement cannot proceed in a single step, but no meaningful intermediates have been located in-between these two species. Opening the internal PC bond of 8e(e) finishes the rearrangement reaction with the formation of pentaphosphaferrocene 9a(s) and releasing its relative energy of −210.2 kJ/mol with respect to 8a(s). The calculated structure parameters of 9a(s) and its diamagnetism agree very well with the experimental data of 9, if the tert-butyl substituents are taken into consideration [11,32]. We regard this as a good test for the applied model.

B3LYP*/SHAsvp calculated ground state structures of 8d(t) (Erel: 64.9 kJ/mol), 8e(t) (Erel: 43.5 kJ/mol) and pentaphosphaferrocene 9a(s) (Erel: −210.2 kJ/mol).
The complete rearrangement process can be rationalized by the schematic drawings of Scheme 6. 8a(s) – 8d(t) can be regarded as formal Fe(0) complexes, but 8e(t) and 9a(s) are Fe2+ compounds with two anionic ligands. It seems to be worth mentioning, that the majority of the complicated rearrangement process takes place in the regime of the triplet states. The specific iron-P-heterocycle interaction grants a low singlet – triplet activation energy in most cases. As only 16 VE are required for the triplet species, the intermediates gain structural flexibility if compared to the related singlet 18 VE states. That helps adopting the necessary structural changes from intermediate to intermediate. A point of concern remains in the moment. The first step that converts 8a(s) into 8b(t) seems to be relatively high in energy for a reaction, which takes place for its tert-butyl derivative around or even below room temperature, but the influence of the substituents is under investigation. The structural and electronic flexibility of complexes that contain iron atoms and unsaturated P-heterocycles or P-alkynes seems to be almost unlimited in the light of these studies.
Complex 8c(t) exhibits an open side of the molecule that should be suitable for reactions with additional ligands and its 16 VE count points to the same direction. In the case of an excess of P-alkyne 3 with respect to the iron source 5, 3 will definitely attack 8c(t) if it is present in solution at all. This consideration was followed by the same theoretical methods as outlined above [28]. As suggested, a P-alkyne binds side on at the iron atom of 8c(t) to form 16a(t). This initiates a cage forming process. Two antiparallel PC single bonds connect the former four and six membered ring ligands of 16b(t) and squeeze the iron metal atom out of the central position between these rings. This process generates extra space in the coordination sphere of the metal atom for adding another P-alkyne. Additional electron density from the P-alkynes allows the metal to move further out of the cage in construction. The triphosphacyclohexa-1,3-diene moiety of 16b(t) rearranges in a way that generates a π-complexed 2-phosphaallyl unit of 16c(s). Cyclic addition of the two P-alkyne ligands of 16c(s) requires two principal steps to yield the 1,3-diphosphete ligand of 16d(s) without significant changes of the Fe(P5C5H5) cage fragment structure. The final step of the reaction sequence deals with an nucleophilic attack of a P-lone pair on a CH unit of the 1,3-diphosphete ligand to finish with the formation of the CH version 11a(t) of PCR-heptamer iron complex 11 (Scheme 7). This step is just a reversed version of the interligand PC bond formation step 8b(t) → 8c(t), where the 1,3-diphosphete ligand played the role of the nucleophile.

As observed for the rearrangement reaction 8a(s) → 9a(s), singlet and triplet states play a role for the addition of two P-alkynes to intermediate 8c(t) to form 11a(t) in the end. For 16a, the triplet state 16a(t) is stabilized by −7.1 kJ/mol with respect to its singlet analogue 16a(s), 16b(t) is stabilized by −29.3 kJ/mol, 16c(s) by −2.9 kJ/mol, 16d(s) by 29.7 kJ/mol, and the final product 11a(t) is stabilized by −33.5 kJ/mol with respect to 11a(s). After correction for the additional two P-alkyne units, a relative stabilization energy of −205.6 kJ/mol was determined for 11a(t) with respect to 8a(s). As a consequence, 11a(t) has to be viewed as almost isoenergetic with pentaphosphaferrocene 9a(s). That explains the pronounced thermal and oxidative stability of 11, which allows even P-oxidation without decomplexation of the metal or significant structural changes with the exception for the two additional oxygen atoms. The calculated structural parameters of molecular 11a(t) are close to the experimental X-ray data of 11 if the different substituents are taken into consideration [19]. This demonstrates the quality of the model applied even for the triplet states.
The rearrangement reaction sequence 8a(s) → 9a(s) and the formation of 11a(t) are directly linked by the common intermediate 8c(t). Besides the relative concentrations of iron starting material 5 and P-alkyne 3, the relative activation energies for the competing steps 8c(t) → 8d(t) and 8c(t) → 16a(t) control the access to the two reaction channels. The activation energies have not been determined for the model compounds, but the relative energies of 16a(t) and 8d(t) are in favor with −40.6 kJ/mol for 16a(t) after correction for their different composition. If this relation would account for the experimental accessible tert-butyl derivatives, a significant preference for the formation of heptamer complex 11 would have resulted. Experiments proof the contrary. Pentaphosphaferrocene 9 dominates strongly. Again, the non-negligible influence of the tert-butyl substituents will contribute their part to that. On the other hand, the extensive DFT calculations allowed connecting so different structures as 9 and 11 to a common part of the complex reaction sequences that lead to the final products.
4 Conclusions
What can be concluded from these combined experimental and theoretical studies? Chemical reactions between 2,4,6-tri-tert-butyl-1,3,5-triphosphinine 7 or its precursor compound tert-butylphosphaalkyne 3 with reactive Fe(0) complexes resulted in completely unexpected products, which found explanation only after very deep theoretical studies. First of all, the combination of 7 as an excellent π-acceptor with an electron-rich Fe(0) creates a unique situation for the electronic balance between these two components. Either a complete intramolecular electron transfer Fe → triphosphinine occurs, or the π-bound triphosphinine is activated by the metal to open the gate to a totally unexpected reactivity, that includes the exchange of ring elements between two parallel oriented P-heterocyclic π-ligands of a sandwich complex. As the results of advanced DFT calculations are not directly helpful for experimental chemists to understand such a process in a sense, which allows transformation of the gained knowledge into a concept for further studies, a closer view on the crucial steps of the reaction sequence that rearranges the proposed intermediate [(η4-2,4-di-tert-butyl-1,3-diphosphete)(η6-2,4,6-tri-tert-butyl-1,3,5-triphosphinine)Fe] 8 to form hexaphosphaferrocene 9 as the thermodynamically stable end product seems to be helpful.
As indicated by our earlier studies, a nucleophilic attack of a P-lone pair of one π-ligand on the electrophilic centers of the other P-heterocyclic π-ligand is reasonable [30]. Chemical reactivity of a P-lone pair of a coordinated unsaturated P-heterocycle far outside its main direction has been identified in more cases [33] and can be related to its high s-character which grants sufficient nucleophilicity of the lone pair in a wide range of angles around the P-atom. Due to different ring sizes, a P-lone pair of the 1,3-diphosphete ligand may overlap with the π*-orbitals at the positions of all ring elements of the 1,3,5-triphosphinine ligand of 8a, depending on the conformational state of the complex. That includes the phosphorus atoms but PP interligand interaction would cause a closer contact between the tert-butyl substituents of the two ring ligands. This orientation is therefore regarded as a competing process with a higher activation barrier. The version the other way round, where a P-lone pair of the 1,3,5-triphosphinine acts as a nucleophile with respect to the 1,3-diphosphete ligand (8a′) does not work at all. The radius of the cyclic π-electron system of the 1,3-diphosphete ligand is smaller than that ob the π-bound 1,3,5-triphosphinine and cannot be reached by the P-lone pairs of that ligand. (Scheme 8). As we learned from the calculations, the formation of the interligand PC bond after converting 8a(s) into 8c(t) represents the crucial step for removing a CR fragment from the 1,3,5-triphosphinine and integrating it into the 1,3-diphosphete to form the 1,3-diphospholyl and 1,2,4-triphospholyl ligands of 9a, respectively.
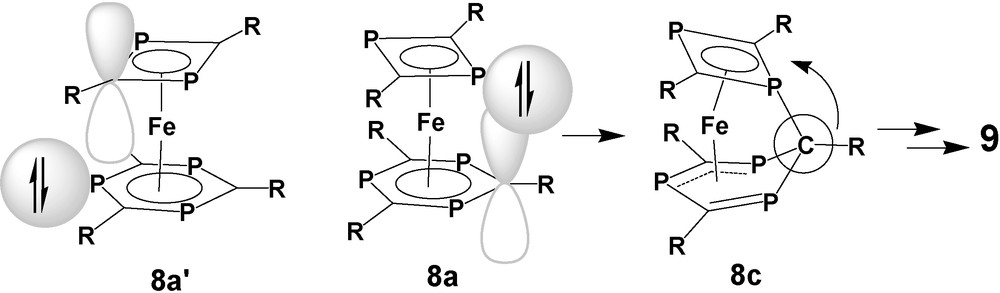
As a general essence of this study, we can state that a P-lone pair of the 1,3-diphosphete ligand may act like a harpoon to hit a CR fragment of the 1,3,5-triphosphinine, pull it out of the ring, and integrate it into its own structure.
Acknowledgements
This work was gratefully supported by the Deutsche Forschungsgemeinschaft DFG and the Fonds der Chemischen Industrie. We also thank COST action CM0802 “PhoSciNet” for supporting this study.
Appendix A Supplementary data
The complete set of calculation data of this study may be obtained free of charge by e-mail from the authors of this study: ulrich.zenneck@chemie.uni-erlangen.de or clark@chemie.uni-erlangen.de.